
Abstract
IgG4-related disease (IgG-RD) is capable of causing great morbidity and even mortality if the condition remains undiagnosed or poorly treated yet is typically a treatment-responsive disorder. Glucocorticoids have not been studied rigorously, and practices with regard to dosing and duration of treatment remain largely empiric. In addition, IgG4-RD patients are often particularly susceptible to and intolerant of the deleterious effects of glucocorticoid therapy. B cell depletion with anti-CD20 monoclonal antibodies appears to be a rapid, effective means of obtaining disease control and limiting patients’ glucocorticoid exposure, but this option is frequently available. Other therapies targeting the B cell lineage may also be efficacious, and one is under study. The means by which depletion or inhibition of B cells and their progeny ameliorate IgG4-RD are coming into focus now through careful mechanistic studies of samples from treated patients.
The mechanistic understanding of IgG4-RD will bring an array of specific targets for therapeutic intervention. Plasmablast-directed therapy with a CD19 monoclonal antibody is currently in clinical trials. CD4+ cytotoxic T lymphocytes and fibrosis, both observed nearly universally in the tissue of IgG4-RD patients, present two unexploited vulnerabilities in controlling and even reversing the effects of the disease. Further development of such therapies is a major goal for the next few years.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- IgG4-related Disease
- Obtain Disease Control
- Glucocorticoid
- Tubulointerstitial Nephritis
- Autoimmune Pancreatitis
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
IgG4-related disease (IgG4-RD) is an immune-mediated, fibroinflammatory condition that can affect nearly any organ system [1]. The biliary tract is often targeted, typically in association with type 1 autoimmune pancreatitis. The first line of therapy for IgG4-RD in general and for hepato-pancreatico-biliary disease specifically has been glucocorticoids. Glucocorticoids generally induce remission in a high proportion of patients with IgG4-related hepato-pancreatico-biliary disease but have not been investigated thoroughly in randomized controlled trials. The application of systemic glucocorticoids to patients fitting the typical demographic profile of IgG4-RD—namely, middle-aged to elderly individuals, often prone to other comorbidities—is often problematic. Moreover, given the predilection of patients with IgG4-related sclerosing cholangitis to have simultaneous pancreatic dysfunction, treatment with glucocorticoids poses other challenges in the context of glucocorticoid-induced diabetes.
To date, although disease-modifying anti-rheumatic drugs (DMARDs) are often used in the hope of reducing glucocorticoid dependency, there is a paucity of data to suggest that these medications actually provide any benefit beyond that which is observed with glucocorticoids alone. All of the above considerations underscore the importance of exploring new approaches to the treatment of IgG4-related sclerosing cholangitis. Particularly desirable would be the development of medical therapies rooted in a firm understanding of disease pathophysiology. In this regard, the potential for devising new treatment approaches is bright, for even over the short period of time in which IgG4-RD and its associated hepato-pancreatico-biliary disease have been known to exist, much has been learned about the pathophysiology of this condition.
In this chapter, we consider possibilities for new medical treatment approaches to IgG4-related sclerosing cholangitis, based on the current understanding of its pathophysiologic features. We begin with an overview of the current state of IgG4-RD treatment as it relates to glucocorticoids and non-biologic DMARDs.
Glucocorticoids in IgG4-RD
Glucocorticoids are typically employed in both remission induction and remission maintenance modes in IgG4-RD.
Glucocorticoids for the Induction of Remission
Glucocorticoids are highly effective agents for establishing prompt disease control in IgG4-RD. Patients’ response to glucocorticoids—generally on the order of 40 mg/day of prednisone—is swift and leads to disease remission in the majority of cases, particularly if the drugs are employed in moderately high doses for a period of 4–8 weeks. A retrospective, multicenter study in 25 IgG4-RD patients in France demonstrated that a starting daily dose of prednisone of approximately 47 mg, equating to 0.67 mg/kg for a 70 kg patient, was effective in controlling the disease in 90% of patients [2]. The investigators in that study defined treatment response as the presence of at least two of the following features: improved clinical status, reduction in serum IgG4 concentration, and improved radiologic findings. Even higher response rates have been reported in autoimmune pancreatitis [3,4,5]. Another retrospective study examined the effect of prednisone in 30 patients with IgG4-related sclerosing cholangitis and found that 97% of patients experienced either improvement or resolution of strictures and liver function tests on treatment [6]. Such studies support the use of glucocorticoids as a cornerstone of remission induction efforts [7]. A number of important caveats to the use of glucocorticoids exist, however. These are discussed below, under Glucocorticoid-Related Side Effects.
Glucocorticoids for Remission Maintenance
A widely used regimen for the initiation of glucocorticoids is a 2–4-week course followed by a gradual taper [7]. Some studies have employed a taper of 5 mg per week discontinuation [2, 3, 6]. Another regimen includes tapering by 10 mg every 2 weeks until the achievement of a daily dose of 20 mg, continuing 20 mg/day for 2 weeks, and then continuing to taper by 5 mg every 2 weeks discontinuation [7]. Whereas Japanese clinicians often continue prednisone at a low to moderate dose (2.5–10.0 mg daily) for up to several years, the practice in North America is to taper the glucocorticoid completely off within 2–3 months [8].
Glucocorticoid-Related Side Effects
Two major issues with regard to glucocorticoid therapy are pertinent. First, baseline comorbidities and frailties often make IgG4-RD patients poor candidates for long-term glucocorticoid therapy. A substantial proportion of patients with IgG4-RD, usually a disease of middle-aged to elderly individuals, suffer at baseline from obesity, glucose intolerance, hypertension, osteoporosis, and other relative contraindications to prolonged glucocorticoid courses. Moreover, autoimmune pancreatitis often leads to endocrine as well as to exocrine insufficiency, further complicating glucocorticoid treatment.
A single-arm prospective trial of glucocorticoid treatment alone from Japan maintained patients on doses of prednisone between 5 mg/day to somewhat higher than 10 mg/day. The duration of follow-up in that trial was 1 year. That trial reported disease control in 67% of patients, but 28% developed either new diabetes or exacerbations of previously known diabetes, and there were a variety of other serious complications of long-term glucocorticoid treatment, including infections [9]. In the cohort of patients from France described above, 67% of patients experienced side effects from glucocorticoid therapy [2]. Thus, the comorbid conditions of each patient and the potential for glucocorticoid intolerance must be considered on an individual basis when deciding on the suitability of treatment, as well as the initial dose and duration of glucocorticoid therapy. To date, no study has a starting dose of prednisone calculated to control the disease and then followed prospectively through a prescribed prednisone taper to discontinuation of the medicine.
The second major point of relevance with regard to glucocorticoids is that although only a minority of patients fail to respond to glucocorticoid treatment, a large percentage relapse during or after the glucocorticoid taper. Between 30% and 60% of patients relapse within 3 months of discontinuing glucocorticoid monotherapy in the absence of remission maintenance therapy [3, 5]. Even with low-dose maintenance glucocorticoids studied retrospectively in patients with autoimmune pancreatitis, 23% relapsed while on treatment [5]. Thus, the substantial risk of adverse effects from glucocorticoids and their failure to provide sustained disease control at doses that are tolerable from the perspective of safety are major inducements to the search for new therapies.
Conventional DMARDs in IgG4-RD
A group of international IgG4-RD experts collaborating on a 2015 Consensus Guidance Statement on the Management and Treatment of this condition concluded that few data support the use of conventional steroid-sparing agents in IgG4-RD [7]. In the largest study of therapy published to date, in fact, no benefit of adding a DMARD to glucocorticoids was observed in terms of relapse-free survival [10]. The enthusiasm of these authors for DMARD therapy as potential steroid-sparing agents is low.
A Consideration of IgG4-RD Pathophysiology
Overview
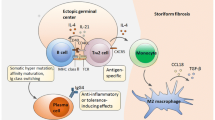
The pathophysiology of IgG4-RD involves a series of interactions between various cells of the B and T cell lineages, including B cells, plasmablasts, plasma cells, CD4+ CTLs, and follicular helper T (Tfh) cells—in addition to communications among myofibroblasts, macrophages, and eosinophils. The sum of these pathways leads to the histopathological findings characteristic of IgG4-RD in essentially every organ system: a lymphoplasmacytic infiltrate, frequent mild to moderate tissue (as well as peripheral) eosinophilia, obliterative phlebitis (and occasionally arteritis), and storiform fibrosis.
Concept of a Th2-Mediated Pathophysiology is Out of Favor
The original notion that IgG4-RD is a Th2-mediated disease has now been largely debunked. Circulating CD4+ GATA3+ T lymphocytes produce stereotypic Th2 cytokines (IL-4, IL-5, IL-13) only in patients who have longstanding atopic histories antecedent to their clinically evident IgG4-RD [11]. Moreover, the expansions of Th2 cells taken from the blood of patients with IgG4-RD are polyclonal, a reflection of their lifetime exposure to environmental allergens rather implicating a response to a specific triggering antigen [11].
Plasmablasts and the Cells Driving the Class Switch: T Follicular Helper Cells
Plasmablasts, defined by cell surface expression of CD19, CD27, and CD38 but negative for CD20, are dramatically elevated in the peripheral blood of patients with IgG4-RD who have not received treatment [12, 13]. These plasmablasts are oligoclonally expanded, express IgG4, and show intense somatic hypermutation [12]. They also respond swiftly to B cell depletion and demonstrate clonal divergence upon reconstitution. A subset of Tfh cells appears to drive the class switch within germinal centers through the elaboration of IL-4.
The Linchpin: A CD4+ Cytotoxic T Lymphocyte?
Both the peripheral blood and affected tissues of patients with IgG4-RD demonstrate oligoclonal proliferations of a novel CD4+ effector-memory T cell [14]. These effector-memory T cells demonstrate both modified Th1 cytotoxic lymphocyte signatures and myeloid cells signatures [14]. These cytotoxic T lymphocyte (CD4 CTL) cells demonstrate striking oligoclonal expansion by next-generation sequencing [14]. The implication is that in any given patient, IgG4-RD is likely to be triggered by an antigen—or perhaps a collection of antigens, different from patient to patient—that stimulates the CD4 CTL expansion observed. These CD4+ CTLs have been shown to elaborate a number of powerful cytokines known to mediate fibrosis, namely, transforming growth factor beta, interferon gamma, and interleukin-1 beta. These cells could therefore serve as the primary driver of the storiform fibrosis that is such an important part of the pathology of IgG4-RD.
What of the IgG4 Molecule Itself?
Orthodox thinking about IgG4-RD has always held that IgG4 differs from other IgG subclasses by its relative inability to fix complement (at least via the classical pathway of complement activation) or bind Fc receptors and that it is therefore better suited to a counterregulatory immune response. Concordant with this traditional thinking about IgG4, once postulated to be at the center of the disease with regard to pathophysiology, this molecule is now believed (somewhat ironically) to subserve a far less important role than the one originally conceived for it in IgG4-RD. The principal role of IgG4 in IgG4-RD appears to lie not in an association with the primary immune response but rather in an ineffectual effort to suppress the primary response.
In summary, the current model of pathophysiology not only explains in large measure the known efficacy of certain medications (e.g., glucocorticoids and rituximab), it also provides the rationale for other potential treatments. In this context, we discuss current and future treatment options for patients with IgG4-RD.
B Cell-Targeted Therapy in IgG4-RD
The discovery of oligoclonally expanded plasmablasts in patients with IgG4-RD [12] and their correlation with disease activity [13] elucidate further how targeting cells of the B cell lineage might work in IgG4-RD (and other diseases). Plasmablasts, circulating plasma cells that arise from activated CD20+ B cells, are antibody-secreting cells and typically develop into tissue-based plasma cells. Rituximab functions via antibody-dependent cell-mediated cytotoxicity, leading directly to B cell depletion, thereby eliminating plasmablasts’ progenitors. Despite lacking CD20, plasmablasts decline quickly following rituximab administration. The decline of these cells—and perhaps more importantly their recurrent rise over time in some patients—correlates better with disease activity than does the serum IgG4 concentration [12].
The potential utility of rituximab in IgG4-RD was demonstrated initially in case series [15, 16] and then in a prospective, open-label trial involving 30 patients [17]. Seventy-seven percent of the patients enrolled achieved the primary outcome in the trial, defined as a decline in the IgG4-RD responder index (IgG4-RD RI) of ≥2 points, no disease flares before 6 months, and no glucocorticoid use between months 2 and 6. Twenty-six of the 30 patients enrolled were treated without glucocorticoids, yet 29 of 30 a therapeutic response. Moreover, 47% achieved a complete remission at 6 months, defined by an IgG4-RD RI of 0 and no additional glucocorticoid treatment [17].
In addition to interfering with antigen presentation by plasmablasts, B cell depletion may also achieve its effect through the reduction of immune complex formation. The potential importance of immune complex formation as a possible disease mechanism has yet to be studied thoroughly in IgG4-RD. This may be particularly relevant for those patients with hypocomplementemia and the associated manifestation of tubulointerstitial nephritis (TIN) [10]. The phenomenon of immune complex formation in IgG4-related disease remains incompletely understood, yet seems to be operative in some organ manifestations—particularly IgG4-related TIN. IgG4 does not bind complement well under most circumstances but other IgG subclasses that are often elevated to a lesser but still substantial extent in IgG4-RD. As examples, elevations in IgG1 and IgG3 might easily account for this observation. Moreover, the mannose-binding lectin pathway of complement activation is a possible mechanism whereby IgG4 could also trigger this phenomenon itself.
Treatment with rituximab usually leads to symptomatic improvement within 1 month, a swift decline in serum IgG4 concentrations, and the ability to discontinue glucocorticoids entirely within a few weeks in most patients [15,16,17]. Blood plasmablasts have some utility for monitoring disease activity and gauging the need for potential retreatment [13] but are more likely to be elevated to a striking degree in patients who have never been treated before.
Plasmablasts as a Target of Treatment
Therapies targeting plasmablasts may offer a more specific approach to treating IgG4-RD. XmAb5871, a monoclonal antibody (homodimer) with a high-affinity variable region binding to CD19 and an enhanced Fc domain that binds to the FcγRIIb inhibitory receptor of B cells, is currently in phase II development for IgG4-RD treatment. This nondepleting anti-CD19 therapy has been studied in phase 1 trials and mechanistic studies in both rheumatoid arthritis [18] and systemic lupus erythematosus [19]. The rapid on/off effect of Xmab5871, its fully humanized structure, and its status as a nondepleting antibody may pose potential advantages over rituximab.
Future Therapeutic Directions
CD4+ CTL-Directed Treatments
Oligoclonally expanded CD4+ effector-memory T cells with a cytotoxic phenotype (CD4+ CTLs) have been identified and characterized recently in IgG4-RD [14]. The clonal expansion, pro-fibrotic phenotype, and correlation to disease activity of these cells are consistent with a central role in the pathophysiology of IgG4-RD. These cells express SLAMF7, IL-1β, TGF-β1, granzyme B, and perforin. Despite their lack of CD20 expression, the concentrations and percentages of the overall T cell pool of these novel CD4+ CTLs decreased substantially following rituximab administration. The responsiveness to CD20-targeted B cell depletion is theoretically related to the interference of T and B cell collaboration as plasmablasts present antigen and activate effector-memory CD4 CTLs at the site of inflammation.
Anti-fibrosis Therapies
Some IgG4-RD patients have a substantial burden of fibrosis even at the time of diagnosis. The fibrotic features are unlikely to respond to the currently available therapies and are therefore in great need of therapies designed specifically to address fibrosis. Some data indicate that the fibrosis of IgG4-RD may in many cases be at least partially reversible. A decrease in both circulating markers of fibrosis and myofibroblast activation in the affected tissue following rituximab therapy has been observed [20]. Evidence also comes from both studies of posttreatment tissue samples in the laboratory [20] and from serial clinical evaluations—particularly chest imaging—of patients following the institution of treatment. The impact on fibrosis of Xmab5871 and potential future therapies such as those directed against the CD4+ CTL remain uncertain at the moment, but such effects will be a key aspect of the evaluation of any new treatment agent.
Conclusion
The rapid progress in understanding the pathophysiology of IgG4-RD has led to several exciting mechanism-based therapies for IgG4-related sclerosing cholangitis. These include B cell depletion, a first-in-class homodimer targeting both CD19 and FcγRIIb, and the possibility of directing therapy against a novel CD4+ CTL that may be at the heart of this condition. Other potential therapeutic approaches will certainly emerge as our understanding of the pathophysiology of IgG4-RD becomes even more detailed.
References
Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012;366(6):539–51.
Ebbo M, Daniel L, Pavic M, et al. IgG4-related systemic disease: features and treatment response in a French cohort: results of a multicenter registry. Medicine (Baltimore). 2012;91:49–56.
Raina A, Yadav D, Krasinskas A, et al. Evaluation and Management of Autoimmune Pancreatitis: experience at a large US Center. Am J Gastroenterol. 2009;104:2295–306.
Chari S, Smyrk T, Levy M. Diagnosis of autoimmune pancreatitis: the Mayo Clinic experience. Clin Gastroenterol Hepatol. 2006;4:1010–6.
Kamisawa T, Shimosegawa T, Okazaki K. Standard steroid treatment for autoimmune pancreatitis. Gut. 2009;58:1504–7.
Ghazale A, Chari S, Zhang L, et al. Immunoglobulin G4-associated cholangitis: clinical profile and response to therapy. Gastroenterology. 2008;134:706–15.
Khosroshahi A, Wallace ZA, Crowe JL, Akamizu T, Azumi A, Carruthers MN, et al. International consensus guidance statement on the management and treatment of IgG4-related disease. Arthritis Rheum. 2015;67:1688–99.
Kamisawa T, Okazaki K, Kawa S, et al. Amendment of the Japanese consensus guidelines for autoimmune pancreatitis, 2013 III. Treatment and prognosis of autoimmune pancreatitis. J Gastroenterol. 2014;49:961–70.
Masaki Y for the All-Japan IgG4-RD Team; abstract presentation at the 2014 Second International Symposium on IgG4-Related Disease and Associated Conditions.
Hart PA, Topazian MD, Witzig TE, Clain JE, Gleeson FC, Klebig RR, et al. Treatment of relapsing autoimmune pancreatitis with immunomodulators and rituximab: the Mayo Clinic experience. Gut. 2013;62:1607–15.
Mattoo H, Della-Torre E, Mahajan VS, Stone JH, Pillai S. Circulating Th2 memory cells in IgG4-related disease are restricted to a defined subset of subjects with atopy. Allergy. 2014;69:399–402.
Mattoo H, Mahajan V, Della-Torre E, et al. De novo oligoclonal expansions of circulating plasmablasts in active and relapsing IgG4-related disease. J Allergy Clin Immunol. 2014;134:679–87.
Wallace ZS, Mattoo H, Carruthers M, Mahajan VS, Della Torre E, Lee H, Kulikova M, Deshpande V, Pillai S, Stone JH. Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Ann Rheum Dis. 2015;74:190–5.
Mattoo H, Mahajan VS, Maehara T, Deshpande V, Della-Torre E, Wallace ZS, et al. Clonal expansion of CD4+ cytotoxic T lymphocytes in patients with IgG4-related disease. J Allergy Clin Immunol. 2016;138(3):825–38.
Khosroshahi A, Carruthers MN, Deshpande V, et al. Rituximab for the treatment of IgG4-related disease lessons from 10 consecutive patients. Medicine. 2012;91:57–66.
Khosroshahi A, Bloch D, Deshpande V, et al. Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG-related systemic disease. Arthritis Rheum. 2010;62:1755–62.
Carruthers M, Topazian M, Khosroshahi A, et al. Rituximab for IgG4-related disease: a prospective, open-label trial. Ann Rheum Dis. 2015;74:1171–7.
Chu SY, Yeter K, Kotha R, Pong E, Miranda Y, Phung S, Chen H, et al. Suppression of rheumatoid arthritis B cells by XmAb5871, an anti-CD19 antibody that coengages B cell antigen receptor complex and Fcγ receptor IIb inhibitory receptor. Arthritis Rheum. 2014;66(5):1153–64.
Horton HM, Chu SY, Ortiz EC, Pong E, Cemerski S, Leung IW, Jacob N, et al. Antibody-mediated coengagement of FcγRIIb and B cell receptor complex suppresses humoral immunity in systemic lupus erythematosus. J Immunol. 2011;186(7):4223–33.
Della-Torre E, Feeney E, Deshpande V, Mattoo H, Mahajan V, et al. B-cell depletion attenuates serological biomarkers of fibrosis and myofibroblast activation in IgG4-related disease. Ann Rheum Dis. 2015;74(12):2236–43.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Science+Business Media Singapore
About this chapter

Cite this chapter
Perugino, C.A., Stone, J.H. (2019). Pathophysiology-Based Approaches to Treatment. In: Kamisawa, T., Kim, MH. (eds) IgG4-Related Sclerosing Cholangitis. Springer, Singapore. https://doi.org/10.1007/978-981-10-4548-6_22
Download citation
DOI: https://doi.org/10.1007/978-981-10-4548-6_22
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-4547-9
Online ISBN: 978-981-10-4548-6
eBook Packages: MedicineMedicine (R0)