
Abstract
Purpose
Following curative treatment for localised renal cell carcinoma (RCC), up to 30% of patients develop tumour recurrence. Prognostic scores are essential to guide individualised surveillance protocols, patient counselling and potentially in the future to guide adjuvant therapy. In metastatic RCC, prognostic scores are routinely used for treatment selection in clinical practice as well as in all major trials.
Methods
We performed a literature review on the current evidence based on prognostic factors and models for localised and metastatic RCC.
Results
A number of prognostic factors have been identified, of which tumour node metastasis classification remains the most important. Multiple prognostic models and nomograms have been developed for localised disease, based on a combination of tumour stage, grade, subtype, clinical features, and performance status. However, there is poor level of evidence for their routine use. Prognostic scores for patients with metastatic RCC receiving targeted treatments are used routinely, but have limited accuracy. Molecular markers can improve the accuracy of established prognostic models, but frequently lack external, independent validation.
Conclusion
Several factors and models predict prognosis of localised and metastatic RCC. They represent valuable tools to provide estimates of clinically important endpoints, but their accuracy should be improved further. Validation of molecular markers is a future research priority.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Renal cell carcinoma (RCC) is the most common malignancy of the kidney, with more than 300,000 new cases diagnosed each year [1]. Due to the increased use of cross-sectional abdominal imaging, the majority of patients present with early stage disease. Complete surgical resection is the only curative treatment for localised RCC. Partial nephrectomy is the standard of care for clinical T1a tumours and provides excellent cancer control with optimal preservation of renal function [2], while total nephrectomy is preferred for patients with clinical T1b–T4 tumours. Following curative treatment for localised RCC, up to 30% of patients develop tumour recurrence after being considered disease-free [3,4,5]. An accurate prediction of the individual likelihood of recurrence based on prognostic factors is essential to counsel patients, individualise surveillance, and select patients for adjuvant clinical trials. In the absence of approved adjuvant therapies providing an overall survival (OS) benefit, the current paradigm is for close surveillance to detect recurrent disease at an earlier stage [6]. However, if such adjuvant treatment option becomes available, the most accurate prognostic system available will ensure correct allocation of patients to adjuvant therapy whilst ensuring the avoidance of toxicity of patients who will not gain any benefit.
Advances in molecular sciences have improved our understanding of RCC biology and have led to significant evolvement of the treatment landscape for metastatic RCC (mRCC). Inhibitors of the vascular endothelial growth factor (VEGF) pathway, the mammalian target of rapamycin pathway, and immune checkpoints have demonstrated improvements in objective response rates, progression-free survival (PFS), and OS compared with standard of care. A number of clinical prognostic factors have been identified based on clinical trials and retrospective multivariable analyses, leading to the creation of prognostic models assigning patients to favourable-, intermediate-, and poor-risk categories. These models are the basis for all major trials and became the standard for patient counselling and treatment selection in clinical practice.
This review provides an overview on existing prognostic factors and prognostic models for localised and metastatic RCC.
Methods
A non-systematic literature search was conducted using Medline. The database was searched without time limit on 20 December 2017 using the terms (“factors” OR “models”) AND (“prognostic” OR “prognosis” OR “predictive”) in conjunction with “Carcinoma, Renal Cell”[Mesh] AND “Adult”[Mesh], revealing 2068 articles. Auto-alerts in Medline were also searched and reference lists of selected manuscripts were checked manually for eligible original articles. There was no language restriction. A list of articles judged to be relevant was circulated by the first author, and all authors reached a consensus for final inclusion of studies in this review.
Results
Definitions
A prognostic factor provides information about disease outcome irrespective of treatment and in the absence of any treatment and thereby the natural course of the disease [7]. In contrast, a marker is predictive if the effect of a given treatment is different for marker-positive and marker-negative patients [7]. Prognostic and predictive markers should be surrogates of biological processes, i.e., the pathophysiology of disease states and the effects of an intervention. As we do not have the full picture of those processes for any disease and specifically not for RCC, a marker must be validated and constantly re-evaluated [8]. If the marker or molecule has repeatedly shown to indicate biological processes by predicting clinically relevant outcomes, it can be called a “biomarker” or “biomolecule”.
Except for OS, definitions for time-to-event endpoints in RCC were not standardised until recently when the DATECAN renal cancer group used a modified Delphi method for establishing a consensus [9]. In the first round of this process, there was only 31% consensus for all time-to-event endpoints. After the third round, the group defined primary outcome measures for initially localised RCC as disease-free survival (DFS), metastasis-free survival (MFS) and local regional-free survival. For metastatic disease, the group recommended the use of PFS and RCC-specific survival. These definitions should become standard practice for future clinical trials and retrospective studies, facilitating reporting, interpretation and comparisons.
Prognostic factors
Prognostic factors are historically sub-classified into anatomical, histological, clinical and molecular factors. In general, the use of anatomical and histological prognostic factors is supported by a higher level of evidence than the use of clinical and molecular prognostic factors.
Anatomical prognostic factors
The classic anatomical prognostic factor is the tumour, node and metastasis (TNM) classification, which has been the most commonly used staging system for a number of decades [10]. TNM includes several prognostic features, such as tumour size, invasion of the venous system, invasion of the collecting system, extension into the adrenal gland, extension beyond the renal capsule or Gerota’s fascia (T classification), and spread to regional lymph nodes or distant sites (N and M classification). In all RCC subtypes, prognosis worsens with increasing T, N, and M classification [11]. In patients with mRCC, prognosis is further driven by the number of metastatic sites and the site of metastasis [12, 13]. The use of TNM as prognostic factor is recommended by the European Association of Urology (EAU) RCC guideline panel [14].
Histological prognostic factors
This group of prognostic factors comprises tumour grade, subtype, presence of sarcomatoid or rhabdoid features, microvascular invasion (MVI), and tumour necrosis. Among these factors, use of tumour grade and subtype is recommended by the EAU guidelines [14].
The four-tiered Fuhrman grading [15] is currently the most widely accepted and validated prognostic grading system [16], although simplified two- or three-tiered modifications may be as accurate [17, 18]. The prognostic value, its inter- and intra-observer variability, and thus the applicability of the Fuhrman grading system have been shown to be suboptimal. Therefore, a new grading system was proposed by the International Society of Urological Pathology (ISUP) in 2012 [19] and subsequently recommended for routine clinical use by the World Health Organisation (WHO). In this ISUP grading system, grades 1–3 are defined based on nucleolar prominence, while extreme nuclear pleomorphism, sarcomatoid or rhabdoid differentiation relates to grade 4. It has been suggested for use in clear cell and papillary RCC, while chromophobe RCC should not be graded according to this system [19].
Historically regarded as a single entity, RCC is now recognised as a family of cancers that result from distinct genetic abnormalities with unique morphologic features, but all are derived from renal tubular epithelium. There were significant changes in the most recent WHO classification with several newly defined renal cell tumours [20]; however, the prognostic role of these rare entities is not yet defined. Among the five most common subtypes, it is well established that patients with collecting duct and unclassified RCC have the poorest outcomes [19]. For clear cell, papillary and chromophobe RCC, studies indicate that patients with clear cell RCC treated by surgery have worse prognosis on univariable analysis, but this difference disappears after adjustment for stage and grade [21, 22]. In patients with sarcomatoid features (grade 4), an underlying non-clear cell RCC appears to have worse prognosis than clear cell RCC [23]. Subtyping of papillary RCC into type 1 and type 2 has been suggested by Delahunt and Eble [24] and correlates with more advanced grade [25, 26], but generally loses statistical significance on multivariable analysis [26,27,28]. Furthermore, there is a significant overlap in their immunohistochemical and histopathological features and a morphological continuum between the two types [25]. It has been suggested that grade should be used instead of papillary type [29].
Metastatic spread of RCC occurs through haematogenous and lymphatic routes. While macroscopic tumour invasion into the major vessels has been recognised as a prognostic factor within the TNM system for some decades, MVI has not been acknowledged yet. MVI refers to the presence of tumour cells within microscopic veins or lymphatic vessels. Several retrospective observational studies associated MVI with adverse outcomes [30]. Furthermore, a recent meta-analysis on 14,946 patients showed a 2–3-fold increased risk of recurrence, metastatic spread and RCC-specific death for patients with MVI [31]. There remain several issues with MVI, specifically with the definition of “micro” and the overlap between micro- and macrovascular invasion.
Coagulative necrosis occurs when tumours outgrow their blood supply, reflecting an aggressive biology, rapid proliferation, and progression. It is evaluated microscopically and estimated visually in increments of 5%. The presence of tumour necrosis has been linked with larger tumour sizes, higher TNM stages, venous invasion, higher tumour grades, and poorer survival [32]. Scoring of necrosis according to its extent appears to be superior to a presence/absence classification [33].
Clinical prognostic factors
Numerous clinical prognostic factors have been investigated, including performance status (PS), presenting symptoms, paraneoplastic syndromes, and laboratory values such as calcium, albumin, hemoglobin, and C-reactive protein (CRP). Based on data of multiple retrospective studies in mRCC, prognostic models based on several clinical factors formed the basis for inclusion into major clinical trials and subsequently for routine risk-group assignment and treatment decisions [34, 35]. There is also low-level evidence demonstrating their prognostic ability in non-metastatic disease [3, 36, 37], though they are not used in routine care.
Molecular prognostic factors
The identification of molecular markers has led to new insights into RCC biology and the development of novel targeted therapies. Evidence regarding prognostic markers has been reviewed recently [10]. In brief, protein staining data of proliferation markers such as Ki-67, p53, PTEN as well as factors of the hypoxia-inducible factor pathways such as carbonic anhydrase IX and the VEGF family have been studied extensively [10, 38]. Furthermore, there are emerging data on associations of somatic mutations, gene methylations, gene expression data, germline variations, and immune biomarkers such as CD8 and PD-L1 with prognosis [39,40,41,42,43,44,45,46]. Some of these markers have improved the discrimination of current prognostic models, especially when markers were used in combination; however, none was extensively validated and there remain methodological issues. As such, their routine use in clinical practice is currently not recommended [14].
Prognostic models
As a prognostic factor is not accurate when used alone, factors have been combined in multivariable prognostic models. Synonyms for prognostic models include prognostic (or prediction) index, risk (or clinical) prediction model, and predictive model [47]. In these models, TNM has been supplemented by several additional routinely available factors, such as presenting symptoms, PS, laboratory data (i.e., haemoglobin, cholesterol, lactate dehydrogenase, neutrophil count, platelet count, and C-reactive protein), and coagulative tumour necrosis [3, 33, 48,49,50,51,52,53]. These models contain an outcome variable (dependent, i.e., DFS, MFS, or OS) and two or more explanatory (independent) variables that are being evaluated for their association with clinical outcomes. A nomogram is a simple graphical representation of a multivariable prognostic model, which is based on the equation derived from the regression coefficients of each variable. It can be used for estimating individual probabilities at certain landmark points.
The accuracy of prognostic models is typically evaluated with a measure of discrimination, such as the area under the curve or the concordance index (C-index). Discrimination ranges from 0.5 (chance) to 1 (perfect), but it is unknown how “high is high enough” to justify use of a new model. As such, prognostic models are calibrated and a decision-curve analysis (DCA) can be used for judging whether a model should be used for clinical decision-making [54].
There are several prognostic models for patients at all RCC stages [10], such as the stage, size, grade and necrosis score (SSIGN) score [55, 56], and the Karakiewicz nomogram [57]. Because of inclusion of all stages, they are seldom used in clinical practice and rather represent a research tool. Their endpoint (usually RCC-specific survival) is neither useful to guide decision-making in any disease setting nor can it aid clinical trial design. Of note, the reported rates for discrimination are somewhat superior to that of models for specified disease stages, as they include M classification and thus the strongest prognostic factor. The following paragraphs focus on prognostic models that were specifically designed for localised RCC (N0/+ M0) following surgery and mRCC (M1).
Prognostic models for clinically localised disease
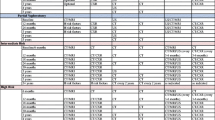
The Leibovich prognostic score [58], the University of California Integrated Staging System (UISS) [59], is the most commonly used postoperative prognostic models for adjuvant trials and can be assigned from routine clinical and pathological data (Table 1). The Leibovich score does include patients with N+ stages, whilst UISS treats those as metastatic. While the Leibovich score was developed for use in clear cell RCC, UISS includes all RCC subtypes. Both the Leibovich score and the UISS were developed on historic TNM definitions, but can be applied to contemporary patients without further conversion except for a subgroup of patients with contiguous extension into the ipsilateral adrenal gland UISS. Neither model includes surgical margin status. While a positive margin is less relevant after partial nephrectomy for T1 tumours [60], it is an important predictor of DFS in patients with advanced T classification [61, 62]. These prognostic models have been compared in terms of discrimination of recurrence and death from RCC. The C-index was around 70% and thus fairly low, although the Leibovich score performed slightly better than the UISS [63]. It merits to be mentioned that the UISS used in ASSURE differs considerably from the original UISS definitions (Table 1). These changes were not validated before the trial was initiated. Separate prognostic models for every RCC subtype were recently proposed by Leibovich et al. [29], but these models were not yet assessed for calibration and net benefit, and are not externally validated.
One of the first prognostic models that were presented in a nomogram was developed by Kattan et al. [64]. The authors studied RFS in 601 patients with clinically localised RCC and developed a nomogram based on symptom classification, subtype, tumour size, and T stage. The overall C-index was 74%. In a further analysis on patients with clear cell RCC, the same group identified tumour necrosis, vascular invasion, and tumour grade as additional prognostic variables. Discrimination in the validation cohort was as high as 82% [65].
Although prognostic models and nomograms are mentioned in current guidelines, their use in routine clinical practice is currently not recommended for localised disease, but they may provide the basis for enrolling patients into adjuvant clinical trials [14]. At present, EAU guidelines recommend risk-based postoperative follow-up imaging based on prognostic models such as the UISS, which appeared to be the most widely used and validated prognostic model to the guideline panel [14]. For low risk patients, the panel proposed an abdominal/renal ultrasound at 6 months and 2 years, and a CT scan of the chest, abdomen and pelvis at 1 and 3 years and bi-yearly thereafter. For intermediate- and high-risk patients, a CT scan of the chest, abdomen, and pelvis was recommended at 6 months, 1, 2, 3 years, and bi-yearly thereafter [14].
Prognostic models for metastatic disease
In 1999, Motzer et al. [66] published the first version of the Memorial Sloan-Kettering Cancer Centre (MSKCC) prognostic model. It was developed on a cohort of 670 patients who were treated within various clinical trials between 1975 and 1996. A poorer Karnofsky PS (< 80%), absence of prior nephrectomy, high lactate dehydrogenase (LDH > 1.5 × ULN), low haemoglobin (< LLN), and high albumin-corrected calcium (> 10 mg/dl) were identified as prognostic factors. The authors generated three risk groups based on the number of adverse prognostic factors: favourable (0 factor), intermediate (1 or 2 factors) and poor (3–5 factors), with median OS of 20, 10, and 4 months, respectively. In an effort to reduce heterogeneity, the same group developed an updated prognostic model by analysing only patients treated with interferon-alpha, which was one of the standard treatments at that time [35]. Here, the variable “nephrectomy” was replaced by “time from diagnosis to treatment” (< 12 vs > 12 months). In the following decade, the 2002 MSKCC model was the standard stratification tool for all major phase III trials leading to registration of approved targeted agents.
The MSKCC model was developed during the cytokine era of mRCC, which was considered a drawback by some authors. Therefore, efforts have been made to establish prognostic models from prospective clinical trial or registry data of patients treated with targeted therapies. Using International Metastatic Renal Cancer Database Consortium (IMDC) data, Heng et al. [34] proposed six prognostic factors, of which four (Karnofsky PS, time from diagnosis to treatment, haemoglobin, corrected calcium) were already included in the 2002 MSKCC model. LDH was removed, while neutrophil and platelet count were added as new variables. Similar to the MSKCC model, a favourable (0 factor), intermediate (1 or 2 factors) and poor prognostic group (3–6 factors) were suggested, which stratified patients according to OS. There were several other initiatives to generate prognostic models in mRCC, which are summarised in Table 2.
Studies comparing discrimination within external cohorts demonstrated C-indices between 64 and 67%, without any relevant differences across models [67]. Data further confirmed that the MSKCC model is applicable to patients in the era of targeted therapy [68]. Even though discrimination is, at best, modest, guidelines give a strong recommendation for the use of prognostic mRCC models without further specification.
At present, current recommendations for management of mRCC are based on IMDC prognostic factors. Cytoreductive nephrectomy may provide an OS benefit in select patients with synchronous mRCC and should not be offered to patients with four or more IMDC prognostic factors at baseline or an estimated survival of < 12 months [13, 69]. Culp et al. [70] developed a prognostic model for patients who underwent cytoreductive nephrectomy and identified high LDH, low albumin, symptoms, liver metastasis, retroperitoneal adenopathy, supradiaphragmatic adenopathy, and clinical tumour classification > or = T3 as predictors of inferior survival; however, this model is currently not validated. Finally, all evidence-based recommendations for systemic treatment are based on IMDC risk group [14], which is dynamic and should be re-assessed during the course of treatment. The IMDC model was developed on treatment naïve patients, but is prognostic for outcomes after second-line treatment as well [71].
Prognostic models with molecular markers
As there is little room for further improvement of conventional prognostic models by adding more routine clinical factors, molecular augmentation of these models has been undertaken by several groups. However, biomolecular approaches to outcome prediction using genomic, transcriptomic, and proteomic signatures are still in an early phase, and none of the published markers is ready for prime time or can be regarded a true “biomarker”. Marker research has multiple challenges, including sample collection and quality, poor clinical information, and small numbers. Lack of validation studies and hurdles to account for tumour heterogeneity are also major issues. Furthermore, there are concerns regarding the applicability of these markers outside of a study/trial setting; however, better selection of patients for trials would improve outcomes with new agents and reduce costs.
Another problem is the application of high-throughput assays with a huge number of markers in a fairly low number of patients. This approach increases the amount of data and complexity of further analyses. High-throughput assays help our understanding of the molecular mechanisms of a tumour, but increase the false discovery rate in prognostic studies. Furthermore, complete and transparent reporting is critical for interpretation of marker studies. The “REporting recommendations for tumour marker prognostic studies” (REMARK) guidelines were released over a decade ago [72], but adherence is low [73].
Brannon et al. [74] stratified clear cell RCC by unsupervised consensus clustering of gene expression microarray data and identified two distinct subtypes within the training set, clear cell type A (ccA) and type B (ccB). In a validation cohort of 177 tumours, patients with ccA had improved disease-specific survival compared to ccB. In further work, the same group developed a 34-gene classifier for localised clear cell RCC (ClearCode34) [75]. The classifier was applied to RNA-sequencing data from 380 samples from the Cancer Genome Atlas and to 157 formalin-fixed samples. ClearCode34 outperformed UISS and the SSIGN score in terms of discrimination. Wei et al. [76] examined 12 data sets using consensus clustering sensitivity analysis. They found that clustering techniques demonstrated relatively unstable tumour classification. As a measure of inter-tumour gene expression heterogeneity, they subsequently developed the continuous CLEAR score (continuous linear enhanced assessment of clear cell RCC) based on an 18-transcript signature. In applying it to multiple external data sets, the authors demonstrated its association with disease-specific survival. Furthermore, lower CLEAR scores were associated with a greater likelihood of response to sunitinib and high dose interleukin-two treatment. Rini et al. [77] studied RNA expression of 732 genes in 942 patients with stage I–III clear cell RCC. Among the 516 genes that were associated with recurrence-free survival, 11 genes of interest and five reference genes were selected for a continuous recurrence score. In an independent validation cohort, the score was associated with recurrence-free survival across all stage categories. In another study using tissue microarray technology, immunohistochemical staining frequencies of Ki-67, p53, VEGFR-1 and VEGF-D were significantly associated with DFS in T1-4 N0 M0 clear cell RCC. A model combining these markers with ECOG PS and T classification reached a C-index of 90%, which was significantly superior to UISS (78%) [78].
Recently, the prognostic ability of ClearCode34 was tested in the setting of systemic therapy for mRCC [79]. On multivariable analyses that adjusted for the effects of IMDC groups, ClearCode34 ccB remained independently associated with poorer OS. The addition of ClearCode34 ccA/ccB to IMDC improved discrimination, but the overall discriminatory power remained poor (63 vs 60%).
Limitations
Current prognostic models have multiple limitations. In general, prognostic models were derived from retrospectively collected data with its inherent biases in primary data collection, absence of quality control and non-standardised treatments and follow-up. Many prognostic models were not validated in independent cohorts or applied and validated in (retrospective) cohorts in which they were not developed. Furthermore, the quality of a prognostic model is typically measured by its ability to predict outcomes, namely by discrimination, calibration and DCA. There is currently no guidance on how well a model should discriminate and how well a model should be calibrated. Furthermore, these statistical measures can be difficult to interpret and do not indicate if a model will improve clinical care or not. A DCA estimates the net benefit of basing clinical decisions on a new prognostic model and compares it to standard strategies or other prognostic models, but the use of threshold probabilities makes assumptions about perceptions of benefits and harms of a certain management approach. Although DCA may help to decide if a prognostic model may be useful in clinical care, it remains a purely statistical method that cannot replace a prospective trial. In this regards, prognostic models should be evaluated in impact trials, which randomises patients to application of the prognostic model (stratified care) or to standard care. Results of impact trials provide an unbiased estimate of whether stratified care improves outcomes. Several impact trials have been conducted (i.e., [80]), but none in RCC.
Conclusions
TNM stage is the most important conventional prognostic factor in RCC. Data indicate that a ceiling has been reached in predicting prognosis based solely on anatomical, histological and clinical factors [81]. Despite significant developments in marker research and statistical methods, no clinically significant advances in prognostic models were seen in the past decade. Molecular prognostic models are under-investigated and should be addressed as a priority by future research. More emphasis should be placed on prognostic score and biomarker validation studies. A hierarchy of validation cohorts should be considered with prospectively collected clinical trials tissues as the optimal source for validation [82].
References
Ferlay J, Soerjomataram I, Dikshit R et al (2015) Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 136:E359–E386
Ljungberg B, Bensalah K, Canfield S et al (2015) EAU guidelines on renal cell carcinoma: 2014 update. Eur Urol 67:913–924
Lucca I, de Martino M, Hofbauer SL et al (2015) Comparison of the prognostic value of pretreatment measurements of systemic inflammatory response in patients undergoing curative resection of clear cell renal cell carcinoma. World J Urol 33:2045–2052
Haas NB, Manola J, Uzzo RG et al (2016) Adjuvant sunitinib or sorafenib for high-risk, non-metastatic renal-cell carcinoma (ECOG-ACRIN E2805): a double-blind, placebo-controlled, randomised, phase 3 trial. Lancet 387:2008–2016
Lam JS, Shvarts O, Leppert JT et al (2005) Postoperative surveillance protocol for patients with localized and locally advanced renal cell carcinoma based on a validated prognostic nomogram and risk group stratification system. J Urol 174:466–472
Bex A, Albiges L, Ljungberg B et al (2017) Updated European Association of Urology guidelines regarding adjuvant therapy for renal cell carcinoma. Eur Urol 71:719–722
Ballman KV (2015) Biomarker: predictive or prognostic? J Clin Oncol 33:3968–3971
Strimbu K, Tavel JA (2010) What are biomarkers? Curr Opin HIV AIDS 5:463–466
Kramar A, Negrier S, Sylvester R et al (2015) Guidelines for the definition of time-to-event end points in renal cell cancer clinical trials: results of the DATECAN project. Ann Oncol 26:2392–2398
Sun M, Shariat SF, Cheng C et al (2011) Prognostic factors and predictive models in renal cell carcinoma: a contemporary review. Eur Urol 60:644–661
Keegan KA, Schupp CW, Chamie K et al (2012) Histopathology of surgically treated renal cell carcinoma: survival differences by subtype and stage. J Urol 188:391–397
Manola J, Royston P, Elson P et al (2011) Prognostic model for survival in patients with metastatic renal cell carcinoma: results from the international kidney cancer working group. Clin Cancer Res 17:5443–5450
Klatte T, Fife K, Welsh SJ et al (2018) Prognostic effect of cytoreductive nephrectomy in synchronous metastatic renal cell carcinoma: a comparative study using inverse probability of treatment weighting. World J Urol 36:417–425
Ljungberg B, Albiges L, Bensalah K, et al (2018) EAU guidelines on renal cell carcinoma. http://uroweb.org/guideline/renal-cell-carcinoma/. Accessed 1 Apr 2018
Fuhrman SA, Lasky LC, Limas C (1982) Prognostic significance of morphologic parameters in renal cell carcinoma. Am J Surg Pathol 6:655–663
Ficarra V, Martignoni G, Maffei N et al (2005) Original and reviewed nuclear grading according to the Fuhrman system: a multivariate analysis of 388 patients with conventional renal cell carcinoma. Cancer 103:68–75
Rioux-Leclercq N, Karakiewicz PI, Trinh Q-D et al (2007) Prognostic ability of simplified nuclear grading of renal cell carcinoma. Cancer 109:868–874
Sun M, Lughezzani G, Jeldres C et al (2009) A proposal for reclassification of the Fuhrman grading system in patients with clear cell renal cell carcinoma. Eur Urol 56:775–781
Delahunt B, Cheville JC, Martignoni G et al (2013) The International Society of Urological Pathology (ISUP) grading system for renal cell carcinoma and other prognostic parameters. Am J Surg Pathol 37:1490–1504
Moch H, Cubilla AL, Humphrey PA et al (2016) The 2016 WHO classification of tumours of the urinary system and male genital organs—part A: renal, penile, and testicular tumours. Eur Urol 70:93–105
Patard J-J, Leray E, Rioux-Leclercq N et al (2005) Prognostic value of histologic subtypes in renal cell carcinoma: a multicenter experience. J Clin Oncol 23:2763–2771
Klatte T, Han K, Said JW et al (2008) Pathobiology and prognosis of chromophobe renal cell carcinoma. Urol Oncol 26:604–609
Nguyen DP, Vilaseca A, Vertosick EA et al (2016) Histologic subtype impacts cancer-specific survival in patients with sarcomatoid-variant renal cell carcinoma treated surgically. World J Urol 34:539–544
Delahunt B, Eble JN (1997) Papillary renal cell carcinoma: a clinicopathologic and immunohistochemical study of 105 tumors. Mod Pathol Off J US Can Acad Pathol Inc 10:537–544
Alomari AK, Nettey OS, Singh D et al (2015) Clinicopathological and immunohistochemical characteristics of papillary renal cell carcinoma with emphasis on subtyping. Hum Pathol 46:1418–1426
Klatte T, Pantuck AJ, Said JW et al (2009) Cytogenetic and molecular tumor profiling for type 1 and type 2 papillary renal cell carcinoma. Clin Cancer Res Off J Am Assoc Cancer Res 15:1162–1169
Ledezma RA, Negron E, Paner GP et al (2016) Clinically localized type 1 and 2 papillary renal cell carcinomas have similar survival outcomes following surgery. World J Urol 34:687–693
Bigot P, Bernhard J-C, Gill IS et al (2016) The subclassification of papillary renal cell carcinoma does not affect oncological outcomes after nephron sparing surgery. World J Urol 34:347–352
Leibovich BC, Lohse CM, Cheville JC et al (2018) Predicting oncologic outcomes in renal cell carcinoma after surgery. Eur Urol 73(5):772–780
Bedke J, Heide J, Ribback S et al (2018) Microvascular and lymphovascular tumour invasion are associated with poor prognosis and metastatic spread in renal cell carcinoma: a validation study in clinical practice. BJU Int 121:84–92
Huang H, Pan X-W, Huang Y et al (2015) Microvascular invasion as a prognostic indicator in renal cell carcinoma: a systematic review and meta-analysis. Int J Clin Exp Med 8:10779–10792
Sengupta S, Lohse CM, Leibovich BC et al (2005) Histologic coagulative tumor necrosis as a prognostic indicator of renal cell carcinoma aggressiveness. Cancer 104:511–520
Klatte T, Said JW, de Martino M et al (2009) Presence of tumor necrosis is not a significant predictor of survival in clear cell renal cell carcinoma: higher prognostic accuracy of extent based rather than presence/absence classification. J Urol 181:1558–1564
Heng DYC, Xie W, Regan MM et al (2009) Prognostic factors for overall survival in patients with metastatic renal cell carcinoma treated with vascular endothelial growth factor-targeted agents: results from a large, multicenter study. J Clin Oncol 27:5794–5799
Motzer RJ, Bacik J, Murphy BA et al (2002) Interferon-alfa as a comparative treatment for clinical trials of new therapies against advanced renal cell carcinoma. J Clin Oncol 20:289–296
Pichler M, Hutterer GC, Stoeckigt C et al (2013) Validation of the pre-treatment neutrophil-lymphocyte ratio as a prognostic factor in a large European cohort of renal cell carcinoma patients. Br J Cancer 108:901–907
de Martino M, Klatte T, Seemann C et al (2013) Validation of serum C-reactive protein (CRP) as an independent prognostic factor for disease-free survival in patients with localised renal cell carcinoma (RCC). BJU Int 111:E348–E353
Ingels A, Hew M, Algaba F et al (2017) Vimentin over-expression and carbonic anhydrase IX under-expression are independent predictors of recurrence, specific and overall survival in non-metastatic clear-cell renal carcinoma: a validation study. World J Urol 35:81–87
de Martino M, Haitel A, Schatzl G, Klatte T (2013) The protease activated receptor 1 gene variation IVSn-14 A > T is associated with distant metastasis and cancer specific survival in renal cell carcinoma. J Urol 190:1392–1397
Stewart GD, O’Mahony FC, Laird A et al (2014) Carbonic anhydrase 9 expression increases with vascular endothelial growth factor-targeted therapy and is predictive of outcome in metastatic clear cell renal cancer. Eur Urol 66:956–963
Sim SH, Messenger MP, Gregory WM et al (2012) Prognostic utility of pre-operative circulating osteopontin, carbonic anhydrase IX and CRP in renal cell carcinoma. Br J Cancer 107:1131–1137
Pantuck AJ, Seligson DB, Klatte T et al (2007) Prognostic relevance of the mTOR pathway in renal cell carcinoma: implications for molecular patient selection for targeted therapy. Cancer 109:2257–2267
Klatte T, Seligson DB, Riggs SB et al (2007) Hypoxia-inducible factor 1 alpha in clear cell renal cell carcinoma. Clin Cancer Res 13:7388–7393
Joseph RW, Kapur P, Serie DJ et al (2014) Loss of BAP1 protein expression is an independent marker of poor prognosis in patients with low-risk clear cell renal cell carcinoma. Cancer 120:1059–1067
Miao D, Margolis CA, Gao W et al (2018) Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 359:801–806
George DJ, Martini J-F, Staehler M et al (2018) Immune biomarkers predictive for disease-free survival with adjuvant sunitinib in high-risk locoregional renal cell carcinoma: from randomized phase III S-TRAC study. Clin Cancer Res Off J Am Assoc Cancer Res. https://doi.org/10.1158/1078-0432.CCR-17-2822
Steyerberg EW, Moons KGM, van der Windt DA et al (2013) Prognosis research strategy (PROGRESS) 3: prognostic model research. PLoS Med 10(2):e1001381. https://doi.org/10.1371/journal.pmed.1001381
de Martino M, Leitner CV, Seemann C et al (2015) Preoperative serum cholesterol is an independent prognostic factor for patients with renal cell carcinoma (RCC). BJU Int 115:397–404
de Martino M, Klatte T, Seemann C et al (2013) Validation of serum C-reactive protein (CRP) as an independent prognostic factor for disease-free survival in patients with localised renal cell carcinoma (RCC). BJU Int 111:E348–E353
Hofbauer SL, Stangl KI, de Martino M et al (2014) Pretherapeutic gamma-glutamyltransferase is an independent prognostic factor for patients with renal cell carcinoma. Br J Cancer 111:1526–1531
Shim SR, Kim SJ, Kim SI, Cho DS (2017) Prognostic value of the glasgow prognostic score in renal cell carcinoma: a meta-analysis. World J Urol 35:771–780
Moreira DM, Gershman B, Lohse CM et al (2016) Paraneoplastic syndromes are associated with adverse prognosis among patients with renal cell carcinoma undergoing nephrectomy. World J Urol 34:1465–1472
Hu H, Yao X, Xie X et al (2017) Prognostic value of preoperative NLR, dNLR, PLR and CRP in surgical renal cell carcinoma patients. World J Urol 35:261–270
Vickers AJ, Cronin AM (2010) Everything you always wanted to know about evaluating prediction models (but were too afraid to ask). Urology 76:1298–1301
Frank I, Blute ML, Cheville JC et al (2002) An outcome prediction model for patients with clear cell renal cell carcinoma treated with radical nephrectomy based on tumor stage, size, grade and necrosis: the SSIGN score. J Urol 168:2395–2400
Parker WP, Cheville JC, Frank I et al (2017) Application of the stage, size, grade, and necrosis (SSIGN) score for clear cell renal cell carcinoma in contemporary patients. Eur Urol 71:665–673
Karakiewicz PI, Briganti A, Chun FK-H et al (2007) Multi-institutional validation of a new renal cancer-specific survival nomogram. J Clin Oncol 25:1316–1322
Leibovich BC, Blute ML, Cheville JC et al (2003) Prediction of progression after radical nephrectomy for patients with clear cell renal cell carcinoma: a stratification tool for prospective clinical trials. Cancer 97:1663–1671
Zisman A, Pantuck AJ, Wieder J et al (2002) Risk group assessment and clinical outcome algorithm to predict the natural history of patients with surgically resected renal cell carcinoma. J Clin Oncol 20:4559–4566
Borghesi M, Brunocilla E, Schiavina R, Martorana G (2013) Positive surgical margins after nephron-sparing surgery for renal cell carcinoma: incidence, clinical impact, and management. Clin Genitourin Cancer 11:5–9. https://doi.org/10.1016/j.clgc.2012.09.010
Karellas ME, Jang TL, Kagiwada MA et al (2009) Advanced-stage renal cell carcinoma treated by radical nephrectomy and adjacent organ or structure resection. BJU Int 103:160–164
Liu NW, Wren JD, Vertosick E et al (2016) The prognostic impact of a positive vascular margin on pT3 clear cell renal cell carcinoma. J Urol 195:264–269
Tan M-H, Kanesvaran R, Li H et al (2010) Comparison of the UCLA Integrated Staging System and the Leibovich score in survival prediction for patients with nonmetastatic clear cell renal cell carcinoma. Urology 75:1365–1370
Kattan MW, Reuter V, Motzer RJ et al (2001) A postoperative prognostic nomogram for renal cell carcinoma. J Urol 166:63–67
Sorbellini M, Kattan MW, Snyder ME et al (2005) A postoperative prognostic nomogram predicting recurrence for patients with conventional clear cell renal cell carcinoma. J Urol 173:48–51
Motzer RJ, Mazumdar M, Bacik J et al (1999) Survival and prognostic stratification of 670 patients with advanced renal cell carcinoma. J Clin Oncol 17:2530–2540
Heng DYC, Xie W, Regan MM et al (2013) External validation and comparison with other models of the International Metastatic Renal-Cell Carcinoma Database Consortium prognostic model: a population-based study. Lancet Oncol 14:141–148
Patil S, Figlin RA, Hutson TE et al (2011) Prognostic factors for progression-free and overall survival with sunitinib targeted therapy and with cytokine as first-line therapy in patients with metastatic renal cell carcinoma. Ann Oncol 22:295–300
Heng DYC, Wells JC, Rini BI et al (2014) Cytoreductive nephrectomy in patients with synchronous metastases from renal cell carcinoma: results from the International Metastatic Renal Cell Carcinoma Database Consortium. Eur Urol 66:704–710
Culp SH, Tannir NM, Abel EJ et al (2010) Can we better select patients with metastatic renal cell carcinoma for cytoreductive nephrectomy? Cancer 116:3378–3388
Ko JJ, Xie W, Kroeger N et al (2015) The International Metastatic Renal Cell Carcinoma Database Consortium model as a prognostic tool in patients with metastatic renal cell carcinoma previously treated with first-line targeted therapy: a population-based study. Lancet Oncol 16:293–300. https://doi.org/10.1016/S1470-2045(14)71222-7
McShane LM, Altman DG, Sauerbrei W et al (2005) REporting recommendations for tumour MARKer prognostic studies (REMARK). Br J Cancer 93:387–391
Mallett S, Timmer A, Sauerbrei W, Altman DG (2010) Reporting of prognostic studies of tumour markers: a review of published articles in relation to REMARK guidelines. Br J Cancer 102:173–180
Brannon AR, Reddy A, Seiler M et al (2010) Molecular stratification of clear cell renal cell carcinoma by consensus clustering reveals distinct subtypes and survival patterns. Genes Cancer 1:152–163
Brooks SA, Brannon AR, Parker JS et al (2014) ClearCode34: a prognostic risk predictor for localized clear cell renal cell carcinoma. Eur Urol 66:77–84
Wei X, Choudhury Y, Lim WK et al (2016) Recognizing the continuous nature of expression heterogeneity and clinical outcomes in clear cell renal cell carcinoma. Sci Rep 7(1):7342. https://doi.org/10.1038/s41598-017-07191-y
Rini B, Goddard A, Knezevic D et al (2015) A 16-gene assay to predict recurrence after surgery in localised renal cell carcinoma: development and validation studies. Lancet Oncol 16:676–685
Klatte T, Seligson DB, LaRochelle J et al (2009) Molecular signatures of localized clear cell renal cell carcinoma to predict disease-free survival after nephrectomy. Cancer Epidemiol Biomark Prev 18:894–900
de Velasco G, Culhane AC, Fay AP et al (2017) Molecular subtypes improve prognostic value of International Metastatic Renal Cell Carcinoma Database Consortium prognostic model. Oncologist 22:286–292
Battle C, Abbott Z, Hutchings HA et al (2017) Protocol for a multicentre randomised feasibility STUdy evaluating the impact of a prognostic model for management of BLunt chest wall trauma patients: STUMBL trial. BMJ Open 7:e015972. https://doi.org/10.1136/bmjopen-2017-015972
Galsky MD (2013) A prognostic model for metastatic renal-cell carcinoma. Lancet Oncol 14:102–103
Simon RM, Paik S, Hayes DF (2009) Use of archived specimens in evaluation of prognostic and predictive biomarkers. J Natl Cancer Inst 101:1446–1452
Beisland C, Guðbrandsdottir G, Reisæter LAR et al (2016) A prospective risk-stratified follow-up programme for radically treated renal cell carcinoma patients: evaluation after 8 years of clinical use. World J Urol 34:1087–1099
Négrier S, Escudier B, Gomez F et al (2002) Prognostic factors of survival and rapid progression in 782 patients with metastatic renal carcinomas treated by cytokines: a report from the Groupe Français d’Immunothérapie. Ann Oncol 13:1460–1468
Choueiri TK, Rini B, Garcia JA et al (2007) Prognostic factors associated with long-term survival in previously untreated metastatic renal cell carcinoma. Ann Oncol 18:249–255
Author information
Authors and Affiliations
Contributions
TK: project development, data collection, data analysis, manuscript writing and editing. SHR: data analysis, manuscript editing. GDS: project development, data analysis, manuscript editing, supervision.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no relevant conflicts of interest.
Research involving human participants and/or animals
The following manuscript is a review of existing data. Therefore, this article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
For this type of study (review), formal consent is not required.
Rights and permissions
About this article

Cite this article
Klatte, T., Rossi, S.H. & Stewart, G.D. Prognostic factors and prognostic models for renal cell carcinoma: a literature review. World J Urol 36, 1943–1952 (2018). https://doi.org/10.1007/s00345-018-2309-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00345-018-2309-4