
Abstract
Angiosarcoma is a rare form of soft tissue sarcoma originating from endothelial tissue, accounting for < 1% of all sarcomas. Primary epithelioid angiosarcomas of the central nervous system (CNS) are even more elusive, with only four reports described in the literature. In this article, we describe the first case in pediatric population, with a brief literature review regarding this entity. A 13-year-old girl presented to emergency services with raised intracranial pressure. MRI demonstrated a heterogenous lesion in the temporal lobe. She underwent emergency craniotomy and subtotal excision of the tumor. Eventually the patient developed multiple infarcts and succumbed post operatively. Pre-operative diagnosis on radiology is difficult considering the rarity of this entity and heterogeneity in radiological appearance. One needs to have a high degree of suspicion to consider angiosarcoma as a radiological differential. Overall prognosis remains poor. Early adjuvant treatment may improve overall survival.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Angiosarcoma is a rare form of soft tissue sarcoma originating from endothelial tissue, accounting for < 1% of all sarcomas [1]. Primary epithelioid angiosarcomas of the central nervous system (CNS) are even more elusive, with only four reports described in the literature [2,3,4,5]. In this article, we describe the first case in pediatric population, with a brief literature review regarding this entity.
Case report
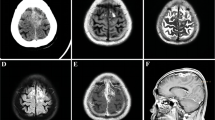
A 13-year-old girl presented to emergency department with symptoms of increased intracranial pressure (ICP) with no focal neurological deficit. Radiological evaluation revealed an intraaxial, irregular solid cystic, heterogeneously contrast-enhancing lesion in right temporal lobe, with mass effect on the brainstem and midline shift toward the opposite side (Fig. 1). There was no perilesional edema. The patient underwent emergency right temporal craniotomy with subtotal excision of the lesion. Intraoperatively, an intraaxial infiltrative tumor with heterogenous consistency containing fibrous and hemorrhagic areas was noted extending from the temporal lobe towards the insula. The tumor was seen involving the middle cerebral artery branches. Many branches of the middle cerebral artery were seen to be shriveled and had no flow. There were multiple thrombosed vessels inside the tumor. Tumor was not vascular and there was minimal blood loss during the surgery. In view of the less vascularity of the lesion, a possibility of fungal lesion was also evaluated. The poor vascularity of the lesion did not favor the diagnosis of a high-grade glioma. Post-operative course was complicated with multiple infarcts and the patient eventually succumbed.

A T2 axial, B T1 contrast axial, and C coronal images reveal intraaxial, irregular solid cystic, heterogeneously contrast-enhancing lesion in right temporal lobe, with mass effect on the brainstem and midline shift toward the left side
The histopathology examination revealed a cellular tumor with large areas of hemorrhage and necrosis (Fig. 2). The individual tumor cells were oval to epithelioid shaped with the presence of intracytoplasmic vacuoles. Focally, immature vasculature formations by the tumor cells were noted. Few eosinophilic hyaline globules were also present. The tumor cells showed nuclear atypia and frequent mitosis. There was a sharp demarcation from the adjacent brain parenchyma with invasion into the underlying cortex along the Virchow-Robin spaces. Upon immunohistochemistry, strong and diffuse positivity was noted for CD31, ERG, and vimentin, with focal positivity for keratin. Tumor cells were negative for GFAP, EMA, desmin, myogenin, OCT-4, SALL-4, S-100, SATB2, and Melan A. INI-1 nuclear staining was retained. The final histological diagnosis was epithelioid angiosarcoma.

A Section from tumor shows a cellular tumor with areas of necrosis (hematoxylin and eosin, × 100). B The tumor cells are lining slit-like spaces containing red blood cells and show moderate nuclear atypia (hematoxylin and eosin, × 400). C The tumor cells show strong membranous CD34 positivity (immunohistochemistry, × 400). D The tumor cells show strong nuclear ERG positivity (immunohistochemistry, × 400)
Discussion
Angiosarcoma is a highly malignant, rare type of sarcoma, originating from endothelial cells. These are known to occur in various soft tissue locations like muscle, skin, and bones. However, rarely are known to arise from skull bones, meninges, or brain [6]. These have been classified by World Health Organization (WHO) under malignant mesenchymal tumors of vascular origin. They are subdivided into classical and epithelioid variety based on histology. They may arise de novo and have a primary intracranial origin or may metastasize from a secondary location to brain. The exact underlying etiology is unknown; some studies have linked it to radiation therapy, thorotrast contrast use, and vinyl chloride exposure [7,8,9].
Angiosarcomas classically occur in the 5th to 7th decade of life. A few cases of pediatric and adolescent age have been reported in the literature [10]. However, no cases of primary pediatric intracranial epithelioid angiosarcoma have been reported in the literature. In the previous series including adults, around 30 cases of primary intracranial (brain and meninges) angiosarcoma have been previously documented, with 4 having epithelioid differentiation [11]. The majority of these lesions have been noted in supratentorial, lobar regions with few present in rare locations like septum pellucidum, pineal gland, and brainstem [3, 11, 12]. Clinical presentation may range from focal neurological deficit or raised ICP symptoms. Usually, rapid clinical worsening is noted, attributable to the rapid growth of the lesion or secondary to hemorrhage within the lesion.
Preoperative diagnosis on radiology is difficult considering the rarity of this entity and heterogeneity in radiological appearance. One needs to have a high degree of suspicion to consider angiosarcoma as a radiological differential. In a review by Lee et al., no common radiological features could be identified among findings reported for over 30 primary intracranial angiosarcomas [11]. Most lesions are identified by heterogenous contrast enhancement with areas of necrosis and hemorrhage. Perilesional edema is seen in majority of cases on T2 weighted imaging; however, this was absent in our index case. Flow voids have also been observed in few cases.
Histopathological evaluation of epithelioid angiosarcoma reveals pleomorphic, polygonal epithelioid cells with atypia. These cells are arranged in sheets along irregular blood vessels. Hemorrhage and necrosis may also be present. They are strongly positive for vimentin, ERG, CD31, and CD34. Focal keratin expression may also be seen in epithelioid variant, which is not seen in classical variant [13]. Proliferative indices like Ki-67 may be used to further quantify the aggressive behavior of the tumor. The differential diagnosis of epithelioid tumors is wide, which include epithelioid sarcoma, epithelioid malignant peripheral nerve sheath tumor, epithelioid angiosarcoma, epithelioid malignant melanoma, epithelioid osteosarcoma, and epithelioid glioblastoma. However, the presence of vacuolated cytoplasm, vascular formations on morphology, and immunopositivity for CD31, CD34, and ERG1 established the diagnosis of epithelioid angiosarcoma in the index case.
Gross total excision with adjuvant therapy in the form of radiotherapy and chemotherapy is the treatment of choice for intracranial angiosarcomas [5]. Due to the rarity of epithelioid differentiation, no separate consensus is present in the literature. Adjuvant radiotherapy in the form of conventional or stereotactic is commonly used to prevent local recurrence [14].
Numerous chemotherapeutic agents like Paclitaxel, Temozolomide, and Bevacizumab have been used, but generally poor overall response has been noted with these [14,15,16]. Hence, role of chemotherapy is inconclusive and may be tailored to the individual case.
Long-term prognosis is variable with factors such as tumor size, location, and extent of resection having significant influence over it [17]. Intraparenchymal lesions have been noted to have a poor prognosis as compared to dural based lesions. In our patient, postoperative course was complicated with developed of multiple infarcts, and eventually, the patient succumbed. Our intraoperative impression ranged from intracranial aspergillosis to high-grade gliomas. Treatment and resection strategies vary widely for these possible etiologies and as surgeon is in a dilemma regarding the extent of resection in such cases. Survival may range from a few months to as high as 105 months, median survival of 1 year [6]. Due to a wide range of survival, additional parameters like proliferative indices should be used for better risk stratification (Table 1).
Conclusion
Primary intracranial angiosarcoma is a rare entity with its epithelioid variant being even rarer. A high index of suspicion is needed to establish its preoperative diagnosis, due to a wide heterogeneity of clinical and radiological presentation. Gross total excision with adjuvant treatment helps in improving overall survival. The intraoperative findings can be variable leading to a diagnostic dilemma in a surgeon’s mind.
References
Scholsem M, Raket D, Flandroy P, Sciot R, Deprez M (2005) Primary temporal bone angiosarcoma: a case report. J Neurooncol 75(2):121–125
Fuse T, Takagi T, Hirose M (1995) Primary intracranial epithelioid angiosarcoma—case report. Neurol Med Chir (Tokyo) 35(6):364–368
Baldovini C, Martinoni M, Marucci G (2013) Epithelioid angiosarcoma of the septum pellucidum. Case Reports in Pathology 2013:1–3
Kuang R, Li S, Wang Y (2023) Primary cerebral epithelia angiosarcoma: a case report. BMC Neurol 23:49
La Corte E, Acerbi F, Schiariti M, Broggi M, Maderna E, Pollo B et al (2014) Primary central nervous system angiosarcoma: a case report and literature review. Neuropathology 35(2):184–191
Gao M, Li P, Tan C, Liu J, Tie X, Pang C et al (2019) Primary central nervous system angiosarcoma. World Neurosurgery 132:41–46
Hoshiai S, Masumoto T, Matsuda M, Sugii N, Sakamoto N, Minami M (2016) Radiation-induced angiosarcoma of the brain. BJR|Case Rep 2(2):20150374
Balamurali G, du Plessis D, Wengoy M, Bryan N, Herwadkar A, Richardson P (2009) Thorotrast-induced primary cerebral angiosarcoma. Neurosurgery 65(1):E210–E211
Bosetti C, La Vecchia C, Lipworth L, McLaughlin J (2003) Occupational exposure to vinyl chloride and cancer risk: a review of the epidemiologic literature. Eur J Cancer Prev 12(5):427–430
Hart J, Mandavilli S (2011) Epithelioid angiosarcoma: a brief diagnostic review and differential diagnosis. Arch Pathol Lab Med 135(2):268–272
Lee C, Shin Y, Choi J (2020) Primary brainstem angiosarcoma mimicking cavernous malformation. World Neurosurgery 139:232–237
Kurian K, Tagkalakis P, Erridge S, Ironside J, Whittle I (2006) Primary intracranial angiosarcoma of the pineal gland: an unusual cause of recurrent intraventricular haemorrhage and superficial haemosiderosis. Neuropathol Appl Neurobiol 32(5):557–561
Miettinen M, Fanburg-Smith J, Virolainen M, Shmookler B, Fetsch J (1999) Epithelioid sarcoma: an immunohistochemical analysis of 112 classical and variant cases and a discussion of the differential diagnosis. Hum Pathol 30(8):934–942
Hackney J, Palmer C, Riley K, Cure J, Fathallah-Shaykh H, Nabors L (2012) Primary central nervous system angiosarcoma: two case reports. J Med Case Rep 6(1)
Ray-Coquard I, Domont J, Tresch-Bruneel E, Bompas E, Cassier P, Mir O et al (2015) Paclitaxel given once per week with or without bevacizumab in patients with advanced angiosarcoma: a randomized phase II trial. J Clin Oncol 33(25):2797–2802
Young R, Woll P (2017) Anti-angiogenic therapies for the treatment of angiosarcoma: a clinical update. Magazine of European Medical Oncology 10(4):190–193
Gaballah A, Jensen C, Palmquist S, Pickhardt P, Duran A, Broering G et al (2017) Angiosarcoma: clinical and imaging features from head to toe. Br J Radiol 90(1075):20170039
Ziegler JW (1975) Primary angiosarcoma of the brain: report of a case. J Am Osteopath Assoc 74(10):957–960
Mena H, Garcia J (1978) Primary brain sarcomas. Light and electron microscopic features Cancer 42(3):1298–1307
Mena H, Ribas J, Enzinger F, Parisi J (1991) Primary angiosarcoma of the central nervous system. J Neurosurg 75(1):73–76
Kirk I, Dominguez R, Castillo M (1992) Congenital primary cerebral angiosarcoma: CT, US, and MR findings. Pediatr Radiol 22(2):134–135
Merimsky O, Lepechoux C, Terrier P, Vanel D, Delord J, LeCesne A (2000) Primary sarcomas of the central nervous system. Oncology 58(3):210–214
Suzuki Y, Yoshida Y, Shirane R, Yoshimoto T, Watanabe M, Moriya T (2000) Congenital primary cerebral angiosarcoma. J Neurosurg 92(3):466–468
Guode Z, Qi P, Hua G, Shangchen X, Hanbin W (2008) Primary cerebellopontine angle angiosarcoma. J Clin Neurosci 15(8):942–946
Author information
Authors and Affiliations
Contributions
Tejasvi Singh Randhawa and Mayur Gopichand Gharat wrote the manuscript draft. Debyajyoti Chatterjee and Chirag Ahuja prepared slides, radiology, figures. Ashish Aggarwal and Raghav Singla edited and finalised the manuscript. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Randhawa, T.S., Aggarwal, A., Chatterjee, D. et al. Pediatric primary intracranial angiosarcoma with epithelioid differentiation: a surgeon’s dilemma. Childs Nerv Syst 40, 267–271 (2024). https://doi.org/10.1007/s00381-023-06114-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-023-06114-9