
Abstract
Background
Lynch syndrome (LS) is associated with germline mutations in one of the mismatch repair genes or EPCAM. The majority of the causative alterations are point mutations. Large genomic rearrangements represent only 5–20%. Hypothetically, the allelic imbalance, like the loss of heterozygosity, may be another high penetrance risk factor.
Case presentation
We describe the case of a patient who developed 5 tumors during her lifetime and with a family history characterized by a high frequency of tumors associated with LS. The proband was tested for mutations and copy number alterations with a panel of hereditary cancer genes and by SNP array. She showed a 187 Kb duplication including EPCAM and the first 7 exons of MSH2, plus two loss of heterozygosity (LOHs) in chromosome 20 and one in chromosome X which include many tumor suppressor genes.
Conclusion
We found a novel large EPCAM-MSH2 duplication associated with LS and the presence of LOHs in regions containing numerous tumor suppressors, raising the hypothesis that these alterations could contribute to cancer susceptibility. Our results underline the importance to deepen the knowledge of molecular mechanisms in order to determine the role in cancer predisposition of novel genetic alterations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Lynch syndrome (LS) is an autosomal dominant disorder due to the presence of germline mutations in the mismatch repair (MMR) genes MSH2, MSH6, MLH1, PMS2, and also EPCAM. The majority of the causative mutations so far identified in LS patients are missense variants, nonsense variants, and small frameshift insertions/deletions [1].
Large genomic rearrangements are less frequent than point mutations, with a relative incidence in LS families that vary from 5 to 20%, most of which are deletions, and are distributed predominantly in MSH2 [2]. Mutations or copy number variations in MMR genes, however, cover only 68% of patients who fulfill the Amsterdam and Bethesda criteria [3, 4].
Allelic imbalance, such as the loss of heterozygosity (LOH), can be considered a possible risk factor. Several chromosomal regions have been linked to colorectal cancer (CRC) susceptibility in terms of copy number loss or gain, in particular in chromosomes 20 and 18 [2, 5]. The presence of many tumor suppressor genes in these regions can probably explain the association with the disease [6].
In this study, we describe the case of a patient harboring an EPCAM-MSH2 duplication never reported in the literature that confirmed the LS diagnosis, and also two LOHs on chromosome 20 and one on chromosome X, which could contribute to the high susceptibility to cancer detected in this family.
Case presentation
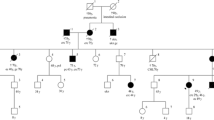
The first family member, recruited by our counseling service at the Cancer Prevention Unit of Morgagni-Pierantoni Hospital in Forlì, was a 52-year-old woman (II-1) (Fig. 1A).

A Pedigree of the family with the EPCAM-MSH2 duplication. Circles represent females and squares represent males. Symbols with a slash indicate deceased individuals. The arrow points to the proband. Under the symbols are reported the current age, the age at cancer onset, and cancer type, when available. B EpCAM expression in tumor and normal tissues: (a) proband (II-1), (b) IRST03 CRC sample (control), (c) IRST02 CRC sample (control), and (d) SKBR3 cell line used as positive control. Staining intensity (SI) is reported inside the single pictures
She was diagnosed with a sigmoid colon adenocarcinoma and an ascending colon adenocarcinoma at the age of 37 and 41, respectively. A year later, she developed an endometrial adenocarcinoma. At the age of 45, she had an invasive duodenal adenocarcinoma of the intestinal type, and at the age of 52, an invasive ductal breast carcinoma, moderately differentiated.
The proband reported in anamnesis that her mother (I-4) died at the age of 77 of an unspecified neoplasia, and that also one maternal and two paternal aunts (I-1, I-2, and I-5) died for unspecified neoplasias.
The proband had two sisters deceased at 43 and 40 years (II-3 and II-5) of colon cancer, a sister deceased at the age of 13 due to unknown causes (II-6), and a sister deceased of pancreatic cancer at 47 years (II-4), whose son (III-3) died for a colon cancer at 27 years. The proband’s brother (II-2) had a colon cancer at the age of 55. The proband’s daughter (III-1) and son (III-2) never had cancer.
The proband was tested for microsatellite instability (MSI) and immunohistochemistry (IHC) expression analysis of MMR as indicated by guidelines for LS [3, 4]. The analysis revealed a high MSI, and the IHC showed the absence of both MSH2 and MSH6 proteins in the tumor tissue specimens of the ascending colon cancer.
The next-generation sequencing (NGS) analysis was performed using the enrichment protocol TruSight Cancer (Illumina) for simultaneous sequencing of a panel of 94 genes involved in the main hereditary cancer syndromes while the copy number variation analysis was performed using the enrichment protocol Hereditary Cancer Solution (HCS) (SOPHiA GENETICS) which analyze 27 most clinically relevant genes associated with LS and intestinal polyposis syndromes (Supplementary Material S1).
The bioinformatic analysis revealed the presence of 4 variants with a frequency in the population lower than 0.01 or unknown (Esp6500, 1000genomes, and Exac03); 3 missense variants in APC (c.3386T>C p.Leu1129Ser), EGFR (c.3419A>G p.Asn1140Ser), and FANCA (c.3430C>T p.Arg1144Trp) genes; and 1 synonymous variant in PTCH1 gene (c.4080C>T p.Ser1360Ser).
According to the American College of Medical Genetics (ACMG) guidelines, only the EGFR and FANCA variants are classified as variants with uncertain significance, while the others are reported as benign or likely benign. None of the variants are reported as pathogenic in ClinVar (www.ncbi.nlm.nih.gov/clinvar/).
The analysis revealed also a copy number variation of all 9 exons of EPCAM and 1–7 exons of MSH2, described as the presence of three copies.
In order to define the size of the duplication, we performed a SNP array profile assay, carried out according to the manufacturer’s protocol (GRCH37/hg19 Plus 2.0 GeneChip arrays, Thermo Fisher). The proband shows a gain of copies in chromosome 2 of 184 Kb (2p21, 47482263-47666308) which includes the EPCAM, MIR559, and MSH2 genes.
The proband also showed two large LOHs in chromosome 20 (20p13p11.1: 3771857-26289925; hmz and 20q11.21q11.23: 29448795-36388176; hmz), and a LOH in chromosome X (Xq11.1q12: 61932503-66974524; hmz).
The copy number variation (CNV) analysis and the SNP array profile were performed also on the proband’s daughter (III-1), while the proband’s son and brother did not give the consent to be tested. The results of the analysis did not reveal the presence of rare variants or of the EPCAM-MSH2 duplication described in II-1, as confirmed by the SNP array profile. She showed a LOH at q11.1 (61932503-67733784) and one in q13.1 (70604117-76584634) of chromosome X.
In order to verify the expression of EpCAM in tumor and normal tissues, we performed an IHC assay using the Ventana Benchmark Ultra Full System. The same FFPE specimens used for MMR expression analysis and cell block derived from SKBR3 cell line (breast cancer) as positive control were used. The EpCAM staining intensity (SI) (i.e., 0, 1+, 2+, 3+) in tumor tissue was evaluated as 3+, while in the normal tissue was 2+/3+ (Fig. 1B).
We compared the tissue expression level of EpCAM of the proband with other 6 CRC samples: one LS, a Muir-Torre syndrome, a Lynch-like syndrome which are two subtypes of the Lynch syndrome, two familial, and one sporadic CRC. All samples showed a level of expression of EpCAM similar to the proband.
Discussion and conclusion
We describe the family history and the genetic characterization of the 54-year-old female patient who developed five primary malignant tumors during her lifetime and which presented a family history characterized by particular recurrence of aggressive neoplasias at a young age.
The proband was carrier of a germline duplication of a 184 Kb region on chromosome 2 that includes the entire EPCAM gene and the first 7 exons of MSH2 gene, never reported previously in the literature. In LS, large genomic rearrangements are less frequent than point mutations, representing only the 5–20% of all alterations. They become even less frequent if we consider only duplications. In our case, the gain of copies of the 7 exons of MSH2 explained the loss of expression of MSH2 protein in IHC and confirmed the LS diagnosis.
The presence of 5 primary tumors in the patient plus the family history suggest the presence of some other factor that increases the susceptibility to tumors besides the malfunctioning of the MMR genes. We excluded the possible involvement of EpCAM protein overexpression in predisposing the onset of tumor because EpCAM expression resulted to be very high in both normal and tumor tissue in all analyzed cases. We found two large LOHs in chromosome 20 and one in chromosome X, which include many tumor suppressor genes involved in carcinogenesis. The two LOHs in chromosome 20 include genes as the proliferating cell nuclear antigen (PCNA), Ras association domain family member 2 (RASSF2), bone morphogenetic protein 2 (BMP2), and ovo-like zinc finger 2 (OVOL2). PCNA plays multiple roles in MMR, including the promotion of MSH2-MSH6 complex binding to the mispaired bases [7] and activates the MutLα endonuclease [8] assuming a role as central coordinator of the MMR. RASSF2 is considered a tumor suppressor gene in CRC [9] because it regulates Ras signaling. In vitro and in vivo studies showed the role of BMP2 as tumor suppressor, because its expression inhibits proliferation and migration in CRC and induces apoptosis [10]. OVOL2 is a tumor suppressor in colon cancer by the inhibition of WNT signaling and tumor progression [11]. Evidence already suggested that the WNT pathway has particular relevance in CRCs because its activation seems to be the key oncogenic driver in most CRCs [12].
The LOH in chromosome X includes AMER1. Recently, some studies are highlighting its possible involvement in cancer development as a tumor suppressor gene, particularly in colon cancer.
The daughter was not a carrier of the EPCAM-MSH2 duplication, but she showed two LOHs in chromosome X. The LOH in q11.1 includes the same genes of the proband LOH plus one (oligophrenin 1 (OPHN1)). The LOH in q13.1 is unique for the daughter; it contains 26 genes but none is known to be a tumor suppressor or related to CRC or LS.
In conclusion, we found a novel EPCAM-MSH2 duplication associated with LS. We also found LOHs in genetic regions that include tumor suppressor genes. The impossibility of recovering the samples of all the family members prevents us from confirming our hypotheses with force, but we can hypothesize that the presence of the three large LOHs can have an effect on the incidence of cancer in this family, maybe amplifying the effects of the causative mutation. Further investigation is needed to confirm it.
This underlines the need to deepen the knowledge of molecular mechanisms in order to determine the role in cancer predisposition of novel genetic alterations.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
InSiGHT-Variant database https://www.insight-group.org/variants/databases/ (archived on 3 May 2017)
Therkildsen C, Jönsson G, Dominguez-Valentin M et al (2013) Gain of chromosomal region 20q and loss of 18 discriminates between Lynch syndrome and familial colorectal cancer. Eur J Cancer 49:1226–1235. https://doi.org/10.1016/j.ejca.2012.11.011
Vasen HF, Watson P, Mecklin JP, Lynch HT (1999) New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 116:1453–1456
Umar A, Richard Boland C, Terdiman JP et al (2004) Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer 18:261–268
Cancer T, Atlas G (2012) Comprehensive molecular characterization of human colon and rectal cancer. Nature 487:330–337. https://doi.org/10.1038/nature11252
Peng Z, Zhou C, Zhang F, Ling Y, Tang H, Bai S, Liu W, Qiu GHL (2002) Loss of heterozygosity of chromosome 20 in sporadic colorectal cancer. Chin Med J 115(10):1529–1532
Flores-Rozas H, Clark D, Kolodner RD (2000) Proliferating cell nuclear antigen and Msh2p-Msh6p interact to form an active mispair recognition complex. Nat Genet 26:375
Genschel J, Kadyrova LY, Iyer RR, Dahal BK, Kadyrov FA, Modrich P (2017) Interaction of proliferating cell nuclear antigen with PMS2 is required for MutLα activation and function in mismatch repair. Proc Natl Acad Sci 114:4930–4935. https://doi.org/10.1073/pnas.1702561114
Akino K, Toyota M, Suzuki H, Mita H, Sasaki Y, Ohe-Toyota M, Issa JP, Hinoda Y, Imai K, Tokino T (2005) The ras effector RASSF2 is a novel tumor-suppressor gene in human colorectal cancer. Gastroenterology 129:156–169. https://doi.org/10.1053/j.gastro.2005.03.051
Zhang Y, Chen X, Qiao MIN et al (2014) Bone morphogenetic protein 2 inhibits the proliferation and growth of human colorectal cancer cells:1013–1020. https://doi.org/10.3892/or.2014.3308
Ye G, Sun G, Jiao P et al (2016) OVOL2, an inhibitor of WNT signaling, reduces invasive activities of human and mouse cancer cells and is down-regulated in human colorectal tumors. 2:659–671. https://doi.org/10.1053/j.gastro.2015.11.041
Schatoff EM, Leach BI, Dow LE (2018) HHS Public Access 13:101–110. https://doi.org/10.1007/s11888-017-0354-9.Wnt
Author information
Authors and Affiliations
Contributions
Conception and design of the study: P.F.; supervision: C.D. and M.G.; writing—original draft preparation: P.F.; writing—review and editing: T.G., U.P, and C.D.; collection and analysis of the clinical data,: D.R., R.M., and F.F.; controls collection: D.M. G.; immunohistochemical analysis: M.M.T.; immunohistochemical result interpretation: P.M.; NGS methodology: C. I., Z.V., and F. A.; NGS data analysis: T.M. and V.S.; SNP array analysis: T. C. and S.V. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was performed in accordance with the principles of Good Clinical Practice and the ethical standards laid down in the Declaration of Helsinki and approved by the IRST Ethical Committee (CE IRST IRCCS-AVR, protocol 3030/2018)
Consent for publication
Written informed consent was obtained from participants of this case report.
Competing interest
The authors declare that they have no competing interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 20.8 kb)
Rights and permissions
About this article

Cite this article
Pirini, F., Tedaldi, G., Danesi, R. et al. Identification of a novel large EPCAM-MSH2 duplication, concurrently with LOHs in chromosome 20 and X, in a family with Lynch syndrome. Int J Colorectal Dis 34, 1999–2002 (2019). https://doi.org/10.1007/s00384-019-03414-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00384-019-03414-y