
Abstract
Osseous manifestations of neurofibromatosis 1 (NF-1) occur in a minority of the affected subjects but may be because of significant clinical impairment. Typically, they involve the long bones, commonly the tibia and the fibula, the vertebrae, and the sphenoid wing. The pathogenesis of NF-1 focal osseous lesions and its possible relationships with other osseous NF-1 anomalies leading to short stature are still unknown, though it is likely that they depend on a common mechanism acting in a specific subgroup of NF-1 patients. Indeed, NF-1 gene product, neurofibromin, is expressed in all the cells that participate to bone growth: osteoblasts, osteoclasts, chondrocytes, fibroblasts, and vascular endothelial cells. Absent or low content of neurofibromin may be responsible for the osseous manifestations associated to NF-1. Among the focal NF-1 osseous anomalies, the agenesis of the sphenoid wing is of a particular interest to the neurosurgeon because of its progressive course that can be counteracted only by a surgical intervention. The sphenoid wing agenesis is regarded as a dysplasia, which is a primary bone pathology. However, its clinical progression is related to a variety of causes, commonly the development of an intraorbital plexiform neurofibroma or the extracranial protrusion of temporal lobe parenchyma and its coverings. Thus, the cranial bone defect resulting by the primary bone dysplasia is progressively accentuated by the orbit remodeling caused by the necessity of accommodating the mass effect exerted by the growing tumor or the progression of the herniated intracranial content. The aim of this paper is to review the neurosurgical and craniofacial surgical modalities to prevent the further progression of the disease by “reconstructing” the normal relationship of the orbit and the skull.
Similar content being viewed by others
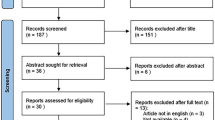

Avoid common mistakes on your manuscript.
Introduction
Neurofibromatosis type 1 (NF-1), also called Recklinghausen disease, is a neurocutaneous-skeletal autosomal-dominant common tumor-predisposing disorder that arises secondary to mutations in the tumor suppressor gene N-F1. It involves multiple systems, including the skin, eyes, brain, and skeleton.
The incidence of NF-1 is approximately 1:3000 births with no gender or race predilection [1].
The diagnosis of NF-1 is currently based on the criteria of the National Institute of Health Consensus Development Conference published in 1987 [2] which take into consideration the characteristic neurofibromas, “café-au-lait” spots, axillary or groin freckling, Lisch nodules, optic pathway gliomas, and skeletal lesions (Fig. 1) [3, 4].

Skeletal lesions. a X-ray of a tibia showing congenital curving and pseudarthrosis. b X-ray of a spine showing short angle scoliosis
The clinical presentation of NF-1 is heterogeneous and deeply related to the formation of tumors in ectoderm and mesoderm tissues [5].
This phacomatosis is caused by mutations in the NF-1 gene, located at 17q11.2, which encodes the tumor suppressor neurofibromin [6,7,8,9,10] which acts as RAS GTPase activating protein (RAS-GAP), thus inactivating Ras pathway.
In humans, neurofibromin mRNA and protein have been detected in osteoblasts, osteoclasts, chondrocytes, fibroblasts, and vascular endothelial cells [1] and its absence or low levels might account for the occurrence of bone anomalies in NF-1. Indeed, loss of neurofibromin, in subjects affected by NF-1, causes a downregulation of osteoblastic activity and an intrinsic bone tissue abnormality [11].
Moreover, Ras pathway is essential for the normal growth of craniofacial structures. Jaws and cranial base are largely derived from the neural crest cells. Indeed, some authors propound that NF-1 can be considered as a pathology of neural crest cells [1].
NF-1-associated osseous lesions or osseous dysplasias include craniofacial and skeletal anomalies such as short stature, osteopenia, osteoporosis, short angle scoliosis, lytic bone lesions, and congenital curving and pseudarthrosis of the tibia (Fig. 1) [1, 10, 12]. Anyway, it is still unclear why the tendency of NF-1 to produce bone dysplasias results in the apparent prevalence of focal lesions that privilege only a few bones. Among these focal lesions, the absence of the greater wing of the sphenoid bone is the most common and almost pathognomonic craniofacial osseous anomaly in subjects with NF-1.
Craniofacial bone dysplasias
The NF-1 gene is supposed to regulate or influence the growth of craniofacial bones, thus contributing to the craniofacial morphology in NF-1 [1, 13,14,15,16]. A number of craniofacial abnormalities in NF-1 have been reported. It includes macrocephaly, sphenoid wing dysplasia, orbital dysplasia, maxillary and mandibular deformities, temporomandibular joint (TMJ) deformities, and dental anomalies [17, 18].
Facial and skull growth can be affected in NF-1 [1, 13, 19]. The first cephalometric study was carried out on a Finnish cohort of NF-1 subjects by Heervä et al. in 2011 [1] and then repeated on a larger white American population including both adults and children with NF-1 by Cung et al. in 2015 [20]. The authors recorded a shorter maxilla, mandible, cranial base (especially anteriorly, p = 0.0001), and diminished facial height in adults. Interestingly, these alterations were not detected in children. Cung and colleagues concluded that the cephalometric differences in adults depended at least in part on the cranial base shortening and accounted for the shorter face, mid-face hypoplasia, reduced facial projection, and smaller jaw. They also suggested that the sphenoid bone shortening could be related to an intrinsic NF-1 bone cell defect, which made the bone more vulnerable to a possible “second hit” in leading to sphenoid wing dysplasia. Indeed, the sphenoid wing dysplasia becomes commonly evident on the clinical examination only in the first 2 years of age.
The role of plexiform neurofibromas in affecting the facies growth and symmetry was stressed by Friedrich et al. in a study based on lateral cephalometry published in 2017 [21]. The authors pointed out on the large deviations of facial measures in patients with NF-1. They did not find significant variations in subjects with NF-1 with only disseminated cutaneous lesions whereas detected significant differences from healthy volunteers in patients with plexiform neurofibromas. These differences depended clearly on the number of trigeminal nerve branches involved by the tumor. The authors also confirmed the necessity of considering the possible presence of a plexiform neurofibromas in all the NF-1 patients presenting with facial asymmetry as they had suggested in a previous study in which they reported jaw malformations in 28 out of 48 NF-1 patients with plexiform neurofibromas originating from the branches of the trigeminal nerve [22]. Facial asymmetry concerns about 10% of patients with NF-1 [1, 16]. Neurofibromas involving the articular disc of the temporo-mandibular joint have been also reported [23]. Indeed, the presence of plexiform neurofibromas has been associated with a variety of facial bone anomalies such as numerical aberrations and retention of teeth, deformed alveolar ridge, early primary tooth eruption, impacted teeth, supernumerary teeth, missing or displaced teeth, overgrowth of the alveolar process, osseous defects in the alveolus, and periapical cemental dysplasia in women with NF-1 [15, 16, 22, 24,25,26,27,28]. Even increased dental caries has been attributed to NF-1, though this association is debatable [15, 18].
The clinical and radiological examinations of adult NF-1 subjects show a decreased antero-posterio diameter of the maxilla in 75% of NF-1 patients, in whom the maxilla is also often retrognathic due to the shortened anterior cranial base when compared with controls [1, 20].
Elongated coronoid process with a deep sigmoid notch [29], notching of the posterior border of the mandibular ramus [30], hypoplasia of the condyle and zygomatic processes [31] have been reported. Lorson et al. suggested to include the elongated coronoid process as a pathognomonic sign of NF-1 [17]. Radiologically, a wide, branching, and enlargement of inferior alveolar canal, enlargement mental foramen, and a decrease in the mandibular angle have been described [15, 16, 18, 22, 24, 25, 32,33,34].
In spite of the shorter than normal skull base, the volume of the skull vault in NF-1 is generally larger than in healthy persons in a significant proportion of the cases. Approximately 25% of patients with NF-1 tend to have a large head circumference and macrocephaly (occipito-frontal circumference > 2 SD above the mean) [35]. The brain volume is also larger in subjects with NF-1 compared with controls [1]. However, it is not clear whether the large skull growth is the primary cause of the macrocephaly or macrocephaly is secondary to the enlargement of the brain [10, 36]. In some cases, the presence of mild ventriculomegaly makes the physiopathogenetic interpretation of the phenomenon more difficult.
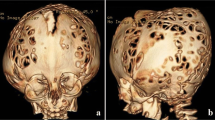
Calvarial defects have been reported in some patients with NF-1 too (Fig. 2) [37].

CT scan imaging. a CT scan showing lytic bone lesions of the vertebrae and mandibular asymmetry. b CT scan of a skull showing calvarial defects
The orbital deformities have been considered uncommon in NF-1 but with the refinement of clinical and radiological diagnosis; nowadays, they are regarded to be relatively common. In 2003, Jacquemin et al. retrospectively reviewed CT and MR imaging abnormalities of the orbit in 31 NF-1 patients, mean age 14 years, and found orbital abnormalities in 24 patients [38]. The most frequent cause was plexiform neurofibromas within the orbit or in relation to the anterior skull base detected in 20 cases; in 13 patients, the orbital abnormalities were due to a distortion of the posterior wall induced by encroachment from an expanded middle cranial fossa; six patients harbored an optic nerve glioma with enlarged optic canal. Enlargement of the orbital rim was noticed in 18 subjects. Other changes such as focal decalcification or remodeling of orbital walls adjacent to plexiform neurofibroma were detected in 18 patients and enlargement of cranial foramina resulting from tumor infiltration of sensory nerves in 16.
Actually, the orbital abnormalities in NF-1 recognize three main causes: the development of an optic nerve glioma, the congenital defect of the sphenoid wing, and the presence of a plexiform neurofibroma. While the orbital deformities associated to optic nerve glioma are nearly always stable due to the absent or slow progression of this tumor, the bone lesions due to plexiform neurofibromas and sphenoid bone dysgenesis are frequently progressive [38]. When a surgical therapy is taken into account, the orbital deformities should be regarded as the combined effect of a primary dysplasia and the secondary response of bone to an expanding mass that can be counteracted only by an appropriate management. In some instances, especially in infants and young children, the orbital abnormalities can regress after the removal of the causative occupying space lesion or the skull base reconstruction.
Sphenoid wing dysplasia
Sphenoid wing dysplasia is the most distinctive craniofacial anomaly in NF-1. It occurs in 5–12% of the cases. Complete agenesis is, however, very rare [39,40,41]. In most cases, the defect of the greater wing of the sphenoid bone is isolated, partial, and unilateral, involving more frequently the left part of the bone, a feature that has been believed to reinforce the hypothesis of its primary and congenital nature. Over 50% of patients who have sphenoid wing defects are NF-1 subjects. Only rarely the sphenoid wing dysgenesis is associated to extensive dysplasia of the skull base [42]. It is congenital though becoming generally clinically apparent post-birth, usually before the age of 2 years [10, 12, 43]. Two main physiopathogenetic interpretations have been propounded. In cases without concurrent causes, the dysgenesis of the sphenoid wing would result from a primary ossification defect with poor mesodermal development and bone formation [10, 20, 41, 44]. In cases with concurrent causes, nearly always plexiform neurofibromas, a multifactorial genesis of the sphenoid wing defect has been hypothesized [41]. According to such an hypothesis that would also explain the progression of the disease, the sphenoid bone dysgenesis would develop secondarily from plexiform neurofibromas in the orbit or in the superficial temporal fossa which would erode or deform the adjacent bony orbit together with local vascular abnormalities due to the tumor itself which can increase the orbital blood circulation and expand the superior orbital fissure. This may develop before birth, in utero, or early childhood [12, 41, 45].
The partial or complete absence of the greater wing of the sphenoid is associated with a prolapse of the temporal lobe in the orbital cavity resulting in progressive facial asymmetry, progressive proptosis, pulsating exophthalmos, restriction of extraocular movement, conjunctival inflammation, and pressure on the optic nerve with risk of blindness [12, 42, 44].
Indeed, the partial absence of the greater wing of the sphenoid or an anterior displacement of the greater sphenoid wing is associated to a widening of the orbital apex and anteroposterior enlargement of the middle cranial fossa (Fig. 3) [41]. An anterior temporal pooling of cerebrospinal fluid, often wrongly reported as an associated arachnoid cyst [41], is usually present and participates to the bone dysplasia progression by its hammer effect that is by amplificating the mechanical effect of CSF pulses. In other words, the presence of the sphenoid wing defect creates a local functional dynamic situation which is similar to that accounting for the progressive herniation of meninges and brain parenchyma in cases of growing cranial fractures. The adjacent bone undergoes thinning and remodeling. Finally, the sphenoid bone defect can become large enough to allow the progressive herniation of the temporal lobe structures. A pulsatile exophthalmos is also created with increasing deviation of the ocular globe and secondary alteration and displacement of the entire orbit. The orbital deformation is usually slow and well tolerated from an ophthalmological point of view. The optic nerve can elongated enormously without a significant impact on the visual function. However, when the proptosis becomes severe, palpebral occlusion may become incomplete leading to potential corneal exposure and damage.

Case of a 3.5-year-old boy referred for a sphenoid wing dysplasia due to NF-1 causing pulsating exophthalmos. Magnetic resonance and CT scan imaging showing a partial dysplasia of the greater wing of the left sphenoid with an expansion of the temporal fossa. A herniation of temporal brain through the sphenoid dysplasia is noted, inducing the exophthalmos (a, d axial view, b, e sagittal view, c, f coronal view). Please note that the use of CT scan in such background of NF-1 should be limited
Management
The management of sphenoid wing dysplasia complicated by ocular globe proptosis is surgical. Currently, there are no clear guidelines. However, as the condition is progressive, an early operation is suggested, preferably to be carried out by a double team that combines neurosurgeons and maxillofacial surgeons, are necessary to prevent further progression of the bone “dysplasia” and further herniation of cerebral structures to prevent or reverse, if present, functional impairment (vision) and to correct cosmetic deformity. The plastic surgeon may be required post-operatively to deal with the exceeding palpebral tissue and to assure the best facial cosmetic result. The surgical procedure aims at reconstructing the cranial and orbit defect and also restoring a barrier between the orbit and the middle cranial fossa without damaging the neural structures. In most severe cases, it could be necessary to repair a excessively thin dura mater and excise damaged nervous tissues encroached in the bone lacuna or an associate plexiform neurofibroma. The many surgical techniques described in the literature [44, 46,47,48,49,50,51,52] might be subdivided in two main approaches: the lateral orbital approach and the intracranial approach. The lateral approach consists of a lateral orbitotomy to enter the orbital cavity and dissect the dura of the temporal lobe off of the periorbita in order to reconstruct the skull base from an anterior view. The procedure may be assisted by an intraoperative computed tomography or neuronavigation to check the positioning of the interposing material used to reestablish the delicate anatomy of the region [44, 46,47,48, 53].
The intracranial approach allows reconstructing the skull base from the interior of the skull allowing a better view of the operatory field and consequently a safer management of the bone defect. With this approach, the retraction of the temporal lobe and the separation of dura from the periorbital tissues are easier than using the orbital approach, and the preservation of the optic nerve is safer [47]. A transient CSF diversion may be needed in order to reduce the intracranial pressure to perform the extradural retraction of the temporal lobe safely and reduce the volume of the CSF pooling usually present at the pole of this lobe [44]. Furthermore, the intracranial approach favors, when necessary, the excision of herniated gliotic temporal lobe tissue and dural grafting, as well as the placement of the material used to create the interposition between the orbital and cranial cavities.
The defect of sphenoid dysplasia can be repaired by using bone grafts, titanium meshes, high-density porous polyethylene implants, or a combination of them [44, 50, 52, 54, 55].
The success of the operation is based on the accurate modeling of the implant utilized to reconstruct the skull base and posterior orbit wall, its stable anchoring, and the correct choice of the implant material.
Traditionally, split bone grafts from iliac crest or calvaria or ribs were used to repair sphenoid wing dysplasia in NF-1. It was for several years the standard technique for craniofacial reconstruction in many cases. Bone grafts have the advantage of being completely tolerable. Ribs have the advantage of being malleable and able to integrate with the surrounding bone tissue; split calvarial bone grafts are easily harvested in the required size and are hard and less absorbable than bone from other sites. However, both types of grafts share the same limitations. Their use increases the operative time and is weighted by the morbidity of the donor site and their reabsorption in a significant percentage of the cases besides the risk of infection [47]. Out of 14 patients with pulsating exophthalmos described by Snyder and coworkers, 11 patients were treated with bone graft only and 4 suffered from recurrence because of implant resorption [48]. In addition, to reabsorption, the bone material is rigid and difficult to sculpt to reconstruct the curved shape of the greater wing of sphenoid and the bony orbital skeleton.
To solve the problem of bone resorption, allogenic materials were introduced namely methyl methacrylate, vicryl, hydroxyapatite, demineralized bone, and titanium (Table 1). Their use has the advantages of avoiding donor site morbidity and graft resorption, reducing surgical time, and the absence of spontaneous remodeling that is a high stability of the construct. Titanium meshes are malleable and can mimic the contour of the anterior and middle cranial fossae floor; their use reduces operative time [49] and prevents recurrence of herniation and proptosis in many cases. Titanium meshes can be also used in association with bone implants or other allogenic materials. Wu et al. described its use with computer-aided design/computer-aided manufactured (CAD-CAM) method [50], and Friedrich et al. utilized a computed cone-beam computed tomography system during the surgery to check the good position of the implant [51]. However, allogenic material carries a risk of infection and development of adhesions; dural herniation or meningoencephalocele through the mesh holes has been reported [44, 47, 49]. In case of revision, it could be risky and challenging for the surgeon to separate titanium mesh and soft tissues [49, 52]. Another drawback is interferences during radiological CT and MRI follow-up [53, 54]. In order to avoid adherence between tissues and titanium mesh or meningoencephalocele through its holes, some techniques have been described using mixed implants. Friedrich et al. described the use of titanium mesh associated with iliac spongiosa bone graft through a lateral orbitotomy under navigator guidance. The defect was therefore repaired extracranially. But it required a revised surgery 6 months later because of turned mesh and resorption of bone graft. More extended mesh was implanted, and iliac spongiosa bone graft was placed on both sides [55]. High-density porous polyethylene implants (MEDPOR®) were also utilized with stable results and without secondary displacement or resorption [49, 56]. Niddam et al. used a 0.85-mm titanium-reinforced porous polyethylene implant sheet, which was modeled intraoperatively according to the orbital cavity anatomy. Porous polyethylene sheet reduces the risk of adhesions and brain herniation through the mesh holes. No screw fixation is necessary, and it decreases the risk of radiologic interference and infection. These implants are biocompatible, resilient, radiolucent, and non-resorbable [49].
Finally, another material that can be used to easily reshape the contour and avoid any adherence is methyl methacrylate that we use in our craniofacial unit to reconstruct the greater wing of the sphenoid in such cases.
All the authors have underlined the use of malleable material apt to create implants that mimic the contour of the anterior temporal fossa and posterior wall of the orbit in the assumption that the shape of this type of implants would assure a better stability. Even a preformed computer-created implants based on preoperative CT scan to better fit with the bone lacuna in the single subject have been considered in this direction. Another technique was described by Di Rocco et al. who, rather than a concave construct covering the skull base defect, use a curved a titanium mesh covered by lyophilized dura with the convex surface against the retracted temporal pole in order to oppose its anterior displacement and compensate for its pulsations [44]. Indeed, the CSF pulses had blamed to favor the reabsorption of the bony implants and further erosion of the margins of the lacuna. This C-shaped titanium mesh covered with liodura, with its posterior convexity over the temporal lobe, will accommodate the CSF pulses because of its elasticity; its lateral borders implanted on lateral and mesial walls of the temporal fossa, the volume of which progressively diminishes in postero-anterior direction, will undergo a self-anchorage under the pressure exerted by the brain over its convex central part. Thus, no screw fixation is necessary and dislocation of the implant resulted to be impossible [44]. The technique was successfully utilized in 4 NF-1 subjects, in two children to correct the malformation and prevent its progression and in two advanced adolescent cases that subsequently could undergo the intervention of the maxillo-facial surgeon for correcting the orbit and facial cosmetic abnormalities. Such concave shape can be also obtained with other materials (methyl methacrylate for instance).
Conclusion
Early diagnosis of NF-1 and a multidisciplinary treatment is important for young patients. The article reviews the craniofacial bone alterations in patients with NF-1. Facial bones in patients with NF-1 are short in the anteroposterior direction. Typical craniofacial characteristics of NF-1 are short mandible, maxilla, and cranial base compared with healthy controls. Sphenoid dysplasia is the most distinctive feature of this syndrome. Repair of the great sphenoid wing using a transcranial approach has become more practiced even if lateral orbital approach has been described. Bone graft material may be taken from the skull or iliac crest. And the bone graft is wired, plated, or screwed into the position of the defect.
Unfortunately, resorption of bone grafts has been a key limitation in the reconstruction of sphenoid wing dysplasia. For such a reason, other techniques have been described such as titanium mesh used alone or in combination with bone grafts or tissue, methyl methacrylate, and high-density porous polyethylene implants.
References
Heervä E, Peltonen S, Pirttiniemi P, Happonen RP, Visnapuu V, Peltonen J (2011) Short mandible, maxilla and cranial base are common in patients with neurofibromatosis 1. Eur J Oral Sci 119:121–127. https://doi.org/10.1111/j.1600-0722.2011.00811.x
(1988) Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol 45:575–578
Ferner RE (2010) The neurofibromatoses. Pract Neurol 10:82–93. https://doi.org/10.1136/jnnp.2010.206532
Prudhomme L, Delleci C, Trimouille A, et al (2019) Severe thoracic and spinal bone abnormalities in neurofibromatosis type 1. Eur J Med Genet 103815. https://doi.org/10.1016/j.ejmg.2019.103815
Molins G, Valls A, Silva L, Blasco J, Hernández-Alfaro F (2017) Neurofibromatosis type 1 and right mandibular hypoplasia: unusual diagnosis of occlusion of the left common carotid artery. J Clin Anesth 42:98–99. https://doi.org/10.1016/j.jclinane.2017.08.028
Wallace MR, Marchuk DA, Andersen LB, Letcher R, Odeh H, Saulino A, Fountain J, Brereton A, Nicholson J, Mitchell A, (1990) Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science 249:181–186. https://doi.org/10.1126/science.2134734
Cawthon RM, Weiss R, Xu GF, Viskochil D, Culver M, Stevens J, Robertson M, Dunn D, Gesteland R, O’Connell P, White R (1990) A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell 62:193–201. https://doi.org/10.1016/0092-8674(90)90253-b
Viskochil D, Buchberg AM, Xu G, Cawthon RM, Stevens J, Wolff RK, Culver M, Carey JC, Copeland NG, Jenkins NA, White R, O’Connell P (1990) Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell 62:187–192. https://doi.org/10.1016/0092-8674(90)90252-a
Marchuk DA, Saulino AM, Tavakkol R, Swaroop M, Wallace MR, Andersen LB, Mitchell AL, Gutmann DH, Boguski M, Collins FS (1991) cDNA cloning of the type 1 neurofibromatosis gene: complete sequence of the NF1 gene product. Genomics 11:931–940. https://doi.org/10.1016/0888-7543(91)90017-9
Elefteriou F, Kolanczyk M, Schindeler A, Viskochil DH, Hock JM, Schorry EK, Crawford AH, Friedman JM, Little D, Peltonen J, Carey JC, Feldman D, Yu X, Armstrong L, Birch P, Kendler DL, Mundlos S, Yang FC, Agiostratidou G, Hunter-Schaedle K, Stevenson DA (2009) Skeletal abnormalities in neurofibromatosis type 1: approaches to therapeutic options. Am J Med Genet A 149A:2327–2338. https://doi.org/10.1002/ajmg.a.33045
Yu X, Chen S, Potter OL, Murthy SM, Li J, Pulcini JM, Ohashi N, Winata T, Everett ET, Ingram D, Clapp WD, Hock JM (2005) Neurofibromin and its inactivation of Ras are prerequisites for osteoblast functioning. Bone 36:793–802. https://doi.org/10.1016/j.bone.2005.01.022
Alwan S, Tredwell SJ, Friedman JM (2005) Is osseous dysplasia a primary feature of neurofibromatosis 1 (NF1)? Clin Genet 67:378–390. https://doi.org/10.1111/j.1399-0004.2005.00410.x
Luna E-B, Janini M-E-R, Lima F et al (2018) Craniomaxillofacial morphology alterations in children, adolescents and adults with neurofibromatosis 1: a cone beam computed tomography analysis of a Brazilian sample. Med Oral Patol Oral Cir Bucal 23:e168–e179. https://doi.org/10.4317/medoral.22155
Jouhilahti E-M, Visnapuu V, Soukka T, Aho H, Peltonen S, Happonen RP, Peltonen J (2012) Oral soft tissue alterations in patients with neurofibromatosis. Clin Oral Investig 16:551–558. https://doi.org/10.1007/s00784-011-0519-x
Visnapuu V, Peltonen S, Alivuotila L, Happonen RP, Peltonen J (2018) Craniofacial and oral alterations in patients with neurofibromatosis 1. Orphanet J Rare Dis 13:131. https://doi.org/10.1186/s13023-018-0881-8
D’Ambrosio JA, Langlais RP, Young RS (1988) Jaw and skull changes in neurofibromatosis. Oral Surg Oral Med Oral Pathol 66:391–396. https://doi.org/10.1016/0030-4220(88)90252-6
Lorson EL, DeLong PE, Osbon DB, Dolan KD (1977) Neurofibromatosis with central neurofibroma of the mandible: review of the literature and report of case. J Oral Surg Am Dent Assoc 1965 35:733–738
Javed F, Ramalingam S, Ahmed HB, Gupta B, Sundar C, Qadri T, al-Hezaimi K, Romanos GE (2014) Oral manifestations in patients with neurofibromatosis type-1: a comprehensive literature review. Crit Rev Oncol Hematol 91:123–129. https://doi.org/10.1016/j.critrevonc.2014.02.007
Koblin I, Reil B (1975) Changes of the facial skeleton in cases of neurofibromatosis. J Maxillofac Surg 3:23–27. https://doi.org/10.1016/s0301-0503(75)80009-9
Cung W, Freedman LA, Khan NE, et al (2015) Cephalometry in adults and children with neurofibromatosis type 1: implications for the pathogenesis of sphenoid wing dysplasia and the “NF1 facies.” Eur J Med Genet 58:584–590. https://doi.org/10.1016/j.ejmg.2015.09.001
Friedrich RE, Lehmann J-M, Rother J, Christ G, zu Eulenburg C, Scheuer HT, Scheuer HA (2017) A lateral cephalometry study of patients with neurofibromatosis type 1. J Cranio-Maxillo-fac Surg Off Publ Eur Assoc Cranio-Maxillo-fac Surg 45:809–820. https://doi.org/10.1016/j.jcms.2017.02.011
Friedrich RE, Giese M, Schmelzle R et al (2003) Jaw malformations plus displacement and numerical aberrations of teeth in neurofibromatosis type 1: a descriptive analysis of 48 patients based on panoramic radiographs and oral findings. J Cranio-Maxillo-fac Surg Off Publ Eur Assoc Cranio-Maxillo-fac Surg 31:1–9. https://doi.org/10.1016/s1010-5182(02)00160-9
Van Damme PA, Freihofer HP, De Wilde PC (1996) Neurofibroma in the articular disc of the temporomandibular joint: a case report. J Cranio-Maxillo-fac Surg Off Publ Eur Assoc Cranio-Maxillo-fac Surg 24:310–313. https://doi.org/10.1016/s1010-5182(96)80065-5
Shapiro SD, Abramovitch K, Van Dis ML et al (1984) Neurofibromatosis: oral and radiographic manifestations. Oral Surg Oral Med Oral Pathol 58:493–498. https://doi.org/10.1016/0030-4220(84)90350-5
Visnapuu V, Peltonen S, Tammisalo T, Peltonen J, Happonen RP (2012) Radiographic findings in the jaws of patients with neurofibromatosis 1. J Oral Maxillofac Surg Off J Am Assoc Oral Maxillofac Surg 70:1351–1357. https://doi.org/10.1016/j.joms.2011.06.204
Lammert M, Friedrich RE, Friedman JM, Mautner VF, Tucker T (2007) Early primary tooth eruption in neurofibromatosis 1 individuals. Eur J Oral Sci 115:425–426. https://doi.org/10.1111/j.1600-0722.2007.00474.x
Tucker T, Birch P, Savoy DM, Friedman JM (2007) Increased dental caries in people with neurofibromatosis 1. Clin Genet 72:524–527. https://doi.org/10.1111/j.1399-0004.2007.00886.x
Visnapuu V, Peltonen S, Ellilä T, Kerosuo E, Väänänen K, Happonen RP, Peltonen J (2007) Periapical cemental dysplasia is common in women with NF1. Eur J Med Genet 50:274–280. https://doi.org/10.1016/j.ejmg.2007.04.001
Dalili Z, Adham G (2012) Intraosseous neurofibroma and concurrent involvement of the mandible, maxilla and orbit: report of a case. Iran J Radiol Q J Publ Iran Radiol Soc 9:45–49. https://doi.org/10.5812/iranjradiol.6684
Krishnamoorthy B, Singh P, Gundareddy SN et al (2013) Notching in the posterior border of the ramus of mandible in a patient with neurofibromatosis type I - a case report. J Clin Diagn Res JCDR 7:2390–2391. https://doi.org/10.7860/JCDR/2013/5952.3534
Avcu N, Kansu O, Uysal S, Kansu H (2009) Cranio-orbital-temporal neurofibromatosis with cerebral hemiatrophy presenting as an intraoral mass: a case report. J Calif Dent Assoc 37:119–121
Kaplan I, Calderon S, Kaffe I (1994) Radiological findings in jaws and skull of neurofibromatosis type 1 patients. Dento Maxillo Facial Radiol 23:216–220. https://doi.org/10.1259/dmfr.23.4.7835527
Uchiyama Y, Sumi T, Marutani K, Takaoka H, Murakami S, Kameyama H, Yura Y (2018) Neurofibromatosis type 1 in the mandible. Ann Maxillofac Surg 8:121–123. https://doi.org/10.4103/ams.ams_135_17
Lee L, Yan YH, Pharoah MJ (1996) Radiographic features of the mandible in neurofibromatosis: a report of 10 cases and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 81:361–367. https://doi.org/10.1016/s1079-2104(96)80338-6
Szudek J, Birch P, Friedman JM (2000) Growth in North American white children with neurofibromatosis 1 (NF1). J Med Genet 37:933–938. https://doi.org/10.1136/jmg.37.12.933
Greenwood RS, Tupler LA, Whitt JK, Buu A, Dombeck CB, Harp AG, Payne ME, Eastwood JD, Krishnan KRR, MacFall JR (2005) Brain morphometry, T2-weighted hyperintensities, and IQ in children with neurofibromatosis type 1. Arch Neurol 62:1904–1908. https://doi.org/10.1001/archneur.62.12.1904
Arrington DK, Danehy AR, Peleggi A, Proctor MR, Irons MB, Ullrich NJ (2013) Calvarial defects and skeletal dysplasia in patients with neurofibromatosis type 1. J Neurosurg Pediatr 11:410–416. https://doi.org/10.3171/2013.1.PEDS12409
Jacquemin C, Bosley TM, Svedberg H (2003) Orbit deformities in craniofacial neurofibromatosis type 1. AJNR Am J Neuroradiol 24:1678–1682
Onbas O, Aliagaoglu C, Calikoglu C, Kantarci M, Atasoy M, Alper F (2006) Absence of a sphenoid wing in neurofibromatosis type 1 disease: imaging with multidetector computed tomography. Korean J Radiol 7:70–72. https://doi.org/10.3348/kjr.2006.7.1.70
Balda V, Krishna S, Kadali S (2016) A rare case of neurofibromatosis type 1. J Dr NTR Univ Health Sci 5:222. https://doi.org/10.4103/2277-8632.191849
Jacquemin C, Bosley TM, Liu D et al (2002) Reassessment of sphenoid dysplasia associated with neurofibromatosis type 1. AJNR Am J Neuroradiol 23:644–648
Tam A, Sliepka JM, Bellur S, Bray CD, Lincoln CM, Nagamani SCS (2018) Neuroimaging findings of extensive sphenoethmoidal dysplasia in NF1. Clin Imaging 51:160–163. https://doi.org/10.1016/j.clinimag.2018.04.017
Friedman JM, Birch PH (1997) Type 1 neurofibromatosis: a descriptive analysis of the disorder in 1,728 patients. Am J Med Genet 70:138–143. https://doi.org/10.1002/(sici)1096-8628(19970516)70:2<138::aid-ajmg7>3.0.co;2-u
Di Rocc C, Samii A, Tamburrini G et al (2017) Sphenoid dysplasia in neurofibromatosis type 1: a new technique for repair. Childs Nerv Syst ChNS Off J Int Soc Pediatr Neurosurg 33:983–986. https://doi.org/10.1007/s00381-017-3408-z
Macfarlane R, Levin AV, Weksberg R, Blaser S, Rutka JT (1995) Absence of the greater sphenoid wing in neurofibromatosis type I: congenital or acquired: case report. Neurosurgery 37:129–133. https://doi.org/10.1227/00006123-199507000-00020
Dale EL, Strait TA, Sargent LA (2014) Orbital reconstruction for pulsatile exophthalmos secondary to sphenoid wing dysplasia. Ann Plast Surg 72:S107–S111. https://doi.org/10.1097/SAP.0000000000000090
Lotfy M, Xu R, McGirt M, Sakr S, Ayoub B, Bydon A (2010) Reconstruction of skull base defects in sphenoid wing dysplasia associated with neurofibromatosis I with titanium mesh. Clin Neurol Neurosurg 112:909–914. https://doi.org/10.1016/j.clineuro.2010.07.007
Snyder BJ, Hanieh A, Trott JA, David DJ (1998) Transcranial correction of orbital neurofibromatosis. Plast Reconstr Surg 102:633–642. https://doi.org/10.1097/00006534-199809030-00005
Niddam J, Bosc R, Suffee TM, le Guerinel C, Wolkenstein P, Meningaud JP (2014) Treatment of sphenoid dysplasia with a titanium-reinforced porous polyethylene implant in orbitofrontal neurofibroma: report of three cases. J Cranio-Maxillo-fac Surg Off Publ Eur Assoc Cranio-Maxillo-fac Surg 42:1937–1941. https://doi.org/10.1016/j.jcms.2014.08.004
Wu C-T, Lee S-T, Chen J-F, Lin KL, Yen SH (2008) Computer-aided design for three-dimensional titanium mesh used for repairing skull base bone defect in pediatric neurofibromatosis type 1. A novel approach combining biomodeling and neuronavigation. Pediatr Neurosurg 44:133–139. https://doi.org/10.1159/000113116
Friedrich RE (2011) Reconstruction of the sphenoid wing in a case of neurofibromatosis type 1 and complex unilateral orbital dysplasia with pulsating exophthalmos. Vivo Athens Greece 25:287–290
Martin MP, Olson S (2009) Post-operative complications with titanium mesh. J Clin Neurosci Off J Neurosurg Soc Australas 16:1080–1081. https://doi.org/10.1016/j.jocn.2008.07.087
Fiala TG, Novelline RA, Yaremchuk MJ (1993) Comparison of CT imaging artifacts from craniomaxillofacial internal fixation devices. Plast Reconstr Surg 92:1227–1232
Fiala TG, Paige KT, Davis TL et al (1994) Comparison of artifact from craniomaxillofacial internal fixation devices: magnetic resonance imaging. Plast Reconstr Surg 93:725–731. https://doi.org/10.1097/00006534-199404000-00011
Friedrich RE, Heiland M, Kehler U, Schmelzle R (2003) Reconstruction of sphenoid wing dysplasia with pulsating exophthalmos in a case of neurofibromatosis type 1 supported by intraoperative navigation using a new skull reference system. Skull Base Off J North Am Skull Base Soc Al 13:211–217. https://doi.org/10.1055/s-2004-817697
Madill KE, Brammar R, Leatherbarrow B (2007) A novel approach to the management of severe facial disfigurement in neurofibromatosis type 1. Ophthal Plast Reconstr Surg 23:227–228. https://doi.org/10.1097/IOP.0b013e31805593f1
Krastinova-Lolov D, Hamza F (1996) The surgical management of cranio-orbital neurofibromatosis. Ann Plast Surg 36:263–269. https://doi.org/10.1097/00000637-199603000-00006
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest related to this manuscript.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Chauvel-Picard, J., Lion-Francois, L., Beuriat, PA. et al. Craniofacial bone alterations in patients with neurofibromatosis type 1. Childs Nerv Syst 36, 2391–2399 (2020). https://doi.org/10.1007/s00381-020-04749-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-020-04749-6