
Abstract
Purpose
Pancreatic tumors are rare in children and limited data are available regarding incidence, treatment, and outcomes. We aim to describe patient and tumor characteristics and to report on survival of these diseases.
Methods
Children with pancreatic tumors were queried from the National Cancer Database (2004–2014). The association between treatment and hazard of death was assessed using Kaplan–Meier method and Cox regression model.
Results
We identified 109 children with pancreatic tumors; 52% were male and median age at diagnosis was 14 years. Tumors were distributed as follows: pseudopapillary neoplasm (30%), endocrine tumors (27%), pancreatoblastoma (16%), pancreatic adenocarcinoma (16%), sarcoma (6%) and neuroblastoma (5%). Seventy-nine patients underwent surgery, of which 76% achieved R0 resection. Most patients (85%) had lymph nodes examined, of which 22% had positive nodes. Five-year overall survival by tumor histology was 95% (pseudopapillary neoplasm), 75% (neuroblastoma), 70% (pancreatoblastoma), 51% (endocrine tumors), 43% (sarcoma), and 34% (adenocarcinoma). On multivariable analysis, surgical resection was the strongest predictor of survival (HR 0.26, 95% CI 0.10–0.68, p < 0.01).
Conclusion
Overall survival of children with pancreatic tumors is grim, with varying survival rates among different tumors. Surgical resection is associated with improved long-term survival.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pancreatic tumors in the pediatric population are extremely rare, with an estimated incidence of 1–2 cases per ten million children and young adults in the United States [1, 2]. Limited series have been reported, and even the largest children’s hospitals only report a handful of cases over a number of decades [3,4,5,6,7]. Pancreatic ductal adenocarcinoma is overwhelmingly the most common histology in adults. However, in children, a wide variety of tumors are encountered, which include both benign and malignant lesions. These neoplasms can be classified as exocrine vs. endocrine, epithelial vs. non-epithelial, cystic vs. solid, and benign vs. malignant lesions; and the pancreatic malignancies encompass a wide range of histologies that includes pancreatoblastoma, cystadenocarcinoma, sarcoma, acinar cell carcinoma, solid pseudopapillary neoplasms, endocrine tumors and ductal adenocarcinoma [1].
Current treatment options for pancreatic tumors include surgical management and medical treatments; however, a surgical approach is preferred as it is associated with long-term survival [8,9,10]. These infrequent tumors are extremely challenging to study due to the heterogeneity of treatments and lack of standardized guidelines. Our previous work with the Surveillance, Epidemiology, and End Results (SEER) database demonstrated that female gender and surgical treatment were independent predictors of survival [1]. Our current analysis examines the patterns of care and survival outcomes of pancreatic tumors in children from the National Cancer Database.
Methods
Data source
The National Cancer Database (NCDB) Participant User File (PUF) for pancreatic tumors in children (2004–2014) was reviewed. The NCDB is a nationwide hospital-based cancer registry sponsored by the American College of Surgeons Commission on Cancer and the American Cancer Society. The NCDB collects de-identified data (including patient demographics, clinical factors, treatments, and outcomes) of > 70% of newly diagnosed cancer cases. The Institutional Review Board at the University of Miami Miller School of Medicine deemed this retrospective study to be exempt from full review.
Patient sample
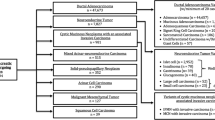
The study population included children (age ≤ 19 years) diagnosed with primary malignant pancreatic tumors. Tumor histology was defined according to the International Classification of Diseases for Oncology, 3rd revision (ICD-O-3) morphologic codes for pseudopapillary neoplasm (8452); neuroblastoma (9490, 9500); pancreatoblastoma (8971); endocrine tumors (8150, 8151, 8152, 8240, 8246, 8249); sarcomas (8806, 8854, 8920, 9364, 9473); and pancreatic adenocarcinoma (8000, 8010, 8140, 8500, 8550). Patients that underwent surgery were identified by surgery for primary site codes including local excision (25), distal pancreatectomy (30), pancreaticoduodenectomy (35, 36, 37), total pancreatectomy (40, 60, 70), and pancreatectomy NOS (80, 90). Extent of resection was recorded as microscopically margin negative (R0), or removal of all macroscopic disease but positive microscopic margins (R1). Cohorts were stratified according to treatment: surgery, chemotherapy, and chemoradiation therapy. Patients with missing survival data (n = 23) were excluded from survival analysis. The NCDB PUF 2014 data dictionary describes other variables used [11].
Statistical analysis
Descriptive statistics were calculated for patients’ characteristics using mean and standard deviation for normally distributed values, median and interquartile range for non-parametric data, frequencies, and percentages. The association of clinicopathologic factors among treatments was analyzed. Categorical variables were compared across groups using Chi-squared (χ2) or Fisher’s exact tests, as appropriate. Continuous variables were assessed using analysis of variance (ANOVA) for normally distributed data, or Kruskal–Wallis test for non-parametric distribution. Our primary endpoint was overall survival (OS), measured from the date of diagnosis until death. The Kaplan–Meier method was used to derive the survival curves estimates across treatments, and the log-rank test was used to make comparisons of the survival rates. Hazard ratios (HR) for mortality with 95% confidence intervals (CI) were calculated using the Cox proportional hazards model. Analyses were adjusted to the following covariates: age, sex, race/ethnicity, insurance status, histology, tumor size, tumor stage, and treatment, to determine any significant effect on OS. Non-significant variables were dropped one at a time, beginning with the least significant, and the models were reassessed. The fact that there were only 24 deaths limited the number of variables that could be used in a multivariable model without impacting model stability. Each analysis’ statistical significance was determined at an alpha level of 0.05 or less. Statistical analyses were carried out using SAS Software version 9.4 (SAS Institute Inc., Cary, NC).
Results
A total of 109 children with malignant pancreatic tumors were identified from 2004 to 2014. Median age at diagnosis was 14 years (IQR 10–16). Males and females were similarly represented (52% and 48%, respectively). Most patients were white non-hispanic (55%) and insured (92%). Neuroblastomas (median age < 1 year) and pancreatoblastomas (median age 6 years) were more common in younger children, whereas adenocarcinomas and pseudopapillary neoplasms were more common in older patients (median age 15 years, p < 0.001). The majority of patients with pseudopapillary neoplasms were female compared to other tumors (85% vs. 39%, p < 0.001) (Table 1).
Clinical and tumor characteristics
Tumors were more commonly located in the head/neck (31%) and tail (29%), followed by the body (13%) of the pancreas. Twenty-seven percent were categorized as overlapping regions of the pancreas. The median tumor size was 5 cm (range 0.2–24). Pseudopapillary neoplasms and endocrine tumors were most commonly located in the tail of the pancreas (37%) whereas adenocarcinomas and pancreatoblastomas more frequently involved overlapping regions of the pancreas (41%). Neuroblastomas and endocrine tumors were significantly smaller than other tumors (3.2 vs. 6.5 cm, p = 0.004). For the cases with tumor grade classification data available, most tumors were well-differentiated (27/40, 68%). The majority of children had stage I disease (39/71, 55%), followed by stage IV (22/71, 31%). Of note, 47% of patients with pancreatic adenocarcinoma had stage IV disease (Table 1).
Treatment characteristics
The majority of patients underwent surgery (79/109, 72%), and most achieved R0 microscopically negative margin resection (60/79, 76%). The most common surgical procedures were distal pancreatectomy (33/79, 42%) and pancreaticoduodenectomy (28/79, 35%). Of children that underwent surgical resection, 85% (67/79) had lymph nodes sampled, and 22% (15/67) had positive nodes. Females were more likely to undergo surgery than males (59% vs. 41%, p = 0.03). Pancreatic adenocarcinoma (71%) and pancreatoblastoma (47%) were less likely to be surgically resected compared to other tumors (p < 0.001). Fifteen of the 30 patients that did not undergo surgery received chemotherapy alone (50%), two patients received chemoradiation (7%), and for 13 patients the treatment was unknown (43%). Of the patients that did not undergo surgery, half had metastatic disease at time of diagnosis (Table 2).
Stage IV patients
There were 22 patients (20%) who were classified with stage IV disease. Histologic type was most often adenocarcinoma (36%) followed by endocrine (32%), pancreatoblastoma (18%), and pseudopapillary (14%). Surgery was performed on seven of these patients (32%), of whom four had endocrine tumors, two had pseudopapillary neoplasms, and one had adenocarcinoma. Overall, 41% of stage IV patients received chemotherapy alone, 18% received surgery alone, 14% received chemotherapy in addition to surgery, and 5% received chemoradiation therapy alone. The treatment status for 23% of these patients was unknown.
Survival analysis
The median follow-up was 38 months (IQR 18–70). Five-year OS for all pancreatic tumors in children was 64%. Males had significantly worse 5-year OS compared to females (50% vs. 79%, p = 0.028); however, this difference was no longer significant (46% vs. 54%, p = 0.401) after excluding pseudopapillary neoplasms from the analysis, which are much more prevalent in females and known to have less aggressive behavior. Patients that underwent surgical resection had improved 5-year OS compared to those that did not have surgery (78% vs. 33%, p < 0.0001); the median OS for children that did not have surgery was 24 months. Five-year OS rates by tumor histology were 95%, 75%, 70%, 51%, 43%, and 34% for pseudopapillary neoplasm, neuroblastoma, pancreatoblastoma, endocrine tumors, sarcoma and pancreatic adenocarcinoma, respectively (Fig. 1).

Overall survival in children with pancreatic tumors by histology
On univariate Cox proportional hazard model, variables associated with worse survival were male sex (HR 2.50, 95% CI 1.07–5.80, p = 0.03), histology of adenocarcinoma or pancreatoblastoma (HR 2.52, 95% CI 1.04–6.10, p = 0.03), and chemotherapy (HR 3.82, 95% CI 1.65–8.86, p < 0.01). On the other hand, surgical resection reduced the hazards of death (HR 0.21, 95% CI 0.09–0.46, p < 0.01). On multivariable analysis, surgery was the only predictor associated with longer survival (HR 0.26, 95% CI 0.10–0.68, p < 0.01) (Table 3).
Discussion
Pediatric pancreatic tumors are exceedingly rare and poorly described in the literature. Even the largest children’s hospitals report an average of only 1–2 patients per year [4, 5]. This study reports a national series of pediatric pancreatic tumors from 2004 to 2014 using the NCDB, which captures more than 70% of newly diagnosed cancer cases in the US. This resulted in identification of 109 cases during this 11-year period. The most recent analysis from the SEER database over a 41-year period (1973–2013) identified 114 cases of malignant pancreatic tumors [12]. This discrepancy in frequency may be due to an increasing incidence of pancreatic cancer or differential reporting between the two databases. We believe the larger number of cases per year in our study is due to the latter, as the NCDB encompasses a wider range of newly diagnosed cancer cases in the US.
In our series, the most common histologies were pseudopapillary neoplasms (30%), endocrine tumors (28%), adenocarcinoma (16%) and pancreatoblastoma (16%). The vast majority of patients with pseudopapillary neoplasms, sarcomas, endocrine tumors, and neuroblastomas underwent surgical resection while those with adenocarcinoma, and surprisingly pancreatoblastoma, were much less likely to be treated surgically. Overlapping lesions were highly prevalent in these patients, which may indicate high disease burden in most of the patients with adenocarcinoma (71%) and nearly half with pancreatoblastoma (43%) with overlapping lesions not pursuing surgery. In addition, our data indicate that nearly half of patients with adenocarcinoma had stage IV disease. Whereas the 5-year overall survival for adenocarcinoma was 34%, there were no survivors with stage IV disease. On the other hand, 23% of patients with pancreatoblastoma were identified as having stage IV disease, with a 5-year overall and stage IV survival for these patients of 70% and 25%, respectively. Although complete staging data were poorly reported for this histologic subtype, it remains unclear why so few cases of pancreatoblastoma (53%) received surgical treatment.
In our cohort, 15% of patients with solid pseudopapillary neoplasms were male, a rate similar to other reported series [13,14,15]. Although there are data to suggest that males with solid pseudopapillary neoplasm may have an atypically aggressive disease [16], this was not demonstrated in the current study. Of the five males with solid pseudopapillary neoplasms, there were three with survival data and all were alive. While 21% of patients with pseudopapillary neoplasms had advanced disease in this study, 5-year overall survival remained 95%, similar to other reports [15, 17]. Given that solid pseudopapillary neoplasms are known to have a more favorable biology and less aggressive clinical course compared to the other malignant neoplasms, we suspected that the much higher incidence of this tumor type in females might be responsible for the significantly improved survival observed for female patients in our series. After exclusion of pseudopapillary neoplasms from the survival analysis, we indeed found that 5-year overall survival was no longer significantly different for males and females. As observed in our series, the relatively higher frequency of solid pseudopapillary neoplasms among females may similarly account for the improved survival in female patients that has been reported in other series.
Endocrine tumors were the second most prevalent tumors. Interestingly, more than half of patients (57%) underwent surgical resection even in the presence of metastatic disease, and 23% received adjuvant chemotherapy. While surgery may have been indicated in these patients for compressive symptoms from local mass effect or symptoms of hormone secretion refractory to medical therapy [18], such information is not recorded in the NCDB.
Complete surgical resection is the only potential cure for patients with malignant pancreatic tumors; and therefore, patients with localized disease tend to be eligible for surgery. In certain tumor types (solid pseudopapillary neoplasm, pancreatoblastoma, endocrine tumors), resection even in the setting of metastatic disease may be beneficial [3, 19]. Therefore, it is not surprising that survival was significantly greater for patients who had surgery. On univariate analysis, surgical resection reduced the hazard of death (HR 0.21, 95% CI 0.09–0.46, p < 0.01) and was the only predictor associated with prolonged survival on multivariable analysis. However, given the lack of granular data in our study to understand why certain patients underwent surgery and others did not, it remains unclear if surgical intervention or simply eligibility for surgery is truly associated with improved survival.
There are limitations to this study that should be considered. Although this is one of the largest cohorts of pancreatic tumors in children, small sample size and high underreporting (12–25%) for some variables challenge the ability to generate robust estimates for survival. Similarly, the lacking clinical data (such as the presence of genetic syndromes, biomarkers, fine needle aspiration or biopsy information) could have helped to explain the approach to treatment. Lastly, recurrence information is not available in the database, which could have better defined oncologic outcomes for this patient population.
In conclusion, malignant pancreatic tumors are an uncommon and heterogeneous group of neoplastic lesions in the pediatric population. Surgical management with or without medical therapy is the preferred treatment for these tumors, often even in the setting of metastatic disease. In general, we found that a surgical approach was associated with improved long-term survival although the exact indications for surgery were unknown. While malignant tumors of the pancreas encompass a broad spectrum of diseases, building on series such as ours will hopefully provide sufficient clinical data for the foundation of consensus guidelines for the management of these very rare, heterogeneous neoplasms in children.
References
Perez EA, Gutierrez JC, Koniaris LG, Neville HL, Thompson WR, Sola JE (2009) Malignant pancreatic tumors: incidence and outcome in 58 pediatric patients. J Pediatr Surg 44(1):197–203
Brecht IB, Schneider DT, Kloppel G, von Schweinitz D, Barthlen W, Hamre MR (2011) Malignant pancreatic tumors in children and young adults: evaluation of 228 patients identified through the Surveillance, Epidemiology, and End Result (SEER) database. Klin Padiatr 223(6):341–345
Shorter NA, Glick RD, Klimstra DS, Brennan MF, Laquaglia MP (1967) Malignant pancreatic tumors in childhood and adolescence: the Memorial Sloan-Kettering experience, 1967 to present. J Pediatr Surg 37(6):887–892
Rojas Y, Warneke CL, Dhamne CA, Tsao K, Nuchtern JG, Lally KP et al (2012) Primary malignant pancreatic neoplasms in children and adolescents: a 20 year experience. J Pediatr Surg 47(12):2199–2204
Yu DC, Kozakewich HP, Perez-Atayde AR, Shamberger RC, Weldon CB (2009) Childhood pancreatic tumors: a single institution experience. J Pediatr Surg 44(12):2267–2272
Jaksic T, Yaman M, Thorner P, Wesson DK, Filler RM, Shandling B (1992) A 20-year review of pediatric pancreatic tumors. J Pediatr Surg 27(10):1315–1317
Nasher O, Hall NJ, Sebire NJ, de Coppi P, Pierro A (2015) Pancreatic tumours in children: diagnosis, treatment and outcome. Pediatr Surg Int 31(9):831–835
Sacco Casamassima MG, Gause CD, Goldstein SD, Abdullah F, Meoded A, Lukish JR et al (2016) Pancreatic surgery for tumors in children and adolescents. Pediatr Surg Int 32(8):779–788
Muller CO, Guerin F, Goldzmidt D, Fouquet V, Franchi-Abella S, Fabre M et al (2012) Pancreatic resections for solid or cystic pancreatic masses in children. J Pediatr Gastroenterol Nutr 54(3):369–373
Marchegiani G, Crippa S, Malleo G, Partelli S, Capelli P, Pederzoli P et al (2011) Surgical treatment of pancreatic tumors in childhood and adolescence: uncommon neoplasms with favorable outcome. Pancreatology 11(4):383–389
National Cancer Data Base Participant User File Data Dictionary (2014) https://www.facs.org/~/media/files/quality%20programs/cancer/ncdb/puf%20data%20dictionary%20version%20puf%202014.ashx. Accessed 26 Jan 2020
Mylonas KS, Nasioudis D, Tsilimigras DI, Doulamis IP, Masiakos PT, Kelleher CM (2018) A population-based analysis of a rare oncologic entity, malignant pancreatic tumors in children. J Pediatr Surg 53(4):647–652
Choi SH, Kim SM, Oh JT, Park JY, Seo JM, Lee SK (2006) Solid pseudopapillary tumor of the pancreas: a multicenter study of 23 pediatric cases. J Pediatr Surg 41(12):1992–1995
van den Akker M, Angelini P, Taylor G, Chami R, Gerstle JT, Gupta A (2012) Malignant pancreatic tumors in children: a single-institution series. J Pediatr Surg 47(4):681–687
Wright MJ, Javed AA, Saunders T, Zhu Y, Burkhart RA, Yu J et al (2019) Surgical resection of 78 pancreatic pseudopapillary tumors: a 30-year single institutional experience. J Gastrointest Surg. https://doi.org/10.1007/s11605-019-04252-7
Lin MY, Stabile BE (2010) Solid pseudopapillary neoplasm of the pancreas: a rare and atypically aggressive disease among male patients. Am Surg 76(10):1075–1078
Kim MJ, Choi DW, Choi SH, Heo JS, Sung JY (2014) Surgical treatment of solid pseudopapillary neoplasms of the pancreas and risk factors for malignancy. Br J Surg 101(10):1266–1271
Peranteau WH, Palladino AA, Bhatti TR, Becker SA, States LJ, Stanley CA et al (2013) The surgical management of insulinomas in children. J Pediatr Surg 48(12):2517–2524
Martin RC, Klimstra DS, Brennan MF, Conlon KC (2002) Solid-pseudopapillary tumor of the pancreas: a surgical enigma? Ann Surg Oncol 9(1):35–40
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
OP and EAP contributed to the study conception and design and performed the data collection and analysis; OP, AF, CZ, and KR prepared the manuscript; CMT, JES, and EAP provided critical revisions to the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest in this work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Picado, O., Ferrantella, A., Zabalo, C. et al. Treatment patterns and outcomes for pancreatic tumors in children: an analysis of the National Cancer Database. Pediatr Surg Int 36, 357–363 (2020). https://doi.org/10.1007/s00383-020-04617-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00383-020-04617-z