
Abstract
Abscisic acid-activated protein kinases (AAPK) are key components in abscisic acid (ABA) signalling and have been implicated in mediating responses to abiotic stresses in plants. However, the mechanistic involvement of AAPK genes in drought stress responses has not been functionally characterized to date in soybean. In this study, we identified Glycine max AAPK-like kinase (GmAALK) genes. Loss-of-function analysis of GmAALK1 in soybean reveal that GmAALK1-RNAi plants are more drought-susceptible than pRAP17-ccdB control lines under drought stress. In contrast, GmAALK1 overexpression lines show enhanced drought resistance under drought stress as compared to pRAP15-ccdB control plants. A root transcriptome analysis reveals the differential expression of many genes related to physiological and biochemical changes in GmAALK1-RNAi plants when compared with the pRAP17-ccdB control plants with or without drought stress. Twelve candidate drought-responsive, differentially-expressed genes have been selected to understand the role of GmAALK1 in during the drought response. Gene expression analyses revealed that GmAALK1 positively modulates the drought stress response. These studies suggest that GmAALK1 overexpressing roots increase the transcription of ABA-responsive genes, indicating that the GmAALK1 is a positive regulator of ABA-mediated stress-signaling pathways in soybean. The phylogenetic and domain analyses also support that GmAALK1 is an AAPK and that it has a role in the drought response. These results provide a genetic foundation for developing drought-tolerant soybean cultivars via manipulating kinase gene family which will be applicable to additional cultivated crops experiencing drought conditions relating to climate change.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Plants face different abiotic and biotic stresses. Among them, drought is one of the most complex, harmful, and least understood abiotic stresses that damages crops, globally (Halwatura et al. 2017). Soybean (Glycine max) is one of the most important crops cultivated worldwide as vegetable and protein source for humans. However, soybean is adversely affected by drought (Song et al. 2016). To cope with environmental stress, plants can sense the changes in their environment, and respond to those changes through different defense mechanisms (Bohnert et al. 2006). There are many stress response mechanisms that plants have evolved, and critical to these responses are the phytohormone pathways. Abscisic acid (ABA) is one of the most vital phytohormones, acting as a signaling mediator in different plant environmental stress responses (Sah et al. 2016). ABA regulates various developmental process and adaptive responses in plants. ABA also plays important role in seed maturation and germination, seedling growth, transpiration, development inhibition, and senescence. In limited water conditions, increased ABA levels trigger stomatal closure and induce stress tolerance responses (Cutler et al. 2010).
Many components of the ABA signaling pathway, such as the ABA receptor (PYR/PYL/RCAR), protein phosphatase 2Cs (PP2Cs), and ABA response element-binding factors (ABFs), have been identified (Klingler et al. 2010). Protein phosphorylation and dephosphorylation are essential mechanisms in ABA signal transduction (Furihata et al. 2006). Stress signals are recognized and transmitted to different cellular compartments via specific signaling pathways in which protein kinases and phosphatase are crucial elements (Kulik et al. 2011). Plant sucrose nonfermenting-1 (SNF1)-related kinases (RK) (SnRKs) are classified into the Sucrose nonfermenting-1 (SNF1) kinases and AMP-activated protein kinases (AMPK). The SNF1/AMPK family genes are homologs of yeast and mammalian AMPKs. AMPK was originally called mammalian protein kinase (Kulik et al. 2011). The SnRKs are divided into three subfamilies, SnRK1, SnRK2, and SnRK3. SnRK2s belong to a family of SnRKs which constitute a class of serine/threonine protein kinases. Members of the SnRK2 family act as a merging point between ABA-independent and ABA-dependent stress signaling pathways. In addition, the SnRK2s play important roles in many developmental processes such as plant growth and developments in plants including being a positive regulator of abiotic stresses (Mustilli 2002; Dey et al. 2016; Mao et al. 2010). Members of SnRK2s family have been identified in different plant species including Arabidopsis thaliana, Oryza sativa (rice), Zea mays (maize), Triticum aestivum (wheat), Brassica napas (rapeseed), Brassica rapa, Vitis vinifera (grape), and Gossypium hirsutum (upland cotton) (Huai et al. 2008; Zhang et al. 2010; Boneh et al. 2012; Huang et al. 2015; Yoo et al. 2016; Liu et al. 2017).
In Arabidopsis, there are ten members of the SnRK2s family, divided into three subclasses, based on their amino acid sequence similarity (Boudsocq et al. 2004). The subclass I is composed of kinases that are activated by ABA whereas subclass II kinases are either activated or very weakly activated by ABA, depending upon the plant species. However, subclass III is strongly activated by ABA. The amino acid sequences of all SnRK2s can be divided into the N and C terminal regions. The N-terminal domain in highly conserved. The C-terminal domain is regulatory. The C-terminal domain comprises stretches of acidic amino acids, either glutamic acid (E; subclass I) or aspartic acid (D; subclass II and III). Further, the C-terminal regulatory domain contains two subdomains, namely Domain I and Domain II. Domain I (about 30 amino acids starting from kinase domain) is required for activation by osmotic stress, which is independent of ABA, in all SnRK2 family members. Likewise, Domain II (about 40 amino acids and just after Domain I) is required for the ABA response and is specific to ABA-dependent SnRK2s only (Kobayashi et al. 2004; Umezawa et al. 2004; Yoshida et al. 2006; Belin et al. 2006; Yoshida et al. 2006; Kulik et al. 2011). All the SnRK2 members excluding SnRK2.9 were found to be rapidly induced by different osmolytes such as sorbitol, mannitol, sucrose, sodium chloride, and some by ABA. The Arabidopsis Domain II of SnRK2.6/SRK2E/OST1 protein kinase has been shown to regulate the ABA-mediated stomatal closure and is responsible for kinase activation by ABA. However, the Arabidopsis mutant ost1 showed a drought-sensitive phenotype and was defective in ABA-induced stomatal closure (Yoshida et al. 2002). Likewise, the srk2d/e/I triple mutant of Arabidopsis showed a decreased drought tolerance and insensitivity to ABA, as documented by defects in seed germination and seedling growth due to reduced expression of ABA- and stress-inducible genes (Fujita et al. 2013). The knockout of these three Arabidopsis genes from the SnRK2 family almost entirely blocks the ABA responses, which demonstrates that they are essential components of ABA-stress signaling pathways in Arabidopsis (Fujii et al. 2011). The snrk2.2/2.3/2.6 triple knock out mutant flowered early and produced fewer seeds that were insensitive to ABA (Fujii and Zhu 2009). The ABA-activated SnRK2.2, SnRK2.3 and SnRK2.6 play essential roles in controlling seed development and dormancy (Nakashima et al. 2009). The overexpression of SnRK2.8 shows upregulation of the stress-induced genes and increases drought tolerance in Arabidopsis (Umezawa et al. 2004).
In rice, there are ten SnRK2 members, i.e., SAPK1-10 (osmotic stress/ABA-activated protein kinases). All of them are found to be activated by hyperosmotic stress, while SAPK8/9/10 were also induced by ABA (Kobayashi et al. 2004). Recently, the SAPK9 gene member of SnRK2 was shown to promote drought tolerance in rice. Evidence suggested that overexpression of the SAPK9 gene improved drought tolerance, which improves grain yield by modulating cellular osmotic potential, stomatal closure, and stress-responsive gene expression. Domain swapping experiments in rice SAPKs showed that grafting the noncatalytic C-terminal region from SAPK8 (E-254 to M-372) onto the SAPK2 catalytic domain is sufficient to confer ABA responsiveness. There were 11 SnRK2 members identified in maize, designated as ZmSnRK2.1–2.11, and most of them are inducible by one or more abiotic stresses (Tian et al. 2013). Likewise, in wheat the first characterized SnRK2 member, PKABA1, showed induction by ABA, hyperosmotic stress and multiple other environmental factors (Anderberg and Walker-Simmons 1992; Xu et al. 2009). Later, three more SnRK2 members were identified in wheat (i.e., TaSnRK2.3, TaSnRK2.4, and TaSnRK2.8) and found to be involved in abiotic stress tolerance (Mao et al. 2010; Zhang et al. 2010; Du et al. 2013; Tian et al. 2013). Therefore, substantial evidence showed that the SnRK2 protein kinase family members are involved in multiple environmental stress responses. Consequently, all have potential biotechnological utility for the generation of high yielding abiotic stress-tolerant crop plants (Tian et al. 2013). Furthermore, in vitro studies have demonstrated that ABA-activated SnRK2s phosphorylate downstream target proteins in different plant species. Phosphorylation is required for the transcriptional activity of individual target proteins that in turn induce the expression of hierarchically organized downstream genes to mitigate the stress condition. Among the downstream transcription factors of SnRK2s are basic region/leucine zipper motif (bZIP) transcription factors (i.e., TRAB1 from O. sativa), TaABF from T. aestivum, AREB from A. thaliana and RNA-binding proteins such as VfAKIP1 from Vicia faba (faba bean) (Kagaya et al. 2002; Johnson et al. 2002; Li et al. 2002; Furihata et al. 2006).
Previously, we identified the first AAPK in Vicia faba which is a guard cell-specific kinase and showed that AAPK is a positive regulator of ABA signaling (Li and Assmann 1996; Li et al. 2000). To date, five SnRK2 genes (i.e., SPK1, SPK2, SPK3, SPK4, and GmAAPK) have been reported in soybean. While the activation of SPK1 and SPK2 expression has been observed in response to hyperosmotic stress in yeast cells, the demonstration of this activation in plant cells is currently lacking. Conversely, the expression of SPK3 and SPK4 is triggered by conditions of high salinity or dehydration as reported by Kim et al. (1997) and Monks et al. (2001). GmAAPK is induced by ABA and participates in the regulatory process during osmotic stress in soybean (Luo et al. 2006). These results suggest that the genetic engineering of AAPK gene has the potential to enhance drought tolerance in crop plants. Although SnRK2s have been well studied in Arabidopsis and rice and are known to be involved in the abiotic stress response, little is known about SnRK2s in soybean. ABA-activated protein kinases belonging to subclass III of the SnRK2 family have not been functionally characterized by transgenic approaches to date. Thus, the understanding of the molecular function of the subclass III, AAPK gene would be important in developing drought-tolerant transgenic soybean. In addition, such knowledge is useful for designing markers for molecular breeding of drought-tolerant soybean cultivar.
In the analysis presented here, a detailed characterization of the AAPK gene and functional role in drought stress response are presented. To achieve these objectives, transgenic-driven target gene overexpressing and RNA interference (RNAi) target gene silencing roots of AAPK-like kinase genes (i.e., Glyma.17G178800, Glyma.11G058800, and Glyma.05G081900) are produced. The transgenic experiments demonstrated that in contrast to silencing, the overexpression of AAPK-like kinase increases drought tolerance. Furthermore, we studied the expression of AAPK-like kinase genes (AALK) in the root to further extend the knowledge of their roles in the stress response. This analysis is important because the root is essential for maintaining crop yields, especially in drought conditions. RNA sequencing (RNA-seq) analyses have provided a detailed analysis of soybean root transcriptome in response to drought stress, identifying several categories of genes of critical importance that are involved in the drought response.
Materials and Methods
Plant Materials and Growth Conditions
Glycine max [cultivar williams 82/ accession PI 518671] has been selected for this study due to the availability of its genome (Schmutz et al. 2010). The soybean plants were grown in an incubation room at 25 °C with 70% relative humidity with a 12-h photoperiod/day. The light intensity maintained approximately 1000 µmol m−2 s−1 from white fluorescents lights.
Identification of SnRK2 Genes in Soybean and Phylogenetic Analysis
The amino acid sequences of AAPK in Vicia faba (Gene Bank, AF186020.1) and Arabidopsis thaliana were obtained from the literature (Li et al. 2000). To identify the SnRK2 genes in soybean, we used BLAST (version 2.2.26, Bethesda, MD, USA) to look for highly conserved sequences of SnRK2s in the soybean genome database (https://phytozome.jgi.doe.gov/pz/portal.html) (Supplemental Table 1). To investigate the evolutionary relationships, the phylogenetic tree was constructed using MEGA7 version 7.0 (Kumar et al. 2016), with the reliability of the tree evaluated using bootstrap analysis on 1000 replicates.
Gene Construction for the Overexpression of GmAALK Studies
The full-length open-reading frame of the GmAALK genes [Glyma.17G178800 (869 bp), Glyma.11G058800 (916 bp) and Glyma.05G081900 (854 bp)] were amplified by PCR using cDNA of roots and leaves of soybean mRNA. The PCR amplified GmAAPK-like kinase genes were cloned into the pENTR/D-TOPO cloning vector (Invitrogen, Carlsbad, CA) and then sequenced to confirm the correctness of the sequence. LR clonase reactions transferred the GmAALK kinase gene in the pENTR/D-TOPO cloning vector into the pRAP15 vector (13,796 nucleotides in length) for overexpression studies (Matsye et al. 2012). The pRAP15 vectors contain a single Gateway® (Invitrogen, Carlsbad, CA) compatible with attR1-ccdB-attR2 cassette, whose expression is driven by the figwort mosaic virus sub-genomic transcript (FMV-Sgt) promoter. The cassette is designed to drive the overexpression of full-length genes. The pRAP15 vector contains an enhanced green fluorescent protein (eGFP) gene driven by the rolD promoter, such that plants can be identified by examining eGFP expression after transformation. The pRAP15 vector has the tetracycline resistance gene for bacterial selection engineered into a BstEII site that lies outside the left and right borders (Matsye et al. 2012). The eGFP gene product is a visual beacon for screening transformed roots. The eGFP cassette was ligated into a HindIII site of pRAP15. The Gateway® compatible attR1-ccdB-attR2 cassette was engineered into the pRAP15 vector between SpeI (5′) and XbaI (3′) sites. The inserted gene cassette is terminated by the cauliflower mosaic virus 35S terminator. The attR sites are LR bacteriophage λ-derived recombination sites. The ccdB gene is one agent for E. coli selection. The attR cassette is interrupted by a ccdB selectable marker gene that acts as an intron. Therefore, genetic engineering of GmAALK genes into pRAP15 would result in a gene positioned in the correct, directional orientation for overexpression (Matsye et al. 2012). The pRAP15 vector with AALK genes was introduced into Agrobacterium rhizogens strain K599 (K599) (a generous gift from Dr. Walter Ream, University of Oregon) and then transferred into soybean roots by K599-mediated transformation. Roots transformed with empty pRAP15-ccdB plasmid served as a control.
Gene Construction for Silencing of GmAALK Genes Via RNA Interference (RNAi) Approach
To generate an RNAi construct for the GmAALK genes, gene-specific fragments of approximately 300 base pairs long (i.e., Glyma.17G178800, Glyma.11gG58800, and Glyma05G081900) were amplified by PCR. The PCR-amplified fragments were cloned into the pENTR/D-TOPO cloning vector (Invitrogen, Carlsbad, CA). Subsequently, LR clonase reaction was performed to transfer the cloned AALK gene within the pENTR/D-TOPO plasmid into the pRAP17 destination vector (15,540 nucleotides in length) (Klink et al. 2009). The LR clonase reaction results in the insertion of the selected fragment of the AALK genes into sense and antisense orientations that are linked by a chloramphenicol resistance gene. The pRAP17 vector also contains an eGFP gene driven by the rolD promoter that exhibits robust and constitutive root expression. The RNAi constructs with inserted AALK gene fragments in the pRAP17 vector was introduced into K599 that ultimately transfers the expression cassette stably into the soybean root cell chromosomal DNA. Roots transformed with the empty pRAP17-ccdB were used as control (Klink et al. 2009).
Generation of Transgenic Lines
A slightly modified version of the protocol described by Matsye et al. (2012) was used in this experiment. Ten day old soybean seeds grown in sand in the greenhouse at ambient temperature (~ 26–29 °C) were used as as a source for explants for in-planta transformation. The cleaned explants were cut at hypocotyl with a sterile blade to remove the roots with the cut end immediately dipped into Agrobacterium co-cultivation medium. Then, the rootless plants underwent vacuum infiltration for 30 min, with the vacuum subsequently released slowly. Then, the explants were covered with saran wrap and incubated overnight at 28 °C at 50 RPM in a rotary shaker in the dark. After overnight incubation, the cut ends of explants were placed individually ~ 4 cm deep into fresh coarse A3 vermiculite (Palmetto Vermiculite Company, Woodruff, SC) in 50-cell flats in a covered 67.3 × 40.1 × 26.4 cm plastic container (Rubbermaid®; Rubbermaid Home Products; Fairlawn, OH) in an incubation room. The temperature of the incubation room was set at 26 ± 2 °C and having fluorescent white 4100 K lights (32-W bulbs) emitting 2800 lumens used as a light source. After seven days of incubation, the plants were uncovered and transferred to the greenhouse.
Confirmation of Transgenic Plants
The one-month-old putative transgenic plants were used to determine the eGFP reporter expression using the Dark Reader Spot Lamp (Clare Chemical Research). Only transgenic roots show green fluorescence since both pRAP15 and pRAP17 vectors have eGFP within the left and right borders. Only genetically chimeric, composite plants with transformed roots showing green fluorescence were kept. The non-transformed roots were excised with a scissors. After excision of the non-eGFP-expressing roots, the plants were planted into pots containing a 1:1 mixture of sand and soil. The plants were allowed to recover for a week.
Molecular Analysis
Total RNAs were extracted from leaves or roots of soybean plants using the RNeasy Mini Kit (Qiagen) following the manufacturer's procedure. The RNA samples were digested with RNase-free DNase I (Qiagen) to remove genomic DNA contamination. RNA concentrations were determined using NanoDrop 2000 C (Thermo Scientific). The A260/280 values of all RNA samples used in this study ranged from 1.8 to 2.2, and A260/230 ratios of all samples were above 2.0. The RNA quality was also monitored on 1.2% agarose gel electrophoresis. cDNAs were synthesized from the total RNA samples using the SuperScript First-Strand Synthesis System (Invitrogen) with an OligodT primer. Confirmation of overexpression of the GmAALK genes in the transformed plants was done by PCR amplification of the cDNA using eGFP primers as well as gene-specific primers. The Quantitative Real-Time PCR (qRT-PCR) analysis was performed using BioRad CFX96 thermocycler for the confirmation of silenced and overexpression of the GmAALK genes in transgenic plants. The 60 s ribosomal gene primers were used as an endogenous control. The relative gene expression levels were determined as described previously (Livak and Schmittgen 2001). The primers identifiers and sequences are provided (Supplemental Table 2).
Drought Stress Treatment and Estimation of Chlorophyll, Flavonoids, Anthocyanin, Nitrogen Balance Index and Soil Moisture
To investigate the role and mechanisms of GmAALK genes in drought stress tolerance, four-week old transgenic roots-containing composite plants, along with the vector control were subjected to drought stress by withholding their water supply for 7 days. Photos were taken to record the phenotypes, and morphological data measurements were taken. The chlorophyll (Chl) content, anthocyanin (Anth), and flavonoids (Flv) were measured under stress conditions after applying the plant stress using Dualex®Scientific from FORCE-A (Fluorescence and Optoelectronics Research for the Communication between Ecophysiology and Agriculture) as described by Cerovic et al. (2012). Nitrogen balance index (NBI) was also calculated using the Dualex data as the ratio of Chl/Flav. Soil moisture data were collected using soil moisture sensor device (HH2 moisture meter with ML2X- theta probe, Delta-T Devices) every day between 12:00 to 14:00 h after applying the treatments. After seven days of drought treatment, transgenic root samples were collected for RNA isolation.
RNA Isolation, cDNA Library Construction and Sequencing
Three biological replicates were used for all RNA-seq experiments from drought treatments of transgenic knockdown roots along with the control. The total RNA was isolated from the roots of soybean plants using the RNeasy Plant Mini Kit (Qiagen). RNA samples were digested with RNase-free DNase I on column according to manufacturer’s protocol (Qiagen) to remove genomic DNA contamination. RNA quality and integrity were verified using Agilent 2100 Bioanalyzer (Agilent Technologies). The RNA Integrity Number (RIN) values greater than seven were accepted in this experiment. The quantification of the total RNA was also performed by a NanoDrop 2000 (Thermo Scientific) and 1.2% agarose gel electrophoresis. cDNA was generated using Clontech SMARTer cDNA kit (Clontech Laboratories, Inc.) from total RNA isolated from Soybean roots. cDNA was fragmented using Bioruptor (Diagenode, Inc.), profiled using Agilent Bioanalzyer, and subjected to Illumina library preparation using SPRIworks HT Reagent Kit (Beckman Coulter, Inc.). The quality and quantity and the size distribution of the illumine librarires were determined using an Agilent Bioanalzyer 2100. The samples were then sequenced on an Illumina HiSeq2500 which generated paired-end reads of 106 nucleotides (nt.) (Otogenetics). Data were generated and quality checked using FASTQC (Babraham Institute).
Data Processing and Bioinformatics Analysis
The RNA-seq reads were initially processed to remove the adaptor sequences, and low-quality reads using Trim Galore (04.0) (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) on the DNAnexus platform (DNAnexus, Inc.). After filtering, clean reads were used for the downstream bioinformatics analysis. High-quality mRNA-seq reads were aligned to the Glycine max reference genome (Wm82.a2.v1 version) using the Spliced Transcripts Alignment to a Reference (STAR) aligner (version 2.5.0b). STAR has a potential for accurately aligning long reads that are generated by the third-generation sequencing technologies (Dobin et al. 2013). STAR Aligner was run with the default parameters and a reference G. max GTF file using the –G option, and replicates of each treatment/sample were mapped independently to improve alignment sensitivity and accuracy for further analysis. Only uniquely mapped reads were used in the analysis. The gene expression levels were estimated using HTseq software (Anders et al. 2015). Only reads which unambiguously mapped to a single gene were counted, whereas reads aligned to multiple positions or overlapping with more than one gene were discarded (Supplemental Table 3). For each treatment, a pairwise comparison was performed between the control and RNAi silenced samples using the libraries synthesized from three biological replicates of each treatment/condition. The differential gene counts analysis was performed using edgeR (Robinson et al. 2010). A ratio of expression (fold-change) was performed by dividing the values of gene expression under stress treatments and control conditions. False Discovery Rate (FDR) was calculated to identify statistically significant differentially expressed genes and to avoid inflation of type-1 errors. Differentially expressed genes between different treatments were identified using the following criteria: (FDR) < 0.001 and abs (log2FC (fold-change)) ≥ 2.
Functional Classification
Differentially expressed genes were functionally classified using gene ontology (GO) terms (Ashburner et al. 2000). The SEA method of AgriGO tool v2.0 (Tian et al. 2017) with GO annotations associated with the soybean genome (Wm82.a2.v1 version) was used to detect significantly over-represented or under-represented GO terms in a drought vs. control comparisons. To decrease the redundancy, results provided by AgriGO were further analyzed by the Reduced Visualize Gene Ontology (REVIGO) method using SimRel as the semantic similarity measure with the default threshold of 0.7 (Supek et al. 2011). The procedure yielded a GO enrichment for the studied samples per condition.
Pathway and Transcription Factor (TF) Identification and Analysis
KEGG Pathway (http://www.genome.jp/kegg/pathway.html) enrichment analysis was used to identify significantly enriched metabolic or signal transduction pathways in differentially expressed gene sets compared with the whole genome background. The drought stress genes in RNAi silenced lines along with controls were plotted by MapMan (Usadel et al. 2009). Several biological and metabolic pathways were plotted together using the log fold change differences indicated with a green and red schema. The cut-off of the absolute value fold change gene expression was 2. The TFs and transcription-related genes were also mapped and plotted by MapMan. The TF family annotations were obtained from plantTFDB (Jin et al. 2017).
Quantitative Real-Time PCR (qRT-PCR) Validation of Gene Expression
Root tissues from pRAP17-ccdB control and RNAi silenced plants under control conditions and drought stress conditions were used in total RNA extraction, DNase treatment, and in cDNA synthesis as described previously. qRT-PCR was carried out in 96-well plates with CFX Real-Time PCR Detection System (Bio-Rad) using SsoFast™ EvaGreen® Supermix with three independent biological replicates. 20 ng of cDNA determined by NanoDrop 2000 C spectrophotometer (Thermo Scientific) were used for qRT-PCR. The relative quantitative analysis was performed under the following conditions: 95 °C for 3 min and 40 cycles at 95 °C 10 s, 60 °C 30 s and 72 °C for 30 s. The melt curve analysis is ranging from 60 to 95 °C, was used to identify the different amplicons, including non-specific products. To ensure specific products, the agarose gel run was performed with qPCR products. A total of 21 soybean genes that were up- or down-regulated in the RNASeq experiments were assayed. Gene-specific primers were designed using Primer3 software (Supplemental Table 2). Raw data were analyzed according to the 2−ΔΔCT based on Livak and Schmittgen method (Livak and Schmittgen 2001), applying 60 s as an endogenous gene (Le et al. 2012a, b).
Results
Identification of GmSnRK2 Members in Soybean
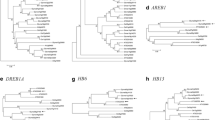
A genome-wide search for AAPK homologs in the soybean genome has been performed. The analysis used the Vicia faba (faba bean) (AF186020.1) as a reference. The analysis identified 22 soybean genes encoding SnRK2 family protein kinases (Supplemental Table 1). Our results is agreement with study conducted by Zhao et al., (2017), who also identified 22 GmSnRK2 genes in the soybean genome. These GmSnRK2 genes were distributed across the 20 chromosomes. For comparison, ten SnRK2 family proteins have been reported to be encoded in rice and Arabidopsis (Kobayashi et al. 2005; Nakashima et al. 2009). A phylogenetic tree was constructed to investigate the evolutionary relationship between the different SnRK2 protein sequences of soybean, rice, and Arabidopsis (Kumar et al. 2016). The neighbor-joining phylogenetic tree suggests that these protein kinases can be divided into three groups, which is consistent with previously reported analyses (Kulik et al. 2011) (Fig. 1). Each subgroup contains members from both Arabidopsis and rice SnRK2, demonstrating that the divergence of these subclasses originated before the divergence of dicots and monocots. The Arabidopsis thaliana was the first plant for which whole genome was sequence in 2000, and it has a genome size of around 134 megabase for the strain Col-0. The Arabidopsis genome has large segmental duplications which cover much of the genome. Duplications are primarily due to at least four different large-scale duplication events that occurred 100 to 200 million years ago (Vision et al. 2000). Similarly, Oryza sativa is a 430 megabase (MB) in size and composed of 12 chromosomes. The rice genome has undergone multiple duplications, including and ancient whole genome duplications. Recent segmental duplications on chromosomes 11 and 12, and massive ongoing individual gene duplications (Yu et al. 2005). Similarly, Glycine max genome contains 1.1 gigabase which is 8 times lareger than Arabidopsis genome. Soybean has 70% more coding genes than Arabidopsis. Soybean gene duplications occurred at approximately 59 and 113 million years ago (Schmutz et al. 2010). Compared to Arabidopsis and rice, soybean has more SnRK2 genes in each group. Groups I and III were the largest with 8 GmSnRK2 members, whereas Group II has only 6 GmSnRK2 members. Vicia faba AAPK, Arabidopsis SnRK2.2/2.3/2.6 and rice SAPK8/9/10 are reported to be activated by ABA and are included in Group III. The protein sequence for Glyma.17G178800 is 81.54% identical to GmAAPK (Glyma.11G058800) from G. max, 77.18% identical to AAPK from Vicia faba, 75.16% identical to AtSnRK2.3/SnRK2I from A. thaliana, and 75.16% identical to AtSnRK2.2/SnRK2D from A. thaliana (Li et al. 2000; Luo et al. 2006; Nakashima et al. 2009). The Glyma.17G178800 has higher homology to AAPK than AtSnRK2.2/2.3. Therefore, the corresponding gene Glyma.17G178800, was named AALK1. We hypothesized that the eight identified GmSnRK2 family members (Glyma.01G183500, Glyma.02G135500, Glyma.05G081900, Glyma.07G178600, Glyma.07G209400, Glyma.11G058800 (GmAAPK), Glyma.17G178800 (GmAALK1), and Glyma.20G009600) might be activated by ABA since they all clustered in the same SnRK2 Group III containing ABA activated genes such as AAPK, OST1, AtSnRK2.2, AtSnRK2.3, and OsSAPK8/9/10 (Fig. 1). SnRK2 proteins have functional domains in their N-or C-terminal regions. To analyze the conserved domains in the SnRK2 protein family, we aligned the C-terminal region of the previously published ABA-activated Group III protein kinases in Arabidopsis and rice, along with AAPK faba bean and the identified eight soybean genes. We found two conserved domains: (1) Domain I is nearly 30 amino acids long starting from the end of the kinase domain, and (2) Domain II is about 40 amino acids long starting from the end of Domain I (Fig. 1), which is consistent with previously published work. According to the previous studies, Domain I is required for ABA-independent activation by osmotic stress, whereas Domain II is needed for the ABA-dependent response (Boudsocq et al. 2004; Kobayashi et al. 2004; Yoshida et al. 2006).

A Phylogenetic Analysis of SnRK2s protein kinase in Arabidopsis thaliana, Oryza sativa and Glycine max by Neighbor-joining (NJ) method. Group I: ABA-independent kinases, Group II: kinases not dependent or weakly dependent on ABA, Group III: ABA-dependent kinases. The tree was built using MEGA 7 and presented using Fig Tree v1.4.3. B Structural analysis of the C-terminal region in ABA-activated protein kinases of Arabidopsis thaliana, Oryza sativa, and Glycine max. The C-terminal region was divided into two parts: Domain-I (dashed blue box) involved in ABA-independent activation in response to osmotic stress and Domain-II (Green box) required for ABA-dependent activation of SnRK2s. Identical amino acid residues are boxed, and similar residues are shaded in gray. Dashes indicate gaps in the sequences to allow the maximal alignment
Overexpressing and Silencing of Abscisic Acid-Activated Protein Kinase Gene (AALK) in Soybean
To investigate the gain-of-function and loss-of-function phenotypes of an AAPK-like kinase gene in soybean through transgenesis, gene overexpression and RNAi-mediated gene silencing (RNAi) constructs were prepared. We cloned G. max genes (i.e., Glyma.17G178800, Glyma.11G058800, and Glyma05G081900), the GmAAPK and two additional AAPK-like (AALK) kinases with the highest homology to the Vicia faba AAPK (Fig. 1). AAPK-like kinase genes were cloned into the pRAP15 vector for overexpression, whereas, for the silencing of these genes, an RNAi construct of GmAALK was generated in the pRAP17 destination vector. Root transformants were selected based on eGFP expression, verified using the Dark Reader Spot Lamp (Supplemental Fig. 1). A qRT-PCR analysis showed high relative transcript abundance (RTA) of eGFP in both overexpression and knockdown lines, which further confirmed the eGFP transgene production in these roots. The eGFP expression of these genes is provided (Supplemental Fig. 1). In addition, the expression and silencing of GmAALK genes was detected by qRT-PCR (Supplemental Fig. 1).
Overexpression of GmAALK1 Results in Increased Tolerance to Water Deficit
To investigate the physiological function of AALK, we generated and selected three independent composite plants having AALK-overexpressing and AALK-RNAi roots for further study, respectively. Among the three AALK genes, Glyma.17G178800 (referred to as GmAALK1) was more susceptible to drought stress response (data not presented here), and thus was selected for further study. There were no obvious phenotype differences among vector control, overexpression and RNAi silenced plants. Four-week-old (after root trimming) transgenic plants, along with control, were subjected to drought stress by withholding water for seven days. pRAP17-ccdB control and GmAALK1-RNAi silenced plants started wilting after four days of stress. However, the GmAALK1-overexpressing composite plants did not show wilting until seven days of drought, indicating that overexpression of GmAALK1 genes in soybean plants results in increased resistance to drought stress (Fig. 2A). We observed that the root system of soybean plants of GmAALK1 overexpressing composite plants developed more new lateral roots than RNAi silenced, and control lines under drought stress (Fig. 2B).

Overexpression of GmAALK1 conferred resistant to drought stress. A control, GmAALK1-RNAi silencing (aalk1-RNAi)and GmAALK1-overexpressing (AALK1-OX) plants were subjected to drought stress on four-week-old plants after seven days of drought stress. B The root system of control, GmAALK1-RNAi silenced and GmAALK1-overexpression lines after seven days of drought stress
The chlorophyll content, anthocyanin, flavonoids, and nitrogen balance index of soybean plants were measured after drought stress (Fig. 3). The Dualex monitor uses the fluorescence and light transmission of a leaf to determine the leaf status. There were significant differences in chlorophyll content and nitrogen balance index between GmAALK1-overexpression and GmAALK1-RNAi silenced plants as compared to control. However, flavonoids and anthocyanin were significantly decreased in GmAALK1-overexpression lines as compared to control and RNAi-silenced plants.

Comparison of physiological indices related to drought stress response for control, GmAALK1-RNAi silencing, GmAALK1-overexpressing lines. A chlorophyll content, B flavonoids, C anthocyanin and D nitrogen balance index measured using Dualex instruments. The values are mean and standard errors of five replications in each line. The bars followed by the same letters did not differ significantly at 5% by Tukey’s test
Differentially Expressed Genes (DEGs) Caused by a Response to Drought Stress
After seven days of imposing drought stress, the expression pattern of transgenic pRAP17-ccdB control roots vs. GmAALK1-RNAi roots of composite plants were evaluated under drought and non-drought conditions. In this study, each condition was represented by three biological replicates. A Venn diagram illustrating the clustering of DEGs into different groups along with the corresponding conditions and highlights the overlap of DEGs between the four pairwise comparisons (Fig. 4C). The comparisons include: Group 1 is the genotype control under drought vs. non-drought conditions (yellow color). Group 2 is the genotype GmAALK1-RNAi under drought vs. non-drought conditions (blue in color). Group 3 is GmAALK1-RNAi vs. pRAP17-ccdB control under drought conditions (green color). Group 4 is GmAALK1-RNAi vs. pRAP17-ccdB control under non-drought conditions (pink color) (Fig. 4C). To reduce the rate of false positives and reliability identify the most significant changes in gene expression, only genes with FDR corrected p-Values < 0.001 and abs (log2FC) ≥ 2 were selected. After applying a stringent criterion described above, 6800 genes in pRAP17-ccdB control samples, and 2,813 genes in the GmAALK1-RNAi samples were differentially expressed under drought vs. non-drought conditions (Fig. 4B). There were 69 genes differentially expressed between GmAALK1-RNAi vs. the pRAP17-ccdB control under drought conditions, whereas 212 genes in GmAALK1-RNAi vs. pRAP17ccdB control under non-drought conditions (Fig. 4A).

Number of up- and down-regulated genes from the four comparisons performed. A Up- and down-regulated genes comparing drought-treated (DT) to untreated (UT) roots in GmAALK1-RNAi silencing line or genotype control. B Up-and down-regulated genes comparing GmAALK1-RNAi silencing line to the genotype control under drought-treated or untreated conditions. C Venn diagram of DEGs which passed the cut-off logFC > 2 in four comparisons performed. Genes shared among the four comparisons are represented by overlapping areas. Yellow represents the number of DEGs resulting from comparing drought treated (DT) to untreated (UT) conditions in genotype control samples, while blue is the number of DEGs from comparing drought treated (DT) to untreated (UT) conditions in GmAALK1-RNAi silencing line. Green represents the number of DEGs from the comparison between the two genotypes under drought-treated conditions [RNAi vs. control (DT)], while pink represents the number of DEGs between the two genotypes under untreated conditions [RNAi vs. control (UT)]
In Group 1, DEG genes for the comparison between drought vs. non-drought conditions in the genetype control sample (Fig. 4C, [in yellow]), contains 6,800 genes with 4977 having increased RTAs (relative transcript abundance), and 1823 having decreased RTAs (Fig. 4B). In Group 1, 4,142 genes or 57.7% of total DEGs were unique among all categories, the largest set of DEGs among all comparisons. In Group 2, the comparison between drought vs. non-drought conditions in GmAALK1-RNAi (Fig. 4C, [in blue]) 2,813 DEGs were divided between the 1,669 genes with decreased RTAs and 1,144 genes with increased RTAs (Fig. 4B). In Group 3, the comparison between GmAALK1-RNAi vs. pRAP17-ccdB control under drought conditions (Fig. 4C, [in green]) out of 69 genes, approximately half of the genes (34) were decreased in their RTAs, and a half (35) were increased in their RTAs (Fig. 4A), and only 27 genes were unique when subtracting DEG genes from other comparisons. Likewise, in Group 4, the comparison between GmAALK1-RNAi vs. pRAP17-ccdB control under non-drought conditions (Fig. 4C, [in pink]). Out of 212 DEGs, only eight genes were increased in their RTAs. The majority of the genes (i.e., 204 genes) were decreased in their RTAs (Fig. 4A), with only 18 unique genes decreased in their RTAs in GmAALK1-RNAi under non-drought conditions. There were 328 genes unique among all comparisons. No genes were found to be in common between all four comparisons (Supplemental Data Set 1).
Highly Differentially Expressed Genes in all Four Comparison Groups
In all groups of DEGs a moderate expression ratio was detected for most of the genes. High fold-changes for a small number of genes were also observed. Therefore, relatively few genes were highly affected, indicating a specificity of the response.
In Group 1, the highest upregulation of gene expression changes were found for several heat shock proteins. These proteins included Glyma.07G200700, Glyma.07G200500, Glyma.02G205600, Glyma.08G068700, Glyma.20G213900, Glyma.08G068800, Glyma.16G206200 (log2FC 11 to 13); and for Glyma.14G156400 (log2FC 13), an alcohol dehydrogenase 1; Glyma.09G237600 (log2FC 12); Glyma.18G260000 (log2FC 11), a nitrate transporter; Glyma.03G113200, Glyma.15G211300, a NAD(P)-binding Rossmann-fold superfamily protein (log2FC 11); and Glyma.09G185500 (log2FC 11), an uncharacterized protein. Among the downregulated genes in Group 1; Glyma.15G260700, Glyma.08G162400 (log2FC -12.94/-11.5), an eukaryotic aspartyl protease family protein; Glyma.10G180100 (log2FC-11.94), an indole-3-acetic acid inducible; Glyma.18G055400 (log2FC-11.64), a peroxidase superfamily protein; Glyma.05G121600 (log2FC -11.4), a vacuolar iron transporter; Glyma.07G150400 (log2FC -11.4), hydrolyse-type esterase superfamily protein; Glyma.08G230100 (log2FC -11.4), a pathogenesis-related protein; Glyma.09G118300 (log2FC -11.4), a MLP-like protein 43; Glyma.15G251300 (log2FC -11.4), a nicotianmine synthase 1; Glyma.18G018900 (log2FC -11), a sulfate transporter; and Glyma.05G019200 (log2FC -11), a cytochrome P450. Glyma.10G126100 with unknown function in Group1 had the second most changes in expression (Supplemental Data Set 1).
In Group 2, the highest differentially expressed genes were Glyma.07G200500 (log2FC 13.64), which encodes a heat shock protein. This gene was followed by Glyma.13G291800 (log2FC 12.75), a late embryogenesis abundant domain-containing protein; Glyma.15G211300 (12.52), a NAD(P)-binding Rossmann-fold superfamily protein, and Glyma.14G063700, Glyma.14G063800 (log2FC 12) a heat shock protein; Glyma.02G028400 (log2FC − 13.67), a matrix metalloproteinase; Glyma.02G064100 (log2FC − 12.48), a ribonuclease 1; Glyma.13G364400 (log2FC − 12.17) a nodulin, and Glyma.15G062800 (log2FC − 12), a cysteine-rich secretory proteins; and Glyma.18G018900 (log2FC − 11.8), a sulfate transporter protein are top five in downregulation category (Supplemental Data Set 1).
In Group 3, the highest level of differential expression of upregulated genes was detected for Glyma.U041300 (log2FC 7.57), which codes for an efflux antiporter; Glyma.U042300 (log2FC 6.90), a heat shock family protein; Glyma.06G220600 (log2FC 6), a nucleolar histone methyltransferase-related protein; and Glyma.U000500 (log2FC 5.82), a bHLH transcription factor. Like-wise the highest expression change for down-regulated DEGs in Group 3 was measured for Glyma.02G252500 (log2FC − 5.46), an uncharacterized protein; Glyma.10G119300 (log2FC − 5.41), a serine endopeptidase family protein; Glyma.03G159000 (log2FC − 5.19), a hydrolase superfamily protein; and Glyma.08G321200 (log2FC − 5.17), an eukaryotic aspartyl protease family protein (Supplemental Data Set 1).
In Group 4, the highest change (log2FC 6) was found for Glyma.03G058800 with unknown functions; Glyma.19G132700 (log2FC 4.6), an uncharacterized protein; Glyma.20G114200 (log2FC 4.5), a cinnamate-4-hydroxylase; Glyma.09G284700 (log2FC 3.5), a peroxidase super family protein are top 5 highly expressed protein. Similarly, Glyma.16G177500 (log2FC − 9.13), a metal transporter; Glyma.16G043000 (log2FC − 8.5), ahydroxyproline-rich glycoproteinfamily protein; Glyma.18G056500 (log2FC − 8.35), a nuclear transport factor 2 family proteins; Glyma.06G319000 (log2FC − 8) plant invertase/pectin methylesterase inhibitor superfamily; and Glyma.11G244800 (log2FC − 9) a pectate lyase superfamily are top 5 downregulated protein (Supplemental Data Set 1).
In this study, we identified 4,142 genes uniquely expressed in the genotype control under drought vs. non-drought conditions (Fig. 4C). Among them, the highly expressed genes, Glyma.09G163000, Glyma.05G065300, Glyma.16G212200, Glyma.09G162700, and Glyma.09G163600, which mostly encodes trypsin and protease inhibitor protein, were upregulated under drought vs. non-drought conditions. Out of 4142 genes included exclusively in the genotype control under drought vs. non-drought conditions, Glyma.14G053700, peroxidase superfamily protein, Glyma.10G021300 thioredoxin superfamily protein, and Glyma.14G032000, Glyma.02G282300 with unknown function were downregulated. Similarly, there are 328 genes found uniquely differentially expressed in Gmaalk1-RNAi lines under drought vs. non-drought conditions (Fig. 4C). Among those 328 genes, many are moderately represented, and some are highly expressed. The highly expressed genes were CAP (cysteine-rich secretory proteins, Antigens 5, and Pathogenesis-related 1 protein) superfamily protein (Glyma.13G251700), protein kinase superfamily protein (Glyma.04G056900) were downregulated whereas Glyma.05G177600, Glyma.11G180400 with unknown function and Glyma.16G173600 (SLAC1 homolog) were up-regulated.
While comparing GmAALK1-RNAi vs. pRAP17-ccdB control under drought conditions, 27 genes were uniquely expressed and most DEGs were efflux antiporter (Glyma.U041300), heat shock family protein (Glyma.U042300), and nucleolar histone methyltransferase-related protein (Glyma.06G220600), were up-regulated whereas, with unknown function (Glyma.02G252500), subtilisin-like serine endopeptidase family protein (Glyma.10G119300) and hydrolases superfamily protein (Glyma.03G159000) were down-regulated (Fig. 4C). In the comparison in GmAALK1-RNAi vs. pRAP17-ccdB control under non-drought conditions, 18 genes were found which are moderately expressed (Fig. 4C). Out of 18 genes, 16 DEGs were downregulated, and only two were upregulated, i.e., RING/U-box superfamily protein (Glyma.15G173800) and cinnamate-4-hydroxylase (Glyma.20G114200) (Supplemental Data Set 1).
Functional Roles of Differentially Expressed Soybean Genes in Response to Drought Stress
To sort the functional categories of DEGs, the singular enrichment analysis (SEA) was performed. After testing the 4,142 unique DEGs in genotype control under drought vs. non-drought conditions, 36 GO terms were significantly enriched such as “metabolic process” (GO:0008152), “oxidation–reduction” (GO:0055114), “regulation of gene expression” (GO:0010468), and “RNA biosynthetic process” (GO:0032774) in the biological processes category; “oxidoreductase activity” (GO:0016491), “hydrolase activity, acting on glycosyl bonds” (GO:0016798), “transcription factor activity” (GO:0003700), “iron ion binding” (GO:0005506), and “catalytic activity” (GO:0003824) in the molecular functions category (Supplemental Data Set 2 A). “Oxidation–reduction” (GO:0055114), and “oxidoreductase activity” (GO:0016491) were the most enriched GO terms in the biological and molecular function categories among the 328 differentially expressed genes in GmAALK1-RNAi lines under drought vs. non-drought conditions (Supplemental Data Set 2 B). “Hydrolase activity” (GO:00167870, and “catalytic activity” (GO:0003824) were the significantly enriched GO terms as molecular functions among 27 differentially expressed genes comparing the GmAALK1-RNAi lines to pRAP17-ccdB control under drought conditions (Supplemental Data Set 2 C). “Transferase activity” (GO:001674) was the only one significantly enriched GO term among 18 differentially expressed genes comparing the GmAALK1-RNAi to pRAP17-ccdB control under non-drought conditions (Supplemental Data Set 2 D). “Oxidation–reduction” (GO:0055114), “response to stress” (GO:0006950), and “response to oxidative stress” (GO:0006979) in the biological process category; “photosynthetic membrane” (GO:00343570), and “photosystem II” (GO:0009523) in cellular process category; “oxidoreductase activity” (GO:0016491), and “metal ion binding” (GO:0046872) are among the functional categories enriched in the 2,436 DEG genes under drought vs. non-drought as compared to GmAALK1-RNAi vs. pRAP17-ccdB control (Supplemental Data Set 2 E; Fig. 5).

Enrichment analyses of functional roles in a common set of genes between GmAALK1-RNAi and control lines under drought/non-drought conditions. Genes were associated with gene ontology terms (biological process, molecular function and cellular component) and compared to the soybean genome (False Discovery Rate [FDR], P-value < 0.05) using AgriGO and REVIGO. Blue bars: differentially expressed genes
Metabolic Pathway Enrichment Analysis of DEGs Under Drought as Compared to Non-drought Conditions
Multiple metabolic-related pathways that respond to drought treatments have been identified in this study. Transcripts related to cell wall proteins arabinogalactan-proteins (AGPs), such as fasciclin-like arabinogalactan-protein, cell wall pectin (such as pectin acetylesterase family protein, cell wall modifications (xyloglucan endotransglucosylase/hydrolases) were mostly downregulated whereas only a few transcripts were up-regulated in control lines as compared to drought treated and non-treated conditions. In GmAALK1-RNAi lines, transcripts associated with cell wall protein (AGPs), cellulose synthesis (cellulose synthase) were downregulated, with only a few transcripts upregulated. Several gene families (polygalacturonase, pectinase, responsive to desiccation RD22) involved in cell wall degradation were downregulated except for two transcripts, i.e., Glyma.05G25370 (polygalacturonase inhibiting protein 1) and Glyma.10G17550 (polygalacturonase, putative) which were up-regulated in control lines under drought vs. non-drought conditions. In the case of GmAALK1-RNAi lines, several gene families (polygalacturonase, pectate lyase family protein, pectinase) involved in cell degration were down-regulated, whereas only one transcripts Glyma.10G17550 (polygalacturonase, putative) was up-regulated (Fig. 6, Supplemental Data Set 3). Most of the DEGs involved in lipid metabolism (i.e., fatty acid synthesis, elongation, desaturation, and degradations) were inhibited in control under drought vs. non-drought conditions.

Distribution of up- (in red) and down- (in green) regulated genes in metabolic pathways in response to drought stress. Drought mediated expression changes in the metabolic pathways in control lines (A) and GmAALK1-RNAi lines (B). The figure was generated using MapMan and showed differential gene expressions that passed the cut off of log2FC > 2
Similarly, in GmAALK1-RNAi, only a few transcripts were identified in lipid metabolism; most of them are downregulated, and only a few transcripts were up-regulated under drought vs. non-drought conditions (Fig. 6, Supplemental Data Set 3). Most of the differentially expressed genes involved in secondary metabolism (including wax, fermentation, flavonoids, lignin, and glycolysis-related genes) were found to be downregulated, whereas only a few up-regulated transcripts were identified under drought vs. non-drought conditions. While 189 DEG transcripts were detected in control, just 93 DEG transcripts were detected in the GmAALK1-RNAi in secondary metabolism (Fig. 6, Supplemental Data Set 4). These results showed knockdown of GmAALK1 gene has adverse effects on the metabolic processes of roots and the down-regulated metabolism may slow down root growth under drought conditions as compared to non-drought conditions.
A total of 69 transcripts were identified in carbohydrate metabolism in pRAP17-ccdB control lines, whereas only 30 transcripts in GmAALK1-RNAi lines were differentially expressed under drought vs. non-drought conditions. Three protein kinase genes associated with fructokinase were downregulated in control (Fig. 6, Supplemental Data Set 5) in control lines. Conversely, raffinose synthases, starch cleavage related genes were up-regulated. However, one transcript encoding starch cleavage (beta-amylase) was down-regulated in control lines. Genes encoding trehalose phosphatase/synthase that are present in trehalose metabolism were downregulated. Similarly, hexokinase, sucrose synthase gene families were down-regulated in control lines under drought vs. non-drought conditions (Fig. 6, Supplemental Data Set 5). There was only one downregulated transcript identified in GmAALK1-RNAi lines for fructokinase; however, galactinol, raffinose synthase, starch cleavage and stachyose synthases related genes were up-regulated under drought vs. non-drought conditions (Fig. 6 Supplemental Data Set 5).
Hormonal Pathway Enrichment Analysis of DEGs Under Drought as Compared to Non-drought Conditions
In this study, a total of 57 differentially expressed transcripts associated with auxin synthesis and signaling pathway genes were identified in pRAP17-ccdB control lines, whereas only 34 transcripts were found in GmAALK1-RNAi under drought vs. non-drought conditions. Among them, genes related to auxin biosynthesis (IAR3, IAA-alanine resistant 3; metallopeptidase) were found to be down-regulated in both GmAALK1-RNAi silenced and control lines as under drought vs. non-drought conditions. Five different auxin transporter genes (PIN1, PIN2, PIN6, PIN7, AUX1) and one amino acid permease were identified in control lines whereas only one auxin transporter genes (AUX1) and one amino acid permease were found downregulated in GmAALK1-RNAi under drought vs. non-drought conditions (Fig. 7, Supplemental Data Set 6). Out of 57 transcripts, 47 transcripts were auxin-responsive genes representing more than 80 percent. Among these, 16 transcripts were up-regulated, and the rest were downregulated in control under drought vs. non-drought conditions. In GmAALK1-RNAi, out of 34 transcripts, 31 were auxin-responsive genes (90% in total auxin-responsive genes) were identified, half of them up-regulated, and half downregulated under drought vs. non-drought (Fig. 7, Supplemental Data Set 6).

Distribution of up- (in red) and down- (in green) regulated genes in hormonal pathways in response to drought stress. Drought mediated expression changes in the hormonal pathways in control lines (A) and GmAALK1-RNAi (B). The figure was generated using MapMan and shows differential gene expressions that passed the cut off of log2FC > 2
A total of 22 transcripts were associated with abscisic acid synthesis and signaling pathways in pRAP17-ccdB control lines whereas only five transcripts were found in RNAi lines under drought vs. non-drought conditions. Among the 22 transcripts, half were associated with abscisic acid synthesis [HVA22J, CCD8 (carotenoid cleavage dioxygenase 8), AAO3 (abscisic aldehyde oxidase 3), AAO2 (abscisic aldehyde oxidase 2), CCD7, NCED4 (nine-cis-epoxy carotenoid dioxygenase 4)]; nine were downregulated, and two were up-regulated under drought vs. non-drought conditions. One transcript (ABA-responsive element element-binding protein 3, AREB3) identified in signal transduction was found to be downregulated in the control lines under drought vs. non-drought conditions. A total of ten transcripts were found in ABA-responsive genes, and half of them were downregulated (ATHVA22A, HVA22A/C/F), and half were up-regulated (ATEM6, GRAM domain, ATHVA22D) in control lines under drought vs. non-drought conditions (Fig. 7, Supplemental Data Set 7). In GmAALK1-RNAi silenced lines, two transcripts identified were involved in abscisic acid synthesis (AAO2, NCED5), and three transcripts were ABA-responsive genes (two Gram domain-containing proteins, and HVA22D), all found to be up-regulated under drought vs. non-drought conditions (Fig. 7, Supplemental Data Set 7).
In our study, 128 transcripts associated with the ethylene pathway were differentially regulated; most of the ethylene signal transduction and synthesis related genes were downregulated in control under drought vs. non-drought conditions. Some universal stress protein family, the ethylene response factor genes, were up-regulated, and some are downregulated under drought vs. non-drought conditions (Fig. 7, Supplemental Data Set 8). Only 56 transcripts were associated with the ethylene pathway in GmAALK1-RNAi under drought vs. non-drought (Fig. 7, Supplemental Data Set 8) conditions. Among them, most are downregulated, and only a few transcripts are up-regulated.
Genes potentially involved in other hormonal pathways were also identified in this study. Jasmonic acid (JA, 34 genes), gibberellin (GA, 27 genes), salicylic acid (SA, six genes), cytokinin (15 genes) and brassinosteroid (BA, 10 genes) were found under drought vs. non-drought (Fig. 7, Supplemental Data Set 8) conditions. Out of 15 cytokinin genes, only cytokinin-independent 1 (CKI1) was up-regulated, whereas rest were down-regulated. In GA and SA all the identified transcripts were downregulated. Out of 34 transcripts in JA, nine transcripts (lipoxygenase 3 (LOX3), CYP74A, OPR1(12-oxophytodienoate reductase 1), OPR2, OPR3) were up-regulated, and rest were downregulated in pRAP17-ccdB control samples. Likewise, jasmonic acid (JA, 34 genes), gibberellin (GA, 27 genes), salicylic acid (SA, six genes) and cytokinin (4 genes) were identified in GmAALK1-RNAi silenced lines in drought treated vs. untreated comparisons (Fig. 7, Supplemental Data Set 8). In cytokinin pathways, CKI1 were up-regulated and the remaining three (ATIPT5, CKX3 (cytokinin oxidase 3), CKI2) were downregulated in RNAi lines. Genes OPR1, OPR2, OPR3, and CYP74A were up-regulated in JA pathways, whereas the rest of the transcripts were downregulated in GmAALK1-RNAi lines.
TFs Showing Differential Expression Under Drought as Compared to Non-drought Conditions
Many transcription factors exhibited altered expression patterns in soybean roots underlying drought stress treatment (Table 1). A Venn-diagram illustrating the partitioning of TFs (628 TFs) onto four comparison groups under drought as well as non-drought conditions (Supplemental Fig. 2). In pRAP17-ccdB control lines, a large number of TFs (63.5%) were identified as differentially expressed under drought vs. non-drought conditions. Among them, 322 TFs were downregulated, and only 77 TFs were up-regulated (Supplemental Fig. 2). 29% of identified TFs were overlapped between pRAP17-ccdB control and GmAALK1-RNAi silenced lines. Only a few TFs were identified only in GmAALK1-RNAi silenced lines. In GmAALK1-RNAi lines only 30 TFs were identified which represents 4.8% of total identified TFs. Among them, 17 were downregulated and only 13 TFs were up-regulated. Further, a classification analysis was performed on identified TFs (Supplemental Data Set 9). Only 4 TFs were identified in GmAALK1-RNAi vs. pRAP17-ccdB control under drought conditions. Among them, three TFs (Glyma.06G050300: Zinc finger; Glyma.07G178500: G2-like; Glyma.09G240000: WRKY) were downregulated and Glyma.U000500 (bHLH) was up-regulated (Supplemental Data Set 9). However, a total of 13 TFs were identified in Gmaalk1-RNAi vs. pRAP17-ccdB control under non-drought conditions (Supplemental Fig. 2), all being downregulated. Among them, Glyma.02G157300 belongs to MYB_related family and was highly downregulated. In GmAALK1-RNAi lines, 217 TFs were identified; among them, 87 were up-regulated, whereas 127 were downregulated when comparing drought vs. non-drought conditions.
The TFs regulated explicitly in control, and GmAALK1-RNAi silenced lines under drought vs. non-drought conditions were selected for further gene family analysis (Supplemental Data Set 9). The majority of TFs belong to these two categories. Genes belonging to the ERF (ethylene response factor), MYB (Myeloblastosis), bHLH (basic helix-loop-helix), C2H2, NAC, WRKY, GRAS, bZIP, HDZIP, G2-like and Dof family are the most represented among the differentially expressed TFs in pRAP17-ccdB control lines under drought vs. non-drought conditions. ERF has the highest number (93) of differential transcripts followed by MYB (61), bHLH (55), C2H2 (47), NAC (38) and WRKY (37) (Supplemental Fig. 2, Supplemental Data Set 9). Genes belonging to the bHLH (basic helix-loop-helix), MYB (Myeloblastosis), WRKY (is a class of DNA-binging proteins), ERF (Ethylene responsive transcription factor), and NAC family represents most of the differentially expressed TFs in GmAALK1-RNAi silenced lines under drought treated vs. non-treated conditions. ERF has the highest number (36 TFs) of transcripts, followed by MYB, WRKY, NAC, and bHLH.
A qRT-PCR Validation of Differentially Expressed Transcripts Identified by RNA Seq Analyses
To validate the RNA seq expression data and its reliability, 21 differentially expressed genes were selected for qRT-PCR analysis (Supplemental Data Set 2). To compare these two different methods, the relative expression change from the qPCR was transformed into fold change and compared with the RNA seq fold change value. The 21 selected genes for this comparison included genes associated with metabolism, stress response, and transcription factors.
After comparing the results between qPCR and RNA-seq, we observed that the expression patterns in qRT-PCR and RNA-seq analyses were very similar. Correlation between RNA-seq and qRT-PCR was evaluated using log2 expression levels. The qRT-PCR measurements were highly correlated with the RNA seq results (y = 0.7143x + 0.0344, R2 = 0.7499) (Supplemental Fig. 3). The comparison of data between different methods showed that our RNA seq data quantification was accurate. Thus, RNA seq quantification results represent a good estimate of gene expression changes of the soybean root response to drought stress.
Drought-Responsive Genes are Up-Regulated in Soybean Plants by Overexpressing GmAALK1
In order to elucidate the molecular mechanism of drought tolerance mediated by GmAALK1 in soybean, drought-related candidate genes in GmAALK1-overexpressing lines under drought treatment were analyed by qRT-PCR (Fig. 8). GmRAB18 and GmRD29 are known as ABA marker genes (Hauser et al. 2017). Both genes (Glyma.09G185500, Glyma.09G139600) are up-regulated in GmAALK1-overexpression lines under drought treatment. Similarly, Rab family GTPase gene (Glyma.08g345300) was highly up-regulated in GmAALK1-overexpression lines under drought treatment. Likewise, Glyma.05G219400 (which encodes a heat shock 70 kDa protein 5), Glyma.15g034500 (MYB transcription factor MYB82), Glyma.01g119600 (encoding a stress-induced protein), Glyma.08G032800 (oxidative stress 3) were also up-regulated in GmAALK1-overexpression line under drought. The NINE-CIS-EPOXYCAROTENOID DIOXYGENASE 3 (NCED3), a gene that encodes a key enzyme for abscisic acid (ABA) synthesis under conditions of dehydration stress (Takahashi et al. 2018), was also up-regulated in GmAALK1-overexpression lines under drought treatment (Fig. 8). In addition, Glyma.02G259300 (encoding a peroxidase superfamily protein), Glyma.05G200400 (encoding a homeodomain-like superfamily protein), Glyma.16G198700 (encoding a MATE efflux family protein), and Glyma.14G201100 (encoding an O-methyltransferase family protein) were up-regulated in GmAALK1-overexpression lines under drought treatments (Fig. 8; Supplemental Data Set 9). We showed 12 genes having upregulation in GmAALK1-overexpression lines are well known to mediate drought tolerance.

Expression of candidate drought-responsive genes in GmAALK1-overexpression and GmAALK1-RNAi lines of soybean under drought condition. Blue color represents the gene expression in GmAALK1-overexpressed lines (OX lines) whereas orange color represents the GmAALK1-RNAi lines (RNAi lines). The bar represent the means and standard errors of three biological replicates
Discussion
GmAALK1 Possesses Typical Features of the SnRK2 Subfamily Group III
The SnRK2s subfamily contains osmotic stress-activated protein kinases and has been well studied in rice and Arabidopsis. Growing evidence indicates that subfamily Group-III are the global regulators of multiple stress signaling pathways. However, subfamily Group-III of SnRK2s are not studied well in soybean. So far, only one study was conducted in this group using cDNA arrays and showed that GmAAPK is induced by ABA, PEG, Ca2+, and Na+, but not cold (4 °C) treatments in soybean leaves. No study has been done in SnRK2s subfamily groupIII for functional characterization by the transgenic approach to date in soybean plants. Thus, we are the first to demonstrate that GmAALK1 is a positive regulator of drought stress in soybean plants by the transgenic approach. Our functional analysis of the soybean SnRK2 family combined with the phylogenetic analysis, including the full members of Arabidopsis, rice, and faba bean, is significant for the study of uncharacterized members of soybean and other plant species. The phylogenetic analysis showed that GmAALK1 belongs to SnRK2 subfamily Group-III. GmAALK1 was clustered together with a well-known member of this group, such as SAPK8/9/10 and SnRK2.2./2.3/2.6 and was reported to be activated by ABA (Kobayashi et al. 2004) (Nakashima et al. 2009). GmAALK1 is evolutionary more related to V. faba AAPK than another subfamily Group III members of SnRK2. Previously, we identified the first AAPK in V. faba which is a guard cell-specific kinase and showed that AAPK is a positive regulator of ABA signaling (Li and Assmann 1996; Li et al. 2000). Therefore, the GmAALK1 SnRK2 protein kinase member of subfamily Group III kinases may have evolved by gaining the capacity to be activated by ABA.
The previous study indicates that the subfamily Group III of SnRK2s are regulators of multiple stress signaling pathways, and have specific characteristics of functional domains. Domain II is well conserved among Group III of SnRK2 genes in our data set i.e., three AtSnRK2.2/2.3/2.6, three OsSAPK8/9/10, V. faba AAPK and our identified soybean genes (GmAALK1, Glyma.01G183500, GmAAPK, Glyma.05G081900, Glyma.02G135500, Glyma.07G209400, Glyma.07G178600, Glyma.20G009600). These results indicate that these genes may play a role in ABA and osmotic stress response.
Physiological Changes in Transgenic GmAALK1 Soybean Plants Under Drought Stress
Morphological analysis revealed that GmAALK1 is a positive regulator of drought stress in soybean plants. The observed increase and decrease in the drought tolerance ability of GmAALK1-overexpression and GmAALK1-RNAi lines, respectively, clearly indicated that the GmAALK1 plays an active role in drought tolerance in soybean. Previous studies on either overexpression or silencing of the ABA-responsive kinases gene in rice and other plant species have demonstrated similar results of drought tolerance or sensitivity, respectively (Tian et al. 2013; Dey et al. 2016). TaSnRK2.9, gene from Tobacco showed plants tolerance to drought and salt stresses through enhanced ROS scavenging ability, ABA-dependent signal transduction as well as a specific SnRK-ABF interaction (Feng al el. 2019). Likewise, overexpression of a protein kinase gene MpSnRK2.10 (from Malus prunifolia) leads to drought stress response in Arabidopisis as well as apple (Shao et al. 2019).
Environmental stresses often cause physiological parameters changes and secondary metabolite production in plants. The study suggests that in low water potential during soybean vegetative stages the plant will halt or reduce its shoot growth, but the root will continue to grow (Yamaguchi and Sharp 2010). In this study, we also observed, the root system of soybean plants of GmAALK1 overexpressing composite plants developed more new lateral roots than RNAi silenced, and control lines under drought stress. Thus, the understanding of the soybean root response to drought is very critical for the effective management of abiotic stress (Song et al. 2016). Chlorophyll content, anthocyanin, flavonoids and nitrogen balance index are the parameters for evaluating drought stress in our study. The chlorophyll content is one of the crucial parameters for the determining factor for the accumulation of biomass and grain yield, as well as an assessment of drought, heat, and salt-tolerant assays. A significant increase in chlorophyll content was observed in GmAALK1-overexpression lines than GmAALK1-RNAi and control lines under drought stress, revealing that the GmAALK1 overexpression lines had higher photosynthetic capacities. Similar results have been observed in the overexpression of TaSnRK2.3 in common wheat (Tian et al. 2013). Drought stress induces a decrease in the chlorophyll content, which leads to a change in the ratio of chlorophyll and carotenoid and an increase in the proportion of violaxanthin-cyle pigment, which ultimately affects the reduction of photosynthetic rate (Kyparissis et al. 1995).
Further, flavonoids accumulates in plants in response to water limiting conditions and play a key role in protecting against UV-radiations, pathogens, and abiotic stresses (Treutter 2006; Shojaie et al. 2016). In our study, we observed that flavonoids are significantly reduced in GmAALK1-overexpression lines in comparison with GmAALK1-RNAi and control lines under drought stress. These results are similar to other studies (Agati et al. 2012; Fracasso et al. 2016), where they reported that flavonoids genes were up-regulated in response to drought in sensitive genotype whereas downregulated in tolerant genotype. The biosynthesis of “antioxidants” flavonoids increases more in stress-sensitive species than in stress-tolerant species (Agati et al. 2012). The main reason behind this fact is that stress sensitives species display a less efficient “first line” of defense against ROS in the condition of stress and they are therefore exposed to more severe oxidative stress (Tattini et al. 2006; Wolf et al. 2010). Like flavonoids, anthocyanin is reported to accumulate under drought, salt, and UV-B radiation (Nogués et al. 1998; Chunthaburee et al. 2016). In our study, anthocyanin increases in GmAALK1-RNAi and control lines under drought stress in comparison with GmAALK1-overexpression lines. Anthocyanin is thought to minimize the oxidative damage and act as antioxidants by neutralizing ROS directly (Hughes et al. 2005; Kytridis and Manetas 2006).
Carbon (C), and Nitrogen (N) balance is universal and critical for metabolism, growth, and development in cellular organisms (Huang et al. 2016). Nitrogen balance index (NBI) is more often an indicator of C/N allocation changes due to N-deficiency than a measure of leaf nitrogen content per se (Cartelat et al. 2005). NBI is the ratio of chlorophyll content and flavonoids. In our study, NBI is higher in GmAALK1 overexpression lines as compared to GmAALK1-RNAi silenced and control lines under drought stress. The study suggests that the carbon/nitrogen balance may be involved in the regulation of drought-induced leaf senescence (Chen et al. 2015). Thus, the physiological response of GmAALK1 showed that GmAALK1 modulates the drought stress response in soybean plants. To uncover the molecular mechanism of stress response mediated by GmAALK1 soybean, we performed a transcriptome analysis of GmAALK1-RNAi and control lines using the RNA seq approach.
Knockdown of GmAALK1 Affectes Expression of Drought-Responsive Genes Under Drought Condition
This is the first study conducted on transcriptome level of loss of function of AAPK-like in soybean. Our study provides the first large-scale investigation of gene expression changes that occur in GmAALK1-RNAi silenced plants as compared to control under drought treatments. The results presented here show that unique and differential responses of soybean root tissue under drought and normal conditions in GmAALK1-RNAi silenced lines as compared with control lines. In our RNA seq analysis, the responses differed substantially between the GmAALK1-RNAi silenced lines and control lines regarding the number of genes and pathways involved in drought stress response, but also regarding the constitutive expression level of several pathways. Several thousands of genes of differentially expressed genes in soybean plants in response to water deficit were identified as reported by others (Chen et al. 2013, 2016; Rodrigues et al. 2015; Song et al. 2016).
Although the stress level applied was equal, the GmAALK1-RNAi silenced, and control lines responded differently; in control, a significantly higher number of differentially expressed genes was observed than in the GmAALK1-RNAi silenced lines, resulting in a more significant enrichment of GO terms related to drought stress response in control than GmAALK1-RNAi silenced lines. Drought stress caused the massive production of reactive oxygen species (ROS) that cause oxidative stress (Munne-Bosch et al. 2001). In the “response of carbohydrate metabolic process” (GO:0005975), “response to oxidative stress” (GO:0006979), “response to oxidoreductase” (GO:0016491) and “response to stress” (GO:0006950), genes were more downregulated in GmAALK1-RNAi silenced lines as compared to control lines which indicates that RNAi lines are more susceptible to drought stress than control lines. Similar results have been reported by Fracasso et al. (Fracasso et al. 2016), showed that drought tolerant lines have more up-regulated of these genes than sensitive lines. Many metabolic processes were related to carbohydrate metabolism, which could provide most of the energy required for these pathways under drought conditions. In our research, a higher number of carbohydrate metabolism genes were identified in control vs. GmAALK1-RNAi silenced lines which may indicate that GmAALK1-RNAi silenced lines are sensitive more drought.
In our analysis, differntially expressed genes were found to be associated with auxin (IAA, indole-3-acetic acid), ABA and ethylene signaling pathways. A previous study also reported that ABA, auxin, and ethylene hormones were involved in drought-responsive pathways (Le et al. 2012a, b). The expression of genes associated with auxin transporter has been shown to be regulated by ethylene (Růzicka et al. 2007). However, auxin was found to affect the synthesis of ethylene (Tsuchisaka and Theologis 2004). Hence, ethylene achieves a local activation of the auxin signaling pathways and regulates root growth by stimulating the auxin biosynthesis as well as by modulating the auxin transport machinery (Růzicka et al. 2007). In our finding, five different auxin transporter genes (PIN1, PIN2, PIN6, PIN7, AUX1) in control lines and one AUX1 in GmAALK1-RNAi silenced lines were found to be regulated by water deficit stress. In the plant kingdom, carotenoid oxygenases genes are divided into five clades, CCD1, CCD4, CCD6, CCD8, and NCED. NCED is the first rate-limiting enzymes in ABA biosynthesis, which plays a vital role in plant resistance to stress. A previous study reported that ABA treatment increases the CCD7 and CCD8 transcription in soybean and are key gened for the strigolactone pathway (Wang et al. 2013). In our analysis, CCD7, CCD8 and NCED4 genes were found downregulated in control lines. All of the above results give evidence about transcripts associated with hormones that are associated with drought stress.
Transcriptional control is a crucial component of plant response to many environmental stresses (Singh et al. 2002; Song et al. 2016). In our study, several transcription factor families, which include ERF, MYB, bHLH, C2H2, WRKY, NAC were identified in control, and GmAALK1-RNAi silenced lines under drought conditions. Among the identified TFs, MYB, MYC, AP2, and HD-ZIP play a central role in drought tolerance (Liu et al. 1998; Sugano et al. 2003; Shin et al. 2011). bHLH proteins and bZIP TFs that regulate the stress-responsive ABA-signaling pathway have been reported in (Li et al. 2007; Xiang et al. 2008). ARFs and ERFs are also involved in stress responses (Seo et al. 2009; Wang et al. 2012). We thus believe that the presence of these TFs detected through differential gene expression indicates that the various signal molecules act to improve drought tolerance in soybean. Further molecular studies need to be done to evaluate the importance of these TFs in drought conditions and to determine the role of individual genes. Investigating the transcriptional regulatory network of differentially expressed genes involved in drought stress response as compared to control would provide more information for further functional analysis.
Overexpression of GmAALK1 Enhances Expression of Drought-Responsive Genes Under Drought Condition
To understand the comprehensive function of GmAALK1 under drought stress conditions, we analyzed expression profiles of 12 drought-responsive genes, and all showed up-regulation in GmAALK1-overexpression lines (Fig. 8). The drought-responsive genes include GmRD29 (responsive to desiccation, also known as LTI65) and GmNCED3, a gene that encodes a key enzyme for ABA synthesis under dehydration stress (Takahashi et al. 2018). These two genes were up-regulated in GmAALK1-overexpression lines in our study. Similarly, Glyma.08G032800 gene coding for the oxidate stress three protein (OXS3) has been detected in water-deficit stress and has a reported role in protecting the cell against photooxidation (Rodrigues et al. 2015). The GmRAB18 is a well-known drought-and ABA-responsive gene (Rodrigues et al. 2015). Multidrug and toxic compound extrusion (MATE) family proteins have been shown to contribute to the response of abiotic stress especially related to aluminum toxicity in soybean (Liu et al. 2016) and Arabidopsis (Tiwari et al. 2015). Likewise, homeodomain like superfamily protein were involved in the regulation of plant development and response to environmental stresses (Huang et al. 2014). In our study, these 12 genes are mostly downregulated, or relatively low expression in GmAALK1-RNAi silenced lines under drought stress (Fig. 8). These results suggest that GmAALK1 might be a transcriptional activator of drought stress-related genes and that overexpression or downregulation of GmAALK1 affects the expression of these drought-responsive genes.
Conclusions
We have characterized the physiological and molecular function of GmAALK1, an abscisic acid-activated protein kinase-like gene of soybean through developing gain-of-function and loss-of-function by transgenic approach. Overexpression of GmAALK1 gene from soybean and RNAi mediated silencing of endogenous GmAALK1 in soybean revealed that the GmAALK1 positively regulates drought stress tolerance. The study has demonstrated the usefulness of genome wide gene expression analysis for identification of differentially expressed genes between control and GmAALK1-RNAi silenced lines under drought conditions. Based on this, 12 candidate drought-responsive differentially expressed genes were selected for further study in GmAALK1 overexpression lines. The GmAALK1 overexpression lines enhanced the transcription of drought-responsive genes, indicating that the GmAALK1 is a positive regulator of drought stress signaling pathways in soybean. Together, the present findings strengthen our knowledge about the functional role of GmAALK1 as transactivating kinase and probable transcriptional activator under drought stress, which can be utilized as a promising gene-based molecular marker in transgenic breeding for generating crop plants with improved drought tolerance.
Accession Numbers
Sequence data from this article can be found in Phytozome or Genebank databases under the following accession numbers: AtSnRK2.1 (AT5G08590), AtSnRK2.2 (AT3G50500), AtSnRK2.3 (AT5G66880), AtSnRK2.4 (AT1G10940), AtSnRK2.5 (AT5G63650), AtSnRK2.6 (AT4G33950), AtSnRK2.7 (AT4G40010), AtSnRK2.8 (AT1G78290), AtSnRK2.9 (AT2G23030), AtSnRK2.10 (AT1G60940), Glyma.01G183500, SPK1 (Glyma.01G204200), Glyma.02G135500, Glyma.02G208500, SPK4 (Glyma.04G205400), Glyma.05G066700, Glyma.05G081900, Glyma.05G176100, Glyma.05G197700, Glyma.06G160100, Glyma.07G178600, Glyma.07G209400, Glyma.08G005100, Glyma.08G133600, SPK3(Glyma.08G188300), SPK2 (Glyma.11G038800), GmAAPK (Glyma.11G058800), Glyma.12G169800, Glyma.14G176700, Glyma.17G148800, GmAALK1 (Glyma.17G178800), Glyma.20G009600, OsSAPK1(LOC_Os03g27280), OsSAPK2 (LOC_Os07g42940), OsSAPK3 (LOC_Os10g41490), OsSAPK4 (LOC_Os01g64970), OsSAPK5 (LOC_Os04g59450), OsSAPK6 (LOC_Os02g34600), OsSAPK7 (LOC_Os04g35240), OsSAPK8 (LOC_Os03g55600), OsSAPK9 (LOC_Os12g39630), OsSAPK10 (LOC_Os03g41460), AAPK (AAF27340.1).
Data Availability
The newly generated RNA-seq data discussed in this article can be found in National Center for Biotechnology Information Short Read Archive sequence database unde the accession number PRJNA641330. Other supporting data are included as supplemental files.
References
Agati G, Azzarello E, Pollastri S, Tattini M (2012) Flavonoids as antioxidants in plants: location and functional significance. Plant Sci 196:67–76
Anderberg RJ, Walker-Simmons MK (1992) Isolation of a wheat cDNA clone for an abscisic acid-inducible transcript with homology to protein kinases. Proc Natl Acad Sci USA 89:10183–10187
Anders S, Pyl PT, Huber W (2015) HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169
Ashburner M, Ball CA, Blake JA et al (2000) Gene ontology: tool for the unification of biology. Nat Genet 25:25–29
Belin C, Franco PO, de Bourbousse C, Chaignepain S, Schmitter JM, Vavasseur A, Giraudat J, Barbier-Brygoo H, Thomine S (2006) Identification of features regulating OST1 kinase activity and OST1 function in guard cells. Plant Physiol 141:1316–1327
Bohnert HJ, Gong Q, Li P, Ma S (2006) Unraveling abiotic stress tolerance mechanisms - Getting genomics going. Curr Opin Plant Biol 9:180–188
Boneh U, Biton I, Schwartz A, Ben-Ari G (2012) Characterization of the ABA signal transduction pathway in vitis vinifera. Plant Sci 187:89–96
Boudsocq M, Barbier-Brygoo H, Laurière C (2004) Identification of nine sucrose nonfermenting 1-related protein kinases 2 activated by hyperosmotic and saline stresses in Arabidopsis thaliana. J Biol Chem 279:41758–41766
Cartelat A, Cerovic ZG, Goulas Y et al (2005) Optically assessed contents of leaf polyphenolics and chlorophyll as indicators of nitrogen deficiency in wheat (Triticum aestivum L.). F Crop Res 91:35–49
Cerovic ZG, Masdoumier G, Ghozlen NB, Latouche G (2012) A new optical leaf-clip meter for simultaneous non-destructive assessment of leaf chlorophyll and epidermal flavonoids. Physiol Plant 146:251–260
Chen LM, Zhou XA, Li WB et al (2013) Genome-wide transcriptional analysis of two soybean genotypes under dehydration and rehydration conditions. BMC Genomics 14:687
Chen D, Wang S, Xiong B, Cao B, Deng X (2015) Carbon/Nitrogen Imbalance associated with drought-induced leaf senescence in sorghum bicolor. PLoS ONE 10:e0137026
Chen W, Yao Q, Patil GB et al (2016) Identification and comparative analysis of differential gene expression in soybean leaf tissue under drought and flooding stress revealed by RNA-Seq. Front Plant Sci 7:1044
Chunthaburee S, Dongsansuk A, Sanitchon J, Pattanagul W, Theerakulpisut P (2016) Physiological and biochemical parameters for evaluation and clustering of rice cultivars differing in salt tolerance at seedling stage. Saudi J Biol Sci 23:467–477
Cutler SR, Rodriguez PL, Finkelstein RR, Abrams SR (2010) Abscisic acid: emergence of a core signaling network. Annu Rev Plant Biol 61:651–679
Dey A, Samanta MK, Gayen S, Maiti MK (2016) The sucrose non-fermenting 1-related kinase 2 gene SAPK9 improves drought tolerance and grain yield in rice by modulating cellular osmotic potential, stomatal closure and stress-responsive gene expression. BMC Plant Biol 16:158
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21
Du X, Zhao X, Li X, Guo C, Lu W, Gu J, Xiao K (2013) Overexpression of TaSRK2C1, a wheat SNF1-related protein kinase 2 gene, increases tolerance to dehydration, salt, and low temperature in transgenic tobacco. Plant Mol Biol Rep 31:810–821
Feng J, Wang L, Wu Y, Luo Q, Zhang Y, Qiu D, Han J, Su P, Xiong Z, Chang J, Yang G, He G (2019) TaSnRK2.9, a sucrose non-fermenting 1-related protein kinase gene, positively regulates plant response to drought and salt stress in transgenic tobacco. Front Plant Sci 9:2003
Fracasso A, Trindade LM, Amaducci S (2016) Drought stress tolerance strategies revealed by RNA-Seq in two sorghum genotypes with contrasting WUE. BMC Plant Biol 16:115
Fujii H, Zhu JK (2009) Arabidopsis mutant deficient in 3 abscisic acid-activated protein kinases reveals critical roles in growth, reproduction, and stress. Proc Natl Acad Sci USA 106:8380–8385
Fujii H, Verslues PE, Zhu JK (2011) Arabidopsis decuple mutant reveals the importance of SnRK2 kinases in osmotic stress responses in vivo. Proc Natl Acad Sci USA 108:1717–1722
Fujita Y, Yoshida T, Yamaguchi-Shinozaki K (2013) Pivotal role of the AREB/ABF-SnRK2 pathway in ABRE-mediated transcription in response to osmotic stress in plants. Physiol Plant 147:15–27
FurihataT MK, Fujita Y, Umezawa T, Yoshida R, Shinozaki K, Yamaguchi-Shinozaki K (2006) Abscisic acid-dependent multisite phosphorylation regulates the activity of a transcription activator AREB1. Proc Natl Acad Sci USA 103:1988–1993
Halwatura D, McIntyre N, Lechner AM, Arnold S (2017) Capability of meteorological drought indices for detecting soil moisture droughts. J Hydrol Reg Stud 12:396–412
Hauser F, Li Z, Waadt R, Schroeder JI (2017) SnapShot: Abscisic acid signaling. Cell 171:1708-1708.e0
Huai J, Wang M, He J, Zheng J, Dong Z, Lv H, Zhao J, Wang G (2008) Cloning and characterization of the SnRK2 gene family from Zea mays. Plant Cell Rep 27:1861–1868
Huang X, Duan M, Liao J, Yuan X, Chen H, Feng J, Huang J, Zhang HS (2014) OsSLI1, a homeodomain containing transcription activator, involves abscisic acid related stress response in rice (Oryza sativa L.). ScientificWorldJournal 2014:809353
Huang Z, Tang J, Duan W, Wang Z, Song X, Hou X (2015) Molecular evolution, characterization, and expression analysis of SnRK2 gene family in Pak-choi (Brassica rapa ssp. chinensis). Front Plant Sci 6:879
Huang A, Sang Y, Sun W, Fu Y, Yang Z (2016) Transcriptomic analysis of responses to imbalanced carbon: nitrogen availabilities in rice seedlings. PLoS ONE 11:e0165732
Hughes NM, Neufeld HS, Burkey KO (2005) Functional role of anthocyanins in high-light winter leaves of the evergreen herb Galax urceolata. New Phytol 168:575–587
Jin J, Tian F, Yang DC, Meng YQ, Kong L, Luo J, Gao G (2017) PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res 45:D1040–D1045
Johnson RR, Wagner RL, Verhey SD, Walker-Simmons MK (2002) The abscisic acid-responsive kinase PKABA1 interacts with a seed-specific abscisic acid response element-binding factor, TaABF, and phosphorylates TaABF peptide sequences. Plant Physiol 130:837–846
Kagaya Y, Hobo T, Murata M, Ban A, Hattori T (2002) Abscisic acid-induced transcription is mediated by phosphorylation of an abscisic acid response element binding factor, TRAB1. Plant Cell 14:3177–3189
Kim SY, Chung HJ, Thomas TL (1997) Isolation of a novel class of bZIP transcription factors that interact with ABA-responsive and embryo-specification elements in the Dc3 promoter using a modified yeast one-hybrid system. Plant J 11:1237–1251
Klingler JP, Batelli G, Zhu JK (2010) ABA receptors: the START of a new paradigm in phytohormone signalling. J Exp Bot 61:3199–3210
Klink VP, MacDonald MH, Martins VE, Park SC, Kim KH, Baek SH, Matthews BF (2008) MiniMax, a new diminutive Glycine max genotype with a rapid life cycle, embryogenic potential and transformation capabilities. Plant Cell Tissue Organ Cult 92:183–195
Klink VP, Kim KH, Martins V, MacDonald MH, Beard HS, Alkharouf NW, Lee SK, Park SC, Matthews BF (2009) A correlation between host-mediated expression of parasite genes as tandem inverted repeats and abrogation of development of female Heterodera glycines cyst formation during infection of Glycine max. Planta 230:53–71
Kobayashi Y, Yamamoto S, Minami H, Kagaya Y, Hattori T (2004) Differential activation of the rice sucrose nonfermenting1-related protein kinase2 family by hyperosmotic stress and abscisic acid. Plant Cell 16:1163–1177
Kobayashi Y, Murata M, Minami H, Yamamoto S, Kagaya Y, Hobo T, Yamamoto A, Hattori T (2005) Abscisic acid-activated SNRK2 protein kinases function in the gene-regulation pathway of ABA signal transduction by phosphorylating ABA response element-binding factors. Plant J 44:939–949
Kulik A, Wawer I, Krzywińska E, Bucholc M, Dobrowolska G (2011) SnRK2 protein kinases—Key regulators of plant response to abiotic stresses. OMICS J Integr Biol 15:859–872
Kumar S, Stecher G, Tamura K (2016) MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
Kyparissis A, Petropoulou Y, Manetas Y (1995) Summer survival of leaves in a soft-leaved shrub (Phlomis fruticosa L., Labiatae) under Mediterranean field conditions: avoidance of photoinhibitory damage through decreased chlorophyll contents. J Exp Bot 46:1825–1831
Kytridis VP, Manetas Y (2006) Mesophyll versus epidermal anthocyanins as potential in vivo antioxidants: evidence linking the putative antioxidant role to the proximity of oxy-radical source. J Exp Bot 57:2203–2210
Le DT, Aldrich DL, Valliyodan B et al (2012a) Evaluation of candidate reference genes for normalization of quantitative RT-PCR in soybean tissues under various abiotic stress conditions. PLoS ONE 7:e46487
Le DT, Nishiyama R, Watanabe Y, Tanaka M, Seki M, Ham LH, Yamaguchi-Shinozaki K, Shinozaki K, Tran LSP (2012b) Differential gene expression in soybean leaf tissues at late developmental stages under drought stress revealed by genome-wide transcriptome. PLoS ONE 7:e49522
Li J, Assmann SM (1996) An abscisic acid-activated and calcium-independent protein kinase from guard cells of fava bean. Plant Cell 8:2359–2368
Li J, Watson MB, Assmann SM (2000) Regulation of abscisic acid-induced stomatal closure and anion channels by guard cell AAPK kinase. Science 287:300–303
Li J, Kinoshita T, Pandey S, Ng CKY, Gygi SP, Shimazaki K, Assmann SM (2002) Modulation of an RNA-binding protein by abscisic-acid-activated protein kinase. Nature 418:793–797
Li H, Sun J, Xu Y, Jiang H, Wu X, Li C (2007) The bHLH-type transcription factor AtAIB positively regulates ABA response in Arabidopsis. Plant Mol Biol 65:655–665
Liu Q, Kasuga M, Sakuma Y, Abe H, Miura S, Yamaguchi-Shinozaki K, Shinozaki K (1998) Two transcription factors, DREB1 and DREB2, with an EREBP/AP2 DNA binding domain separate two cellular signal transduction pathways in drought-and low-temperature-responsive gene expression, respectively, in Arabidopsis. Plant Cell 10:1391–1406
Liu J, Li Y, Wang W, Gai J, Li Y (2016) Genome-wide analysis of MATE transporters and expression patterns of a subgroup of MATE genes in response to aluminum toxicity in soybean. BMC Genomics 17:223
Liu Z, Ge X, Yang Z, Zhang C, Zhao G, Chen E, Liu J, Zhang X, Li F (2017) Genome-wide identification and characterization of SnRK2 gene family in cotton (Gossypium hirsutum L). BMC Genet 18:54
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and. Methods 25:402–408
Luo GZ, Wang YJ, Xie ZM, Gai JY, Zhang JS, Chen SY (2006) The putative Ser/Thr protein kinase gene GmAAPK from soybean is regulated by abiotic stress. J Integr Plant Biol 48:327–333
Mao X, Zhang H, Tian S, Chang X, Jing R (2010) TaSnRK2.4, an SNF1-type serine/threonine protein kinase of wheat (Triticum aestivum L.), confers enhanced multistress tolerance in Arabidopsis. J Exp Bot 61:683–696
Matsye PD, Lawrence GW, Youssef RM, Kim KH, Lawrence KS, Matthews BF, Klink VP (2012) The expression of a naturally occurring, truncated allele of an α-SNAP gene suppresses plant parasitic nematode infection. Plant Mol Biol 80:131–155
Monks DE, Aghoram K, Courtney PD, DeWald DB, Dewey RE (2001) Hyperosmotic stress induces the rapid phosphorylation of a soybean phosphatidylinositol transfer protein homolog through activation of the protein kinases SPK1 and SPK2. Plant Cell 13:1205–1219
Munne-Bosch S, Jubany-Mari T, Alegre L (2001) Drought-induced senescence is characterized by a loss of antioxidant defences in chloroplasts. Plant Cell Environ 24:1319–1327
Mustilli AC (2002) Arabidopsis OST1 protein kinase mediates the regulation of stomatal aperture by abscisic acid and acts upstream of reactive oxygen species production. Plant Cell 14:3089–3099
Nakashima K, Fujita Y, Kanamori N et al (2009) Three arabidopsis SnRK2 protein kinases, SRK2D/SnRK2.2, SRK2E/SnRK2.6/OST1 and SRK2I/SnRK2.3, involved in ABA signaling are essential for the control of seed development and dormancy. Plant Cell Physiol 50:1345–1363
Nogués S, Allen DJ, Morison JIL, Baker NR (1998) Ultraviolet-B radiation effects on water relations, leaf development, and photosynthesis in droughted pea plants. Plant Physiol 117:173–181
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140
Rodrigues FA, Fuganti-Pagliarini R, Marcolino-Gomes J, Nakayama TJ, Molinari HBC, Lobo FP, Harmon FG, Nepomuceno AL (2015) Daytime soybean transcriptome fluctuations during water deficit stress. BMC Genomics 16:505
Růzicka K, Ljung K, Vanneste S, Podhorská R, Beeckman T, Friml J, Benková E (2007) Ethylene regulates root growth through effects on auxin biosynthesis and transport-dependent auxin distribution. Plant Cell 19:2197–2212
Sah SK, Reddy KR, Li J (2016) Abscisic acid and abiotic stress tolerance in crop plants. Front Plant Sci 7:571
Schmutz J, Cannon SB, Schlueter J et al (2010) Genome sequence of the palaeopolyploid soybean. Nature 463:178–183
Seo PJ, Xiang F, Qiao M, Park JY, Lee YN, Kim SG, Lee YH, Park WJ, Park CM (2009) The MYB96 transcription factor mediates abscisic acid signaling during drought stress response in Arabidopsis. Plant Physiol 151:275–289
Shao Y, Zhang X, van Nocker S, Gong X, Ma F (2019) Overexpression of a protein kinase gene MpSnRK2.10 from Malus prunifolia confers tolerance to drought stress in transgenic Arabidopsis thaliana and apple. Gene 692:26–34
Shin D, Moon SJ, Han S et al (2011) Expression of StMYB1R-1, a novel potato single MYB-like domain transcription factor, increases drought tolerance. Plant Physiol 155:421–432
Shojaie B, Mostajeran A, Ghannadian M (2016) Flavonoid dynamic responses to different drought conditions: amount, type, and localization of flavonols in roots and shoots of Arabidopsis thaliana L. Turk J Biol 40:612–622
Singh K, Foley RC, Oñate-Sánchez L (2002) Transcription factors in plant defense and stress responses. Curr Opin Plant Biol 5:430–436
Song L, Prince S, Valliyodan B et al (2016) Genome-wide transcriptome analysis of soybean primary root under varying water-deficit conditions. BMC Genomics 17:57
Sugano S, Kaminaka H, Rybka Z, Catala R, Salinas J, Matsui K, Ohme-Takagi M, Takatsuji H (2003) Stress-responsive zinc finger gene ZPT2-3 plays a role in drought tolerance in petunia. Plant J 36:830–841
Supek F, Bošnjak M, Škunca N, Šmuc T (2011) Revigo summarizes and visualizes long lists of gene ontology terms. PLoS ONE 6:e21800
Takahashi F, Suzuki T, Osakabe Y, Betsuyaku S, Kondo Y, Dohmae N, Fukuda H, Yamaguchi-Shinozaki K, Shinozaki K (2018) A small peptide modulates stomatal control via abscisic acid in long-distance signalling. Nature 556:235–238
Tattini M, Remorini D, Pinelli P, Agati G, Saracini E, Traversi ML, Massai R (2006) Morpho-anatomical, physiological and biochemical adjustments in response to root zone salinity stress and high solar radiation in two Mediterranean evergreen shrubs, Myrtus communis and Pistacia lentiscus. New Phytol 170:779–794
Tian S, Mao X, Zhang H, Chen S, Zhai C, Yang S, Jing R (2013) Cloning and characterization of TaSnRK2.3, a novel SnRK2 gene in common wheat. J Exp Bot 64:2063–2080
Tian T, Liu Y, Yan H, You Q, Yi X, Du Z, Xu W, Su Z (2017) AgriGO v2.0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res 45:W122–W129
Tiwari M, Sharma D, Singh M, Tripathi RD, Trivedi PK (2015) Expression of OsMATE1 and OsMATE2 alters development, stress responses and pathogen susceptibility in Arabidopsis. Sci Rep 4:3964
Treutter D (2006) Significance of flavonoids in plant resistance: a review. Environ Chem Lett 4:147–157
Tsuchisaka A, Theologis A (2004) Unique and overlapping expression patterns among the Arabidopsis 1-amino-cyclopropane-1-carboxylate synthase gene family members. Plant Physiol 136:2982–3000
Umezawa T, Yoshida R, Maruyama K, Yamaguchi-Shinozaki K, Shinozaki K (2004) SRK2C, a SNF1-related protein kinase 2, improves drought tolerance by controlling stress-responsive gene expression in Arabidopsis thaliana. Proc Natl Acad Sci USA 101:17306–17311
Usadel B, Poree F, Nagel A, Lohse M, Czedik-Eysenberg A, Stitt M (2009) A guide to using MapMan to visualize and compare Omics data in plants: a case study in the crop species, Maize. Plant Cell Environ 32:1211–1229
Vision TJ, Brown DG, Tanksley SD (2000) The origins of genomic duplications in Arabidopsis. Science 290:2114–2117
Wang Y, Wan L, Zhang L, Zhang Z, Zhang H, Quan R, Zhou S, Huang R (2012) An ethylene response factor OsWR1 responsive to drought stress transcriptionally activates wax synthesis related genes and increases wax production in rice. Plant Mol Biol 78:275–288
Wang RK, Wang CE, Fei YY, Gai JY, Zhao TJ (2013) Genome-wide identification and transcription analysis of soybean carotenoid oxygenase genes during abiotic stress treatments. Mol Biol Rep 40:4737–4745
Wolf L, Rizzini L, Stracke R, Ulm R, Rensing SA (2010) The molecular and physiological responses of physcomitrella patens to ultraviolet-B radiation. Plant Physiol 153:1123–1134
Xiang Y, Tang N, Du H, Ye H, Xiong L (2008) Characterization of OsbZIP23 as a key player of the basic leucine zipper transcription factor family for conferring abscisic acid sensitivity and salinity and drought tolerance in rice. Plant Physiol 148:1938–1952
Xu ZS, Liu L, Ni ZY, Liu P, Chen M, Li LC, Chen YF, Ma YZ (2009) W55a encodes a novel protein kinase that is involved in multiple stress responses. J Integr Plant Biol 51:58–66
Yamaguchi M, Sharp RE (2010) Complexity and coordination of root growth at low water potentials: Recent advances from transcriptomic and proteomic analyses. Plant Cell Environ 33:590–603
Yoo MJ, Ma T, Zhu N, Liu L, Harmon AC, Wang Q, Chen S (2016) Genome-wide identification and homeolog-specific expression analysis of the SnRK2 genes in Brassica napus guard cells. Plant Mol Biol 91:211–227
Yoshida R, Hobo T, Ichimura K, Mizoguchi T, Takahashi F, Aronso J, Ecker JR, Shinozaki K (2002) ABA-activated SnRK2 protein kinase is required for dehydration stress signaling in Arabidopsis. Plant Cell Physiol 43:1473–1483
Yoshida R, Umezawa T, Mizoguchi T, Takahashi S, Takahashi F, Shinozaki K (2006) The regulatory domain of SRK2E/OST1/SnRK2.6 interacts with ABI1 and integrates abscisic acid (ABA) and osmotic stress signals controlling stomatal closure in Arabidopsis. J Biol Chem 281:5310–5318
Yu J, Wang J, Lin W, Li S, Li H, Zhou J, Ni P, Dong W, Hu S, Zeng C, Zhang J (2005) The genomes of Oryza sativa: a history of duplications. PLoS Biol 3:e38
Zhang H, Mao X, Wang C, Jing R (2010) Overexpression of a common wheat gene Tasnrk2.8 enhances tolerance to drought, salt and low temperature in Arabidopsis. PLoS ONE 5:e16041
Zhao W, Cheng YH, Zhang C, Shen XJ, You QB, Guo W, Li X, Song XJ, Zhou XA, Jiao YQ (2017) Genome-Wide Identification and Characterization of the GmSnRK2 Family in Soybean. Int J Mol Sci 18:1834
Acknowledgements
SS was supported by an MAFES (Mississippi Agricultural and Forestry Experiment Station) Director’s Doctoral Fellowship awarded to JL and KR. This material is based upon work that is supported by the National Institute of Food and Agriculture, U.S. Department of Agriculture, Hatch/Multi-State Project (Multistate No. NC1200; Project No. MIS-153230) under Accession No. 1014489. This research was partially supported by MAFES Strategic Research Initiative grants to JL and VPK. GVP acknowledges the support from the USDA-Agricultural Research Unit through the Big Data: Biocomputing, Bioinformatics, and Biological Discovery, Project Number: 6066-21310-004-25-S and from the 2018 MAFES Strategic Research Initiative at Mississippi State University.
Funding
National Institute of Food and Agriculture,MIS-153230,Jiaxu Li,Accession No. 1014489,Jiaxu Li,Agricultural Research Service,6066-21310-004-25-S,George V. Popescu,Mississippi Agricultural and Forestry Experiment Station,Mississippi State University
Author information
Authors and Affiliations
Contributions
JL, VPK and SS: designed the project; SS: performed all the experiments; SS, GVP, KRR, VPK and JL: analyzed the results; SS and JL: wrote the manuscript; SS, VPK and JL: revised the article. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interests.
Additional information
Handling Editor: Ashish Srivastava.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sah, S.K., Popescu, G.V., Reddy, K.R. et al. The Glycine Max Abscisic Acid-Activated Protein Kinase-Like Kinase 1 (GmAALK1) Modulates Drought Stress Response. J Plant Growth Regul (2024). https://doi.org/10.1007/s00344-024-11287-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00344-024-11287-x