
Abstract
Purpose
Somatic mutations on H3 histone are currently considered a genetic hallmark for midline pediatric high-grade gliomas (HGGs). Yet, different tumor histologies have been occasionally described to carry these mutations. Since histone modifications can lead to major epigenetic changes with direct impact on prognosis and treatment, we thought to investigate the occurrence of H3F3A K27M and G34R/V mutations in a cohort of pediatric tumors which included HGGs, low-grade gliomas, ependymomas, medulloblastomas, and a series of rare brain tumor lesions of different histologies.
Methods
A total of 82 fresh-frozen pediatric brain tumor samples were evaluated. PCR or RT-PCR followed by Sanger sequencing for the exon 2 of H3F3A (containing both K27 and G34 hotspots) were obtained and aligned to human genome. Loss of trimethylation mark (H3K27me3) in H3F3A/K27M-mutant samples was confirmed by immunohistochemistry.
Results
We found H3F3A/K27M mutation in 2 out of 9 cases of HGGs; no H3F3A/K27M mutations were detected in low-grade gliomas (27), ependymomas (n = 10), medulloblastomas (n = 21), or a series of rare pediatric brain tumors which included meningiomas, dysembryoplastic neuroepithelial tumors (DNETs), central nervous system (CNS) germ-cell tumors, choroid plexus tumors, cortical hamartoma, subcortical tubers, and schwannomas (n = 15). H3F3A/G34R/V mutation was not observed in any of the samples.
Conclusions
Our investigation reinforces the low frequency of H3F3A somatic mutations outside the HGG setting. Interestingly, an atypical focal brainstem glioma carrying H3F3A K27M mutation that showed protracted clinical course with late-onset tumor progression was identified.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acquired histone modifications resulting in tumor epigenetic imbalance were described as a genetic hallmark of high-grade gliomas (both anaplastic astrocytoma and glioblastoma) occurring in children and adolescents [1]. Somatic mutations in H3.3 were reported to be strongly related to high-grade gliomas (HGGs) in children, mainly at brainstem/midline locations (H3.3 K27) or at brain hemispheres (H3.3 G34) [1, 2]. Mutations at position 34 of H3.3 (G34R/V/W) have also been identified in cases of chondroblastomas and other bone tumors [3]. H3.1 K27M (HIST1H3B/C gene), H3.3 K27M, and G34R mutations are mutually exclusive and strongly related to glioma anatomic locations [3, 4]. There is also a strong relation between these mutations and pediatric glioma prognostication [5], with dismal prognosis observed for K27-mutant HGGs.
Due to the relevance of H3 somatic mutations for pediatric glial tumors, the last edition (2016) of the World Health Organization (WHO) classification of tumors of the central nervous system carved a new diagnostic group termed as “diffuse midline glioma (DMG) H3 K27M mutant.” Although H3 mutations were initially identified as a genetic signature for HGG in children and adolescents, documentation of these histone mutations in other types of pediatric brain tumors of different grades and histologies was reported [6,7,8,9], with possible impact on tumor aggressiveness and targeted treatment strategies. Moreover, recent reports have also described somatic H3 K27M mutations in HGG samples derived from adults [10, 11], pointing to a broader involvement of H3 mutations in brain cancer. In view of the importance of these epigenetic mechanisms for the ontogeny, classification, and prognostication of brain tumors in children and adults, we thought to explore H3F3A K27M and G34R/V mutations in a cohort of predominantly pediatric brain cancer of different histological subtypes, including several lesions of rare histologies.
Methods
Patients and tumor specimens
A total of 82 brain cancer samples, occurring at the pediatric age (from 0 to 21 years of age; mean age at diagnosis = 8.5 years), were evaluated. Tumor histologies were as follows: low-grade gliomas (LGG) (n = 27); HGGs (n = 9; including 2 diffuse midline gliomas); ependymomas (EPN) (n = 10); medulloblastomas (MB) and other embryonal tumors (n = 21); and central nervous system (CNS) tumors of rare histologies, 1 of them with paired specimen at relapse (n = 15). Table 1 summarizes clinical information on cases.
All patients reported here were clinically followed at the Pediatric Oncology Unit of the Clinical Hospital–Faculty of Medicine of Ribeirão Preto. Tumor samples were collected and cryopreserved during standard surgical procedures for treatment, after informed consent was obtained from participants/legal guardians. Ethical approval of the institutional review board of Clinical Hospital–Faculty of Medicine of Ribeirão Preto was obtained for this study (CAAE# 14154819.6.0000.5440).
Mutation analysis of H3F3A
Microdissected fresh-frozen tumor samples were processed to extract either total DNA or RNA, in order to optimize material (tumor sample fragment from tiny samples) and resources and to cope with other ongoing researches at the laboratory. DNA extraction was performed with the DNeasy Blood and Tissue kit (Qiagen, USA) following the manufacturer’s guidelines. Total RNA was extracted using the TRIzol® reagent (Invitrogen Inc., USA) or AllPrep DNA/RNA/Protein kit (Qiagen, USA), according to the manufacturers’ specifications. Both DNA and RNA were quantified in a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific Inc., USA). Total RNA was used for cDNA synthesis using the High Capacity® kit (Applied Biosystems, USA) according to the manufacturer’s instructions. Complete PCR and RT-PCR conditions are available at supplemental material 1.
Fragments amplified by PCR or RT-PCR were sequenced using the ABI PRISM DNA Sequencer (Perkin-Elmer, Applied Biosystems), by the reagent “Big Dye™” Terminator Cycle Sequencing Ready Reaction Kit, according to the manufacturer’s instructions. Sequenced products were analyzed and paired with a reference human genome using the SnapGene Viewer 4.2.11 software.
Immunohistochemistry
Protein expression of lysine 27 trimethylation in histone H3.3 (H3k27me3) was performed by immunohistochemistry in all midline tumor cases where FFPE were available for IHC analysis (16 out of 19 cases of pediatric midline gliomas). Histological sections of 4 μm thick were submitted to antigenic recovery in sodium citrate buffer (pH 6.0). The staining was performed with the primary anti-H3K27me3 antibody, dilution 1:400, overnight (07–449, Millipore) properly diluted with 1% BSA. The detection system used was the EnVision™ polymer (Dako, Glostrup, Denmark), following the manufacturer’s recommendations. Immunostaining patterns of the samples were analyzed by a single pathologist (F.P.S.). Positivity criteria followed [12], where it was considered positive when more than 80% of the cells were stained for H3K27me3. Non-tumoral cells of the tumor microenvironment, including endothelial and immune cells, also served as positive internal controls. Images form representative cases were captured using NIS Elements program under a light microscope (Nikon). The association between IHC results for H3k27me3 and H3K27M status was investigated through Fisher’s exact test, considering significant for a p value < 0.05.
Results
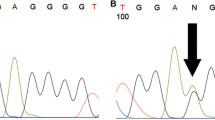
Positive samples for H3F3A K27M mutation accounted for 22.2% (2/9) for HGG analyzed and 10.5% (2/19) of all of pediatric midline gliomas. Electropherograms demonstrating tumor genomic sequences of the one case without mutation and two positive cases are shown in Fig. 1A–C. Clinical details on cases are also presented below. Consistently to what expected, the H3K27M negative cases showed strong staining to H3K27me3 (Fig. 1D), while for H3F3A K27M-mutated samples, the two cases lost histone H3 trimethylation marker (H3K27me3) by immunohistochemistry (Fig. 1E–F). A high concordance rate between H3K27M mutation status and IHC staining for H3K27me3 was confirmed (p = 0.0083), reinforcing the value of this exam as a strong surrogate marker for H3K27M tumor-mutation status (supplemental figure 1). Patchy and mosaic areas of immunostaining for H3K27me3 in tumor cells were observed in 3 samples. None of the studied samples was positive for H3F3A G34 mutation. As expected, no positive H3F3A hotspot mutations (K27 and G34) were found at the cohort that encompassed a large set of rare pediatric brain tumors. Tumors that were evaluated in this group were as follows: meningioma (n = 1), atypical meningioma (n = 5), central nervous system (CNS) germ-cell tumors (n = 2), dysembryoplastic neuroepithelial tumors (DNET) (n = 1), grade I choroid plexus papilloma (CPP; n = 1), grade III choroid plexus carcinoma (CPC; n = 1), cortical hamartoma (n = 1), subcortical tubers (n = 1), primary inflammatory myofibroblastic tumor (IMT) of the CNS (n = 1), and schwannoma (n = 1).

Mutation analysis of H3F3A in cases of 1 and 2 showing A>T mutation. Sanger sequencing chromatograph of resulting PCR-amplified H3F3A showed a high-grade glioma normal (without mutation) (A) and two cases confirmed c.83A > T transversion in case #1 (B) and case #2 (C). (D) IHC staining of case normal using anti-H3K27me3 antibody; H3K27me3 shows strong nuclear positivity in tumor cell. (E) H3K27me3 staining on case #1 shows loss of the expression of this histone marker in H3F3A K27M–mutant tumors and (F) case #2.
Case #1
The first case corresponded to a 16-year-old male diagnosed with a midline brainstem lesion centered at the pons and cerebellar peduncles, with an exophytic extension to the 4th ventricle. Since magnetic resonance image (MRI) findings were not typical for a diffuse intrinsic pontine glioma (DIPG), the exophytic component was biopsied. Histopathological examination of the tumor showed a diffuse and cellular neuroepithelial neoplasia infiltrating cerebellar tissue, consisting of poorly differentiated glial neoplastic cells with rounded nuclei with vesicular chromatin and moderate pleomorphism in which an oligodendrocytic-like phenotype has often noted. Conspicuous microvascular endothelial proliferation, atypical mitotic figures, and isolated bizarre nuclei could be identified. The immunostaining showed diffuse and strong positivity for p53 and GFAP, and a high anti-Ki67-labeled cell proliferation index of 26%. These pathological findings, taken together, were consistent with a H3 K27M–mutant diffuse midline glioma. Somatic H3F3A K27M mutation was positive on specimen. BRAF V600E mutation and BRAF fusions (KIAA1549-BRAF 15-9) tested negative. He was treated with radiotherapy plus temozolomide, followed by maintenance with temozolomide, but rapidly experienced local and distant (leptomeningeal) progression. The patient eventually died of the disease (DOD) 17 months after diagnosis.
Case #2
This case corresponded to a 3-year-old female diagnosed with a focal lesion mainly centered at the medulla oblongata. The child had a previous diagnosis of epilepsy. MRI findings showed a unilateral extrapontine lesion, with extension to the brainstem, reaching the midline location. The extrapontine lesion extended towards the left middle cerebellar peduncle (Fig. 2A, C). A gross total resection was performed. The first histopathological analysis of the tumor showed a diffuse glioma with small and monomorphic atypical cells exhibiting an astrocytic morphology. No conspicuous microvascular proliferation was observed, and lack of mitotic figures or necrosis was present in the tumor sample analyzed. The glioma cells were negative for p53 and GFAP, and the anti-Ki67-labeled cell proliferation index was 5%. The histological features showed a low-grade diffuse glioma pattern (Fig. 2E–H). The final diagnosis was of a grade II diffuse astrocytoma. Due to the patient’s age, low-grade lesion, and GTR, a wait-and-watch approach with close MRI monitoring was adopted. The child remained well and asymptomatic for a long period but, 4 years later, she presented with tumor recurrence with contrast enhancement at the initial tumor bed (Fig. 2B, D). At this point, H3F3A mutation was retrospectively evaluated at the cryopreserved tumor specimen obtained at diagnosis and revealed to be positive for the K27M mutation. Diagnosis was changed to H3 K27M–mutant diffuse midline glioma. The tumor rapidly progressed despite local radiotherapy and temozolomide. The child ultimately DOD at 60 months after the initial diagnosis. Additional genetic testing did not identify BRAF V600E mutation and BRAF fusions (KIAA1549-BRAF 15-9) on specimen.

MRI findings at diagnosis: (A) and (C) are axial T2-FLAIR slices. (B) and (D) are sagittal post-contrast volumetric T1-w slices. The arrows point the intrinsic pontine portion of the tumor as well as the exophytic portion salient to the fourth ventricle. Note the progression of the lesion between both exams, A–B versus C–D. Histopathological features of a diffuse midline glioma, H3 K27M–mutant, centered at lower brainstem. (E) The exophytic portion of the tumor shows diffuse astrocytic morphology with anaplastic features (H&E, original magnification: × 400). (F) The tumor cells show strong GFAP expression (GFAP immunostaining, × 400). (G) p53 immunohistochemistry is negative (p53 immunostaining, × 400). (H) The Ki67 proliferative labeling (arrowheads) index in the tumor at diagnosis was about 5% (Ki67 immunostaining, × 400)
Discussion
Historically, DIPGs and HGGs in children and adolescents are associated to dismal prognosis, irrespective of several different treatment strategies attempted to date. Due to the relative rarity of their occurrence in children, and lack of affordable routine biopsies (particularly for DIPGs), genetic signature on this tumors remained largely unknown for a long period. Yet, in the last decade, the mutational status of histone H3 evolved as a robust biomarker of clinical relevance for accurate diagnosis and follow-up of pediatric brain cancers [13]. Since midline tumors harboring H3K27M became a new entity in the WHO CNS tumor classification in 2016, somatic histone mutation analysis has been increasingly evaluated in an attempt to assess their frequency in different tumor cohorts and to compare prognosis between wild-type and H3-mutated tumors. Also, H3 mutational status may be helpful in understanding its role for brain tumor ontogeny and to deliver new targeted therapies directed to H3K27M status [14].
Regarding this hospital-based Brazilian cohort, two tumor samples (2.4%; 2/82) with the H3F3A K27M mutations were detected; both cases corresponded to gliomas in pediatric patients (3 and 16 years of age), located in the brainstem. The frequency of H3F3A K27M observed in the present study was much lower than those reported in the literature, which ranged up to 69% in DIPGs and 31% of pediatric GBMs [4, 13]. However, differently from other historical series focusing on midline brain tumors or pediatric gliomas alone, our study intended to evaluate a mixed cohort of brain tumors where only a few samples of midline gliomas were evaluated (n = 19). Regarding HGG, the two samples were located midline (brainstem) and bore H3K27M mutation. Unlike the H3F3A K27M mutation, the G34R/V mutation is described to be more frequent in pediatric gliomas located in the cerebral hemispheres [15]. Among the analyzed samples, 48% (39/82) were located at the supratentorial region, and HGG was the diagnosis in 6 of supratentorial glioma lesions; yet, no case of H3F3A G34R/V mutation was observed in our series. Although HIST1H3B mutations in HGG are much less frequent than those of H3F3A [13], the lower incidence observed in the present study may also be related to the lack of HIST1H3B mutational screening of our samples. Of clinical interest, no somatic mutations on H3F3A K27 and G34 were found at the cohort of rare pediatric brain tumors. The PCR/Sanger strategy is a cost-effective approach to detect somatic point mutations. Yet, sensitivity from the method can be low, depending on the fractional abundance of the mutation at the sample. More sensitive methods such as digital-droplet PCR (ddPCR) [16] or next-generation sequencing (particularly MiSeq) [17] are alternatives to PCR/Sanger, although high costs for equipment and reactions for these novel methods may turn them unaffordable to clinical practice in most developing countries.
In agreement with H3F3A K27M mutational status, we identified a global reduction in H3K27me3 in the two cases described. Few studies have specifically addressed the value of H3K27me3 by IHC in relation to H3K27M mutational status. Similarly to our observations, Huang et al. also found 100% concordance between IHC status and histone K27M mutation in a cohort of 69 pediatric glioma specimens [18]. More recently, Castel et al. characterized a new subgroup of DMG (including DIPG cases) that possesses trimethylation loss of H3K27 without H3K27M somatic mutation detected on specimen [19]. The authors have shown that epigenetic changes culminating with H3K27me3 loss may be independently driven by EZHIP overexpression in gliomas. Their results reinforced that H3K27me3 loss in gliomas may also be related to alternative pathways leading to polycomb repressor complex 2 (PRC2) inhibition beyond H3 K27M tumor mutation. In addition, it remains largely unknown the significance (if any) of H3K7me3 pattern on tumor sample, such as the patchy and mosaic areas of immunostaining in three cases from our cohort. In view of these recent results, H3K27me3 status evaluated by IHC may be an important screening to identify pediatric gliomas that ultimately display either H3K27M or EZHIP overexpression, leading to comparable poor clinical prognosis.
Cancer cells harboring H3.3 K27M mutation have shown reduction in trimethylation levels in H3 lysine 27 [20,21,22,23]. Bender and collaborators [20] demonstrated that K27M mutant H3.3 causes a global suppressive histone mark H3K27me3. The authors suggest that the reduction of this mark is caused by the aberrant mechanism of competitive inhibition of the catalytic subunit of PRC2, the EZH2, by mutant H3K27M. In addition, Li et al. [24] showed that all cases of H3K27M-mutant gliomas (100%; 21/21) presented reduction in H3K27Me3 levels. Similarly, it was observed by Castel and collaborators [25] a loss of H3K27me3 in 95% of DIPG samples (59/62), highlighting the importance of immunohistochemistry for histone H3 in the pathology of this disease. Additionally, Mohammad and colleagues [23] also described that there is a global loss of methylation of H3K27me3, but several genes still retain trimethylation and PRC2 for tumor proliferation, suggesting that EZH2 is a strategic potential therapy for the treatment of these tumors.
Interestingly, our study identified a focal midline lesion at lower brainstem harboring the H3F3A K27M mutation and negative IHC staining for p53 that showed a protracted clinical course. Lack of IHC mark for p53 by IHC is usually related wild-type TP53 on tumor, although complete TP53 deletion at that sample cannot be ruled out by this method. Mutations in TP53 have been associated to clinically aggressive subtype of H3.3 K27M–mutant DIPG in diverse studies [26,27,28]. p53 loss in nestin progenitors of the neonatal mouse brainstem with H3K27M mutant expression increased proliferation of ectopic cell clusters in 72% of mice, whereas in H3K27 wild-type mice did not [22]. Larson et al. [28] also confirmed these data, showing that the induction of H3.3 K27M in mouse model cooperated with Trp53 loss, activating platelet-derived growth factor receptor a (PDGFRa) mutant to accelerate the development of diffuse brainstem gliomas that recapitulated human DIPG gene expression signatures. In one of the current cases of H3F3A K27M reported herein (#2), wild-type TP53 on tumor at diagnosis may have contributed to low aggressive features and atypical clinical course. Similar reports in the pediatric population have been recently described. Hochart et al. [29] reported the first case of a long survival (10 years) of a child presenting with H3F3A K27M PA of the cervical spinal cord that underwent spontaneous malignant transformation to a GBM. Nakano et al. [30] described the case of a 2-year-old female with a lateral ventricular glioma that also harbored H3F3A K27M and BRAF V600E double mutations, although methylation analysis clustered the sample as a grade IV diffuse midline glioma H3 K27M mutant. This patient survived more than 8 years with surgical resection alone. Recently, Baroni et al. pooled data on three unusual cases of brainstem tumors positive for H3 K27M somatic mutations that also had an initial indolent and protracted course [31].
This study presents some limitations that need to be considered while analyzing our findings: this is a single institution–based study, with a limited number of cases. In addition, besides the evaluation of H3F3A K27M and G34R/V in a set of rare pediatric brain tumors, these information on LGG, EPN, and MB, although our results are in accordance to previous reports, are not new. In summary, this heterogeneous cohort of pediatric tumor cases reaffirms the extremely low frequency of H3.3 K27 and G34 mutations outside gliomas. Additionally, we describe the H3.3 K27 and G34 mutational status on other far less frequent pediatric brain lesions, including CPPs, CPCs, meningiomas, IMTs, hamartomas, schwannomas, subcortical tubers, DNETs, and CNS germ-cell tumors, where this information remains scarce. As a future plan, a larger study in collaboration with other Brazilian and South American centers would help us to empower these initial findings.
Data availability
All data generated from this study are presented in this article.
References
Schwartzentruber J, Korshunov A, Liu X-Y, Jones DTW, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DAK, Tönjes M, Hovestadt V, Albrecht S, Kool M, Nantel A, Konermann C, Lindroth A, Jäger N, Rausch T, Ryzhova M, Korbel JO, Hielscher T, Hauser P, Garami M, Klekner A, Bognar L, Ebinger M, Schuhmann MU, Scheurlen W, Pekrun A, Frühwald MC, Roggendorf W, Kramm C, Dürken M, Atkinson J, Lepage P, Montpetit A, Zakrzewska M, Zakrzewski K, Liberski PP, Dong Z, Siegel P, Kulozik AE, Zapatka M, Guha A, Malkin D, Felsberg J, Reifenberger G, von Deimling A, Ichimura K, Collins VP, Witt H, Milde T, Witt O, Zhang C, Castelo-Branco P, Lichter P, Faury D, Tabori U, Plass C, Majewski J, Pfister SM, Jabado N (2012) Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482:226–231. https://doi.org/10.1038/nature10833
Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, Zhang J, Gajjar A, Dyer MA, Mullighan CG, Gilbertson RJ, Mardis ER, Wilson RK, Downing JR, Ellison DW, Zhang J, Baker SJ, St. Jude Children's Research Hospital–Washington University Pediatric Cancer Genome Project (2012) Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 44:251–253. https://doi.org/10.1038/ng.1102
Yamamoto H, Iwasaki T, Yamada Y, Matsumoto Y, Otsuka H, Yoshimoto M, Kohashi K, Taguchi K, Yokoyama R, Nakashima Y, Oda Y (2018) Diagnostic utility of histone H3.3 G34W, G34R, and G34V mutant-specific antibodies for giant cell tumors of bone. Hum Pathol 73:41–50. https://doi.org/10.1016/j.humpath.2017.11.020
Kallappagoudar S, Yadav RK, Lowe BR, Partridge JF (2015) Histone H3 mutations — a special role for H3.3 in tumorigenesis? Chromosoma 124(2):177–189. https://doi.org/10.1007/s00412-015-0510-4
Karremann M, Gielen GH, Hoffmann M, Wiese M, Colditz N, Warmuth-Metz M, Bison B, Claviez A, van Vuurden DG, von Bueren AO, Gessi M, Kühnle I, Hans VH, Benesch M, Sturm D, Kortmann RD, Waha A, Pietsch T, Kramm CM (2018) Diffuse high-grade gliomas with H3 K27M mutations carry a dismal prognosis independent of tumor location. Neuro-Oncology 20:123–131. https://doi.org/10.1093/neuonc/nox149
Nambirajan A, Suri V, Kedia S, Goyal K, Malgulwar PB, Khanna G, Panda PK, Gulati S, Garg A, Sharma MC (2018) Paediatric diffuse leptomeningeal tumor with glial and neuronal differentiation harbouring chromosome 1p/19q co-deletion and H3.3 K27M mutation: unusual molecular profile and its therapeutic implications. Brain Tumor Pathol 35:186–191. https://doi.org/10.1007/s10014-018-0325-0
Ryall S, Guzman M, Elbabaa SK, Luu B, Mack SC, Zapotocky M, Taylor MD, Hawkins C, Ramaswamy V (2017) H3 K27M mutations are extremely rare in posterior fossa group A ependymoma. Childs Nerv Syst 33:1047–1051. https://doi.org/10.1007/s00381-017-3481-3
Okuda T, Hata N, Suzuki SO, Yoshimoto K, Arimura K, Amemiya T, Akagi Y, Kuga D, Oba U, Koga Y, Ohga S, Iwaki T, Iihara K (2018) Pediatric ganglioglioma with an H3 K27M mutation arising from the cervical spinal cord. Neuropathology 38:422–427. https://doi.org/10.1111/neup.12471
Yao K, Duan Z, Wang Y, Zhang M, Fan T, Wu B, Qi X (2019) Detection of H3K27M mutation in cases of brain stem subependymoma. Hum Pathol 84:262–269. https://doi.org/10.1016/j.humpath.2018.10.011
Ebrahimi A, Skardelly M, Schuhmann MU, Ebinger M, Reuss D, Neumann M, Tabatabai G, Kohlhof-Meinecke P, Schittenhelm J (2019) High frequency of H3 K27M mutations in adult midline gliomas. J Cancer Res Clin Oncol 145:839–850. https://doi.org/10.1007/s00432-018-02836-5
Meyronet D, Esteban-Mader M, Bonnet C, Joly MO, Uro-Coste E, Amiel-Benouaich A, Forest F, Rousselot-Denis C, Burel-Vandenbos F, Bourg V, Guyotat J, Fenouil T, Jouvet A, Honnorat J, Ducray F (2017) Characteristics of H3 K27M-mutant gliomas in adults. Neuro-Oncology 19:1127–1134. https://doi.org/10.1093/neuonc/now274
Panwalkar P, Clark J, Ramaswamy V, Hawes D, Yang F, Dunham C, Yip S, Hukin J, Sun Y, Schipper MJ, Chavez L, Margol A, Pekmezci M, Chung C, Banda A, Bayliss JM, Curry SJ, Santi M, Rodriguez FJ, Snuderl M, Karajannis MA, Saratsis AM, Horbinski CM, Carret AS, Wilson B, Johnston D, Lafay-Cousin L, Zelcer S, Eisenstat D, Silva M, Scheinemann K, Jabado N, McNeely PD, Kool M, Pfister SM, Taylor MD, Hawkins C, Korshunov A, Judkins AR, Venneti S (2017) Immunohistochemical analysis of H3K27me3 demonstrates global reduction in group-A childhood posterior fossa ependymoma and is a powerful predictor of outcome. Acta Neuropathol 134:705–714. https://doi.org/10.1007/s00401-017-1752-4
Khuong-Quang D-A, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E, Bartels U, Albrecht S, Schwartzentruber J, Letourneau L, Bourgey M, Bourque G, Montpetit A, Bourret G, Lepage P, Fleming A, Lichter P, Kool M, von Deimling A, Sturm D, Korshunov A, Faury D, Jones DT, Majewski J, Pfister SM, Jabado N, Hawkins C (2012) K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol 124:439–447. https://doi.org/10.1007/s00401-012-0998-0
Chi AS, Tarapore RS, Hall MD, Shonka N, Gardner S, Umemura Y, Sumrall A, Khatib Z, Mueller S, Kline C, Zaky W, Khatua S, Weathers SP, Odia Y, Niazi TN, Daghistani D, Cherrick I, Korones D, Karajannis MA, Kong XT, Minturn J, Waanders A, Arillaga-Romany I, Batchelor T, Wen PY, Merdinger K, Schalop L, Stogniew M, Allen JE, Oster W, Mehta MP (2019) Pediatric and adult H3 K27M-mutant diffuse midline glioma treated with the selective DRD2 antagonist ONC201. J Neuro-Oncol 145:97–105. https://doi.org/10.1007/s11060-019-03271-3
Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DTW, Konermann C, Pfaff E, Tönjes M, Sill M, Bender S, Kool M, Zapatka M, Becker N, Zucknick M, Hielscher T, Liu XY, Fontebasso AM, Ryzhova M, Albrecht S, Jacob K, Wolter M, Ebinger M, Schuhmann MU, van Meter T, Frühwald MC, Hauch H, Pekrun A, Radlwimmer B, Niehues T, von Komorowski G, Dürken M, Kulozik AE, Madden J, Donson A, Foreman NK, Drissi R, Fouladi M, Scheurlen W, von Deimling A, Monoranu C, Roggendorf W, Herold-Mende C, Unterberg A, Kramm CM, Felsberg J, Hartmann C, Wiestler B, Wick W, Milde T, Witt O, Lindroth AM, Schwartzentruber J, Faury D, Fleming A, Zakrzewska M, Liberski PP, Zakrzewski K, Hauser P, Garami M, Klekner A, Bognar L, Morrissy S, Cavalli F, Taylor MD, van Sluis P, Koster J, Versteeg R, Volckmann R, Mikkelsen T, Aldape K, Reifenberger G, Collins VP, Majewski J, Korshunov A, Lichter P, Plass C, Jabado N, Pfister SM (2012) Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22:425–437. https://doi.org/10.1016/j.ccr.2012.08.024
Wang Z, Sun K, Jing C, Cao H, Ma R, Wu J (2019) Comparison of droplet digital PCR and direct Sanger sequencing for the detection of the BRAF V600E mutation in papillary thyroid carcinoma. J Clin Lab Anal 33:e22902. https://doi.org/10.1002/jcla.22902
Arsenic R, Treue D, Lehmann A, Hummel M, Dietel M, Denkert C, Budczies J (2015) Comparison of targeted next-generation sequencing and Sanger sequencing for the detection of PIK3CA mutations in breast cancer. BMC Clin Pathol 15:20. https://doi.org/10.1186/s12907-015-0020-6
Huang T, Garcia R, Qi J et al (2018) Detection of histone H3 K27M mutation and post-translational modifications in pediatric diffuse midline glioma via tissue immunohistochemistry informs diagnosis and clinical outcomes. Oncotarget 9:37112–37124. https://doi.org/10.18632/oncotarget.26430
Castel D, Kergrohen T, Tauziède-Espariat A, Mackay A, Ghermaoui S, Lechapt E, Pfister SM, Kramm CM, Boddaert N, Blauwblomme T, Puget S, Beccaria K, Jones C, Jones DTW, Varlet P, Grill J, Debily MA (2020) Histone H3 wild-type DIPG/DMG overexpressing EZHIP extend the spectrum diffuse midline gliomas with PRC2 inhibition beyond H3-K27M mutation. Acta Neuropathol 139:1109–1113. https://doi.org/10.1007/s00401-020-02142-w
Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DTW, Kool M, Zapatka M, Northcott PA, Sturm D, Wang W, Radlwimmer B, Højfeldt JW, Truffaux N, Castel D, Schubert S, Ryzhova M, Şeker-Cin H, Gronych J, Johann PD, Stark S, Meyer J, Milde T, Schuhmann M, Ebinger M, Monoranu CM, Ponnuswami A, Chen S, Jones C, Witt O, Collins VP, von Deimling A, Jabado N, Puget S, Grill J, Helin K, Korshunov A, Lichter P, Monje M, Plass C, Cho YJ, Pfister SM (2013) Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 24:660–672. https://doi.org/10.1016/j.ccr.2013.10.006
Diehl KL, Ge EJ, Weinberg DN, Jani KS, Allis CD, Muir TW (2019) PRC2 engages a bivalent H3K27M-H3K27me3 dinucleosome inhibitor. Proc Natl Acad Sci 116:22152–22157. https://doi.org/10.1073/pnas.1911775116
Lewis PW, Muller MM, Koletsky MS et al (2013) Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340:857–861. https://doi.org/10.1126/science.1232245
Mohammad F, Weissmann S, Leblanc B, Pandey DP, Højfeldt JW, Comet I, Zheng C, Johansen JV, Rapin N, Porse BT, Tvardovskiy A, Jensen ON, Olaciregui NG, Lavarino C, Suñol M, de Torres C, Mora J, Carcaboso AM, Helin K (2017) EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat Med 23:483–492. https://doi.org/10.1038/nm.4293
Li HN, Shan CG, Fan CZ et al (2019) Clinicopathological characteristics and prognosis of diffuse midline gliomas with histone H3K27M mutation: an analysis of 30 cases. Zhonghua Bing Li Xue Za Zhi 48:192–198. https://doi.org/10.3760/cma.j.issn.0529-5807.2019.03.005
Castel D, Philippe C, Calmon R, le Dret L, Truffaux N, Boddaert N, Pagès M, Taylor KR, Saulnier P, Lacroix L, Mackay A, Jones C, Sainte-Rose C, Blauwblomme T, Andreiuolo F, Puget S, Grill J, Varlet P, Debily MA (2015) Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol 130:815–827. https://doi.org/10.1007/s00401-015-1478-0
Mackay A, Burford A, Carvalho D et al (2017) Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell 32:520–537.e5. https://doi.org/10.1016/j.ccell.2017.08.017
Bozkurt SU, Dagcinar A, Tanrikulu B, Comunoglu N, Meydan BC, Ozek M, Oz B (2018) Significance of H3K27M mutation with specific histomorphological features and associated molecular alterations in pediatric high-grade glial tumors. Childs Nerv Syst 34:107–116. https://doi.org/10.1007/s00381-017-3633-5
Larson JD, Kasper LH, Paugh BS et al (2019) Histone H3.3 K27M accelerates spontaneous brainstem glioma and drives restricted changes in bivalent gene expression. Cancer Cell 35:140–155.e7. https://doi.org/10.1016/j.ccell.2018.11.015
Hochart A, Escande F, Rocourt N, Grill J, Koubi-Pick V, Beaujot J, Meignan S, Vinchon M, Maurage CA, Leblond P (2015) Long survival in a child with a mutated K27M-H3.3 pilocytic astrocytoma. Ann Clin Transl Neurol 2:439–443. https://doi.org/10.1002/acn3.184
Nakano Y, Yamasaki K, Sakamoto H, Matsusaka Y, Kunihiro N, Fukushima H, Inoue T, Honda-Kitahara M, Hara J, Yoshida A, Ichimura K (2019) A long-term survivor of pediatric midline glioma with H3F3A K27M and BRAF V600E double mutations. Brain Tumor Pathol 36:162–168. https://doi.org/10.1007/s10014-019-00347-w
Baroni LV, Solano-Paez P, Nobre L et al (2020) Indolent course of brainstem tumors with K27M-H3.3 mutation. Pediatr Blood Cancer. https://doi.org/10.1002/pbc.28102
Acknowledgments
We thank Dr. Luciano Neder for pathological assistance and expertise on cases.
Funding
FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo – grant no. 2018/20275-8)/Brazil, as part of Scientific Initiation Program (granted by Medical Student Vinicius Fernandes Oliveira)
Author information
Authors and Affiliations
Contributions
E.T.V., V.F.O., and G.R.S. planned the study, conducted data analysis, and drafted the manuscript; F.P.S. performed data analysis and immunohistochemistry; A.C.S. performed image analysis and intellectual content; H.R.M., R.S.O., and L.G.T. proceeded sample collection and revised text for important intellectual content. V.F.O and G.R.S. wrote and organized the data, created the figures/tables, and edited and finalized the manuscript. All authors critically read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethics approval
Tumor collection (brain tumor repository) was performed under the approval of the Institutional Review Board (IRB) (#6591/2007). This study was also Institutional Review Board (IRB) approved (CAAE# 14154819.6.0000.5440).
Consent to participate
Obtained by signed informed consent
Consent for publication
Not applicable
Code availability
Not applicable
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplemental Figure 1.
IHC staining for H3K27me3 on 14 cases of pediatric midline gliomas H3F3A K27 wild-type, showing a retained pattern of this histone trimethylation marker. (JPG 10297 kb).
ESM 2
(DOCX 16 kb).
Rights and permissions
About this article

{kind=link}
Cite this article
Oliveira, V.F., De Sousa, G.R., dos Santos, A.C. et al. Evaluating H3F3A K27M and G34R/V somatic mutations in a cohort of pediatric brain tumors of different and rare histologies. Childs Nerv Syst 37, 375–382 (2021). https://doi.org/10.1007/s00381-020-04852-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-020-04852-8