
Abstract
Glycosyltransferase (GT) enzymes are the members of a large multigene family in plants that can transfer activated sugar molecules to an extensive range of acceptors, such as sterols and secondary metabolites. This glycosylation of plant metabolites helps in the fortifying defense of the plant against different environmental stress. Sterol glycosyltransferase (SGT) is a key member of GT family mainly involved in glycosylation of sterols. Previous report has shown that expression profiling of the WssgtL3.1 gene of Withania somnifera increased dramatically under diverse abiotic and biotic stress. Therefore, the present study aimed to heterologously overexpressed WssgtL3.1 gene in Arabidopsis thaliana to study its role in the mitigation of the adverse effect of Pseudomonas syringae infection. In overexpressing lines, upon pathogen infection, less bacterial growth was observed which might be due to enrichment in SA content. In addition, higher PR1 gene expression, less callose formation, less hydrogen peroxide (H2O2) accumulation, low MDA formation, higher SOD enzyme activity, and greater Fv/Fm were observed in all transgenic lines. Moreover, changes in glycosylation of sterols were observed in all samples; as a result, total sterol content was also found to be higher in each overexpression line. Furthermore, increased expression of squalene synthase (SQS) suggested more sterol biosynthesis in each overexpression lines due to the modulation of SA. Overall, these results suggested that high proportions of free and conjugated sterol contents in transgenic lines due to overexpression of WssgtL3.1 plays a significant role in the enhancement of immunity against P. syringae in A. thaliana plants.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In eukaryotes, sterols molecules are involved in cell membranes with the 3β-OH side chain spanning the hydrophobic core to interact with fatty acyl chains of phospholipids and proteins (Hartmann 1998). Hence, sterols regulate the biological function during the external stimulus and play a significant role in the adaptive responses of plants against various types of environmental stress (Chaturvedi et al. 2011; Ferrer et al. 2017). Biosynthesis of plant sterols takes place in endoplasmic reticulum via mevalonate pathway of isoprenoid metabolism (Fig. 1). Most abundant sterols found in higher plants possess the β-OH group at the C-3 position and are mostly found in free form, i.e., β-sitosterol, stigmasterol, and campesterol (Schaller 2003; Grille et al. 2010; Mishra et al. 2015). Among all this, it has been reported that β-sitosterol and stigmasterol have an important role in maintaining the structure and function of cell membranes, providing the tolerance against various stress, while campesterol acts as precursor of brassinosteroids and is necessary for plant growth and morphogenesis regulation (Grille et al. 2010; Griebel and Zeier 2010; Chaturvedi et al. 2011; Aboobucker and Suza 2019). In addition, the free form of sterol can be conjugated to generate conjugated sterols such as sterile glycosides (SGs), acyl sterile glycosides (ASGs), and sterile esters (SEs) through a number of catalytic changes including glycosylation, esterification, and acylation (Fig. 1) (DeBolt, 2009; Grille et al. 2010; Ferrer et al. 2017). Among these catalytic activities, glycosylation represents the most prominent transformations and not only stabilizes the products but also regulates their physical activities and intracellular distribution (Madina et al. 2007b; Sharma et al. 2007). Sterol glycosyltransferases (sgts), a member of glycosyltransferase family-1 (GT1) (http://www.cazy.org/), are principally concerned in the glycosylation of sterols and play a vital role in metabolic changes of sterols during adaptive responses (Grille et al. 2010; Mishra, 2013). These bound sterol derivatives or sterol-modified counterparts are enzymatically interconvertible with free sterol (FS), which occupy a branch point position in the metabolism of conjugated sterols (Moreau et al. 2002; Ferrer et al. 2017). SGs and ASGs are mainly found in the plasma membrane (PM), and combination of these conjugated sterols with other lipids plays an inevitable role in maintaining the proper membrane structure and functions (Grille et al. 2010; Mishra et al. 2015; Ferrer et al. 2017). However, SEs are stored in cytoplasmic lipid bodies and are believed to function as a reservoir to sustain the levels of FS in cell membranes within the physiological range (Kopischke, 2013; Ferrer et al. 2017). The specific function of FS and glycosylated sterol to these processes remains to be completely understood; however, increasing evidence of recent studies supports the alteration of the ratio of FS and their modified counterparts (SG, ASG, and SE) during adaptive response in plants against various stresses (Mishra et al. 2013; Pandey et al. 2014; Ramirez-Estrada et al. 2017; Castillo, 2019). It has been shown that in the majority of plant species, the amount of SGs and ASGs is relatively less than the total sterol (FS + SG) content, while plants of Solanum genus have been shown extremely high content of SG + ASG fraction (DeBolt et al. 2009; Ramirez-Estrada et al. 2017). Such a high amount of glycosylated sterols is known to involve in protecting cell membrane integrity against the high level of disruptive effects of steroidal glycoalkaloids present in Solanum species (Keukens, 1995; Ramirez-Estrada et al. 2017). Recently, several sgt genes have been cloned from bacteria, fungi, and plants using genetic approaches to analyze conjugated sterol functions which indicate that the SGTs play important role in the synthesis of SGs and ASGs and involved in the intracellular trafficking during plant growth and development in response to various environmental challenges (Warnecke, 1999; Grille et al. 2010; Mishra et al. 2013). In accordance, two cotton Ghsgt genes have been reported for heat shock, code for isozymes with distinct biochemical properties, and a possible differential subcellular localization (Li, 2014). Moreover, tomato sgts have shown enhanced expression in various abiotic stress, i.e., salt, osmotic, cold, as well as in the treatment of abscisic acid and methyl jasmonate (Ramirez-Estrada et al. 2017). Similarly, conjugated sterols with carbohydrate moieties play an important role in the regulation of the Arabidopsis thaliana response to Botrytis cinerea infection (Castillo et al. 2019). Furthermore, SGT gene family of Withania somnifera (Wssgts) showed enhanced enzyme activity and higher expression in the presence of heat stress, wounding, and salicylic acid treatments in W. somnifera (Madina et al. 2007a, 2007b; Chaturvedi et al. 2012). Overexpression of WssgtL1 gene in A. thaliana and tobacco showed enhanced resistance against various abiotic and biotic stress (Mishra et al. 2013; Mishra et al. 2017; Pandey et al. 2014). In addition, ectopic overexpression of WssgtL1 gene in W. somnifera and A. thaliana promotes growth and provides tolerance to abiotic and biotic stresses (Saema et al. 2016; Mishra et al.2017, 2021a). Recently, overexpression of WssgtL3.1 gene in Arabidopsis confirmed its crucial role under salt stress (Mishra et al. 2021a). Considering the importance of SGTs of W. somnifera in plant development and adaptation of plants to various environmental stress, the present study aimed to explore the role of the W. somnifera SGT gene (WssgtL3.1) overexpressed Arabidopsis line in comparison to WT to update the current knowledge about the conjugated sterols in plants and the role of SGT enzyme involved in their biosynthesis during pathogen stress.

Schematic representation of the biosynthesis of conjugated sterols and conjugated sterol metabolism in plants. Free sterols derive from isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP) produced by the mevalonic acid (MVA) pathway. Squalene synthase (sqs) catalyzes the biosynthesis of free sterol, a first pathway specific transformation in sterol metabolism. UDP-glucose:sterol glycosyltransferase enzyme was cloned, is indicated in bold, and showed the interconversion of free sterol to conjugated sterol. Acid hydrolysis and alkaline hydrolysis (saponification) cleavage sites are indicated with the arrow. The dashed arrow shows multiple steps
Materials and Methods
In Silico Analysis
For characterization of WsSGTL3.1 protein (GenBank accession EU342379), a sequence was obtained from the NCBI database (www.ncbi.nlm.nih.gov). The further sequence was analyzed through web tool, ORF-finder, and my-hits online motif scan tool (http://myhits.isb-sib.ch/cgi-bin/motif_scan) for potential open reading frames in a DNA sequence and UGT prosite motifs. Alignment of deduced WsSGTL3.1 amino acid sequence with other plant SGTs amino acids was performed to analyze the conserved domain by using Clustal-W. For multiple alignment, sequences of SGT in A. thaliana (At3g07020, At1g43620), Panax ginseng (AB071962), Avena sativa (Z83832) W. somnifera WsSGTL1 (DQ356887), WsSGTL3.2 (EU342374), WsSGTL3.3 (EU342375), and Oryza sativa (Q5N968) were retrieved from EMBL and NCBI database. Phyre 2 analysis was used to predict the 3D model of WsSGTL3.1 protein and potential binding site (Kelley et al. 2015). The PDB file was obtained with 100% confidence and 70% coverage to the backbone. Further, the phylogenetic analysis was carried out with stress upregulated SGTs, i.e., Catharanthus roseus (BAD29722), Solanum lycopersicum (CAA59450), Medicago truncatula (AAW56091), Solanum tuberosum (AAB48444), Alium cepa (AAP88406), W. somnifera (ABC96116), Nicotiana (TOGT1, TOGT2), and A. thaliana (UGT87A2, UGT73B2, UGT73C1, UGT74E2) using Clustal-X for the relatedness of WsSGTL3.1 amino acid. The analysis was conducted in MEGA 6.0, and evolutionary distances were calculated using the Poisson correction method in phylogenetic analysis.
Transgenic Plant Generation
For isolation of full-length WssgtL3.1 (1.8 kb) gene sequence, total RNA was isolated from young leaves of W. somnifera (Spectrum Plant Total RNA kit, Sigma, USA) and subsequently, cDNA was synthesized (Revert Aid First Strand cDNA synthesis kit Fermentas, USA). Further template cDNA was used to amplify full-length gene through PCR using WssgtL3.1_F1 and WssgtL3.1_R1 primers (Supplementary Table 1). The isolated gene sequence was first cloned into an entry vector by designing primers that contained CACC overhang before ATG at the 5' end of the forward primer, whereas the reverse primer was kept blunt. The PCR products obtained by using these primers were cloned in D-TOPO entry vector (Invitrogen, USA) (Supplementary Fig. S1 a, b). Sequencing confirmed that WssgtL3.1 gene was cloned in entry vector and then transferred to the destination vector by an LR clonase II recombination reaction. The destination vector used was pYL436 which contained a gentamicin resistance gene for selection in plants and CaMV35S promoter for high-level expression (Supplementary Fig. S1 c, d). The destination vector was mobilized into Agrobacterium tumefaciens (GV3101 strain) and confirmed by PCR analysis (Supplementary Figure S1 e). Subsequently, the WssgtL3.1-cloned construct was transformed in A. thaliana ecotype Col-0 (wildtype, WT) by floral dip method (Clough and Bent 1998). All, WssgtL3.1 transgenic lines were screened on ½ MS medium containing 50 µg ml−1 gentamicin antibiotics (Hayford et al. 1988). All screened transgenic lines were further established by PCR analysis (Supplementary Fig. S1 f). Southern analysis was performed by the previously described method by Mishra et al. (2013). Genomic DNA (10 μg) from each genotype was digested with XbaI restriction enzyme, followed by separation on agarose (1%) gel and transferred to Hybond N + membrane (Sigma Chemical Company, St. Louis, MO). For hybridization 1.5 kb fragment of WssgtL3.1 gene, 32P labeled was used as a probe. Relative expression of the WssgtL3.1 gene in each transgenic line was analyzed by real-time PCR and semi-quantitative PCR. All Arabidopsis plants were grown in a growth chamber at 20–22 °C under long-day conditions (16 h light and 8 h dark) with cool white-light illumination (120 µmol m−2 s−1) provided by fluorescent tubes.
Crude Protein Extraction and Glycosyltransferase Activity
For glycosyltransferase activity, crude protein extract from transgenic plants was obtained through the method described by Lim et al. (2003). For it, both transgenic and control leaves (1 g) were ground in liquid N2 and resuspended in 1.5 volumes of extraction buffer (20 mM 2-mercaptoethanol, 1 mM PMSF, 25 mM Tris, pH 6.5, 10% (v/v) glycerol, and 1% polyvinylpolypyrrolidone). Subsequently, the resuspended powder was mixed vigorously and centrifuged at 4 °C, 12,000 rpm for 15 min. After that supernatant was collected (approx. 1 ml) into a new microfuge tube and further centrifuged at 4 °C, 12,000 rpm for 5 min. Subsequently, the resulting supernatant was collected into a new microfuge tube for glycosyltransferase activity. The protein concentration of all samples was determined by Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). For an enzyme assay, 5 mM UDP-glucose was added to the reaction mixture consisting of 50 µl of crude protein extracts (0.2–0.4 mg of total protein), 50 mM Tris/HCl, pH 7.0, and 2 mM sitosterol in a 100 µl reaction. The reactions were then incubated at 30 °C and terminated by addition of 10 μl of trichloroacetic acid (240 mg/ml) after 30 min (Lim et al. 2003). Further, according to the protocol of Madina et al. (2007b), samples were subjected to a TLC plate (Silica gel G-60) using mobile phase of methanol: chloroform (1.5: 8.5, v/v). Vanillin sulfuric acid (1% w/v in 50% sulfuric acid) was used as a spray reagent for the development of TLC plates, and for visualization, plates were heated at 110 °C.
Pathogenicity Assay
For the pathogenicity test, P. syringae virulent strain (vir) [pv. Maculicola PsmES4326] and avirulent (avir) [avrRpt2 (PstDC3000)] strains were used. Fully expanded healthy leaves from 4–5 weeks-old overexpression lines and WT plants were infiltrated (from the abaxial side) with 30 µl of bacterial suspension (1 × 105 CFU) in 1% gelatin solution (Bent et al. 1992; Summermatter et al. 1995). After 3d of infiltration, leaf disks from control and pathogen-treated plants were photographed to visualize the necrotic area. Complete penetration could be visualized by apparent water soaking of the leaves. The bacterial growth was analyzed without leaf sterilization by the method of Katagiri et al. (2002). Leaf disks were collected at different time durations 24 h, 48 h, and 72 h, respectively, and homogenized in 250 µl of 0.85% saline solution. For selection, appropriate dilutions on solid Kings’B medium containing appropriate antibiotics were plated and colony numbers were quantified after 2d to 3d of time durations. For other assays such as callose deposition, measurement of salicylic acid (SA), H2O2 formation, lipid peroxidation, quantitative sterol, and sterol glycoside analysis by HPLC, fully expanded healthy leaves (second whorl position of the rosette) of similar stage of overexpression lines and WT plants were inoculated with moderate bacterial suspension (virulent; 5 × 104 CFU) using surfactant (Silwett-L77). For uniformity, control plants leaves were infiltrated with same amount of 1% gelatin solution.
Callose Deposition
Callose deposition after pathogen inoculation was estimated by Adam and Somerville’s (1996) previously described method. In brief, after 48-h post-inoculation, infiltrated leaves as well as normal leaves were rinsed with 50% ethanol and water, respectively. Afterward leaves samples were stained in freshly prepared 0.01% aniline blue for 30 min (prepared in 150 mM K2HPO4, pH 9.5). All stained samples were mounted with 25% glycerol and examined by epifluorescent illumination (365 nm excitation filter) on a light microscope.
HPLC Analysis for Salicylic Acid Content Before and After Pathogen Infection
Fresh leaves (500 mg) from each WssgtL3.1 overexpression line as well as WT plants (4–5 weeks old) before and after pathogen infiltration (48 h) were used for SA extraction. Fresh leaves were ground with liquid N2 and further homogenized in 2 ml of methanol and formic acid (95:5) mixture. The extract was mixed with 2 M hydrochloric acid and incubated at 80 °C for 60 min. The resulting mixture was centrifuged at 4000 rpm on room temperature. Subsequently, the supernatant was collected in a small round bottom flask and evaporated to dryness in a vacuum rotary evaporator (Buchi, USA), and again resuspended (1 mg/ml) in methanol (Marek et al. 2010). HPLC analysis for SA with reference solution was performed through Shimadzu (Japan) LC-10AT dual-pump system with 0.45 ml min−1 flow rate. The mobile phase of gradient prepared from 1% (v/v) acetic acid in water (Milli Q) and acetonitrile (Mishra et al. 2017).
Quantification of Hydrogen Peroxide (H2O2), Lipid Peroxidation Assay, and SOD Enzyme Activity
Hydrogen peroxide (H2O2) was measured in pathogen-infiltrated leaf samples using the protocol as described by Alexieva et al. (2001). For this assay, the reaction mixture was prepared with 0·5 mL of 100 mM potassium phosphate buffer, 2 mL KI reagent (1 M KI w/v in deionized water), and 0.5 ml leaf extract (extracted in 0·1% trichloroacetic acid (TCA). Then the reaction mixture was kept for 1 h under darkness. Further, absorbance was measured at 390 nm by a spectrophotometer. Hydrogen peroxide content was calculated using known concentrations of H2O2 as standard.
Accumulation of malondialdehyde (MDA) content was estimated in fresh tissues using the protocol of Mishra et al. (2013). For this assay, absorbance was measured spectrophotometrically at 532 and 600 nm. MDA concentration of various samples was calculated by multiplication of the extinction coefficient (€M = 155 mM−1 cm−1) with the subtracted value of OD600 and OD532 of the MDA-TBA complex.
Superoxide dismutase (SOD) enzyme activity was estimated in fresh tissue samples as a function of Nitro blue tetrazolium (NBT) reduction using a spectrophotometer (Beauchamp and Fridovich 1971). For this assay, the Bradford (1976) method was used for protein content determination of each sample. The SOD activity was measured by adding 20 μl supernatant to a freshly prepared reaction mixture consisting of 4.4% (w/v) riboflavin, 57 μm NBT, 10 mM L-methionine, and 0.025% (v/v) Triton-X 100 in 100 mM phosphate buffer. The required amount of enzyme for 50% inhibition of NBT reduction in 2 min at 25 °C called one unit of enzyme activity.
Chlorophyll Fluorescence Imaging After Pathogen Infiltration
Chlorophyll fluorescence parameters were studied in both control and treated plant samples by using Imaging-PAM, M-Series chlorophyll fluorometer (Walz, Effeltrich, Germany), and data were analyzed according to the method described by Maxwell and Johnson (2000). According to Schreiber (2003), the investigated plant is dark adapted and the Kautsky-type fluorescence transient is elicited by sudden exposure to constant light measured together with quenching analysis by saturated light flashes. Fv/Fm indicates the maximum photochemical efficiency of photosystem II (PSII), where Fm is maximum fluorescence of the dark-adapted leaf (under a light saturating flash) and Fv is maximum variable fluorescence (Fm – Fo) that was measured on leaves after 20 min of dark adaptation. The effective quantum yield (YII) of photosystem II (PSII) was calculated using the formula (Fm-Fs)/Fm. Non-photochemical quenching (NPQ) measurement indicates a change in the efficiency of excess excitation energy dissipation as heat. NPQ collectively shows heat dissipation triggered by small thylakoid lumen pH, state transitions of PSII centers, and photoinhibition.
Free and Conjugated Sterols Analysis Through HPLC
Free sterols and conjugated sterols were analyzed quantitatively from acid-hydrolyzed extracts of WssgtL3.1 overexpression lines and WT plants as described by Pandey et al. (2014). Untreated and treated leaves (1 gm) were pulverized in liquid N2 to prepare fine powder for further use in acid hydrolysis. Initially, 10 ml of ethanolic HCL (4 mol l−1) was added to the sample and vigorously shaken. Further, the mixture was refluxed for 1 h at 80 °C and then allowed to cool at room temperature. After that, 10 ml of ethanolic KOH (4 mol l−1) was added to the mixture and vigorously shaken followed by reflux at 70 °C for 1 h. Subsequently, hydrolyzed samples were cooled and extracted three times with 80% methanol. In last, methanolic extracts were evaporated to dryness by vacuum rotary evaporator (Buchi, USA) and was further dissolved in 1 mg/ml of methanol (95%). For the preparation of reference stock, three sterol compounds, i.e., campesterol, β-sitosterol, and stigmasterol (Sigma Aldrich) were weighed in equal amounts and then dissolved in 95% of methanol. Standard solutions were prepared by diluting stock solution with appropriate concentrations, and then 1 µl solution was used for HPLC analysis. Homogenous extracts were obtained through the 0.45 μM filter (Millipore, India). Free sterols were quantified without acid hydrolysis and conjugated sterol quantified with the subtracted value between hydrolyzed sterol contents and free sterols content (Pandey et al. 2014).
For HPLC analysis, we have adopted the previously standardized method with the same reference compounds (Mishra et al. 2013; Pandey et al. 2014). HPLC system (Shimadzu, Japan) equipped with UV-PDA detector and LC-10 system comprising an LC-10 AT dual-pump system were used for quantitative analysis. C18 column (250 × 4.6 mm, 5 μm pore size) (Merck Purospher star® RP) protected by a guard column containing the same packing were used for separation of sterol compounds. For sterol elution, an isocratic solution of acetonitrile and water (95:5, v/v) was used as the mobile phase. The elution of the mobile phase was at a flow rate of 2 ml min−1 by injecting 20 μl of extract. Sterols peaks were identified at 202 nm at 34 °C, and data were integrated with lab solution software (Pandey et al. 2014; Mishra et al. 2020). Sterol content was analyzed by the area of individual peak compared with the reference standard peak of sterol compounds.
Total RNA Isolation and Relative Expression Analysis
Total RNA was isolated using aforesaid described method from 4–5-week-old WssgtL3.1 overexpression lines and WT plants grown under 16 h light/8 h dark, infiltrated with 5 × 104 CFU of virulent strain of P. syringae. Relative expression analysis was done by real-time PCR (qRT-PCR) method using Fast SYBR® Green Master Mix (Applied Biosystems, USA). The actin gene was used for normalization. Primers used in the present study are listed in Supplementary Table 1.
Statistical Analysis
All experimental results were obtained from the means of three independent biological replicates and the results with standard deviation (mean ± SD) or standard error (mean ± SE). A t test was used to analyze the relative expression significance level at p < 0.05 for (*), p ≤ 0.01 for (**), and p ≤ 0.001 or 0.005 for (***). The significance level of H2O2 content, lipid peroxidation assay, and antioxidant activity were analyzed through SPSS 16.0 software (SPSS Inc./IBM Corp. Chicago, USA) using one-way analysis of variance (ANOVA), and means were separated by Duncan’s multiple range test (p < 0.05) (Mishra et al. 2021b).
Results
In Silico Analysis of WsSGTL3.1 Amino Acid Sequence
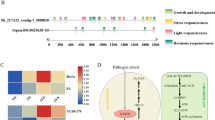
WsSGTL3.1 protein (GenBank accession EU342379) contained an ORF of 1797 bp encoding 599 amino acids followed by a stop codon. The Clustal-W alignment of the deduced amino acid sequence of WsSGTL3.1 showed significant amino acid similarity (46–65%) with A. thaliana, Avena sativa, Oryza sativa and other SGTs of W. somnifera (Fig. 2). This alignment also revealed the presence of two conserved domains, i.e., PSBD (Putative sterol-binding domain) and UGT prosite motifs in WsSGTL3.1 protein and showed significant similarity with other SGTs (Fig. 2). The presence of sterol-binding domain and UGT prosite motifs characteristic in the WssgtL3.1 gene suggested their involvement in sterol modifications. Also, Phyre 2 server predicts the 3D model of WsSGTL3.1 protein based on template ID ‘c5gl5B.’ Analysis of the 3D model structure demonstrates that the WsSGTL3.1 protein has potential ligand-binding sites with sugar-binding domains and sterol-binding domains (supplementary Fig. S2a, b). Further, phylogeny analysis of the WssgtL3.1 gene with the various sgt genes that were upregulated in plants under stress has suggested a strong evolutionary relationship among them (Fig. 3).

Multiple alignments of WsSGTs amino acid sequence with different plant sterol glycosyltransferases using CLUSTAL W. WsSGTL3.1 (EU342379) shows significant amino acid similarity (46–65%) with Avena sativa (Z83832), Oryza sativa (Q5N968), Panax ginseng (AB071962), and other SGTs of W. somnifera (WsSGTL1- DQ35688, WsSGTL3.2-EU342374, WsSGTL3.3 -EU342375) can be retrieved from EMBL database. The green rectangle represents the conserved domain AIIANPPY (Putative sterol-binding domain PSBD) and the orange rectangle shows the conserved domain VVHHGGAG (UGT prosite motif) which are common to the reported SGTs

Phylogenetic analysis of WsSGTL3.1 protein with different sgt genes upregulated under various environmental stress. Neighbor-joining distance tree of W. somnifera SGTs and other plant glycosyltransferases (UGT) belonging to high expression in abiotic and biotic stresses. The amino acid sequences were aligned using Clustal-X. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) is shown at the branches. Evolutionary distances were computed using the Poisson correction method, and analysis was conducted in MEGA 6.0
Development of Overexpressing WssgtL3.1 Lines of A. thaliana
To examine the potential role of WssgtL3.1 gene under biotic stress condition, a total of 12 transgenic lines (G1) was developed and confirmed through PCR. All overexpressing WssgtL3.1 lines were grown up to (G3) generation (under gentamicin selection) for further analysis (Supplementary Fig. S1F). The copy number of T-DNA inserts was analyzed by a Southern blot of genomic DNA of four homozygous overexpression lines, digested by XbaI. Southern hybridization showed different integration patterns in all the lines; thus, it can be said that these lines were derived from independent transformation events (Fig. 4a). Moreover, for the selection of overexpression lines by real-time PCR, relative expression analysis of selected overexpressing lines (T1, T2, and T3) exhibited significant expression of WssgtL3.1 gene (Fig. 4b). Phenotypic observations suggested no significant difference in the rosette area and growth of plants between WT and transgenic lines (Fig. 4c). Apart from this, TLC analysis showed that sterol glycoside was increased in overexpressing lines, compared to WT due to enhanced relative enzyme activities of WsSGTL3.1 in T1, T2, and T3, indicating that the higher expression of WsSGTL3.1 mRNA indeed resulted in its increased enzymatic activities (Fig. 4d).

Molecular characterization and relative enzyme activities of WssgtL3.1-overexpressing A. thaliana (a) Southern blot analysis shows independently transformed lines digested with XbaI and probed with the 1.5 kb gene fragment. Lane 1-5 represents WssgtL3.1 transformants, while lane -6 represents WT plants (b) Phenotypic and morphological observations in 4-week-old WssgtL3.1-overexpressing A. thaliana plants (c) Relative overexpression of WssgtL3.1 gene in A. thaliana lines by qRT-PCR. Actin was used as a housekeeping gene for normalization. (d) Relative enzyme activities of WssgtL3.1 in transgenic plants. Soluble crude extracts from leaves of wild type, and transgenic line T1, T2, and T3 plants were incubated with sitosterol and UDP-glucose, and the reaction mixtures were analyzed by TLC. Sitosterol and sitosterol glycoside were used as reference compounds to identify the compounds
Pathogen Susceptibility of WssgtL3.1 Overexpressing Plants
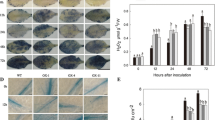
To analyze the effect of overexpression of WssgtL3.1 gene in transgenic A. thaliana, we have examined the susceptibility of each transgenic line against the hemibiotrophic pathogen P. syringae. For the observation of bacterial symptoms, including chlorosis and necrotic lesions in rosette leaves, high concentrations of bacterial inoculums (1 × 105 CFU) were used on 4–5-weeks-old plant leaves. As shown in Fig. 5a, after 3dpi, leaves of WssgtL3.1 transgenic lines were observed less susceptible, whereas WT plants displayed more severe water-soaking or chlorotic symptoms due to virulent inoculation. To observe the detailed disease response, leaves of all transgenic lines with WT plants were infiltrated with a high dose of bacterial suspension (1 × 105 CFU) of virulent and avirulent P. syringae, using surfactant (silwet-L77). The growth curve of both virulent strain and avirulent strain exhibited the actual differences in terms of bacterial growth between overexpression lines and WT plants. Virulent strain bacterial growth in WT plants was significantly more at each time duration compared to all the overexpression lines (Fig. 5b). On contrary, pathogenic bacterial growth was markedly less in transgenic plants at different time points. Due to enhanced bacterial growth, symptomatic WT leaves showed membrane damage, and consequently, enhanced chlorosis and necrotic lesions was observed in WT leaves. In addition, avirulent avrRpt2 (PstDC3000) inoculation on leaves showed no symptomatic lesion, and bacterial growth was also significantly less in each overexpression line at different time durations (Fig. 5c, d). Although avirulent inoculation on WT leaves showed resistance; however, growth curve suggested bacterial growth was found significantly more at all-time durations up to 72 h (Fig. 5d). Moreover, after 48 h of inoculation, aniline blue staining was also demonstrated that the pathogen-infiltrated leaves of the WT formed the maximum deposition of calloses, while all overexpression lines showed a lesser deposition of calloses (Fig. 6).

Pathogen susceptibility assay and determination of bacterial titer in WT and all overexpressing lines of A. thaliana leaves after infection with P. syringae (a) Leaves of overexpression lines and WT were infiltrated with virulent P. syringae (PsmES4326) strain (1 × 105 CFU). Photographed was taken at 72-h post-inoculation (b) P. syringae virulent bacterial growth at different time periods 24 h, 48 h, and 72 h respectively (c) Leaves of overexpression lines and WT were infiltrated with P. syringae avrRpt2 (PstDC3000) pathogen at OD600 = 0.5. Photograph was taken at 72-h post-inoculation. (d) The bacterial growth curve shows less bacterial growth of overexpressing lines at different time periods after inoculation of (1 × 105 CFU) avrRpt2 (PstDC3000). Bacterial growth experiment was repeated three times. Data mean ± SD and significance level at p < 0.01. CFU suggests colony-forming units

Callose formation analysis through aniline blue staining. Challenge inoculated (5 × 104 virulent strain) overexpressing A. thaliana leaves shows less callose formation. Photographs were taken under UV light (wavelength 365 nm) after 48-h post-inoculation (Scale bar, 100 µm)
SA Analysis, H2O2 Accumulation, MDA Measurement, and Antioxidant Activity
SA plays a key role in both locally and systemically induced disease responses. The HPLC–PDA method was used to determine the SA content of WssgtL3.1-overexpressing plant as well as WT leaves. Quantitative analysis showed no significant difference in SA content between transgenic lines and WT plants before pathogen infiltration. However, after 48-h post-inoculation, the SA content of all WssgtL3.1 overexpression lines increased significantly in the range between 2.39 and 2.80 µg/g FW (Fig. 7a). Further, expression analysis of defense marker gene PR1 was analyzed, and it was found that its expression was increased by ~ twofold in each overexpression line compared to WT-treated leaves (Fig. 7b). Furthermore, it is believed that SA accumulation was induced by H2O2; therefore, we have observed the extent of production of H2O2 in overexpression lines and WT plants (Summermatter et al. 1995). The assay showed reduced accumulation of H2O2 (1.5–2.4 µ mol g−1 FW) in each overexpression lines. On the contrary, H2O2 content was higher (3.47 µ mol g−1 FW) in WT pathogenic-infiltrated leaves (Fig. 7c). Enhanced H2O2 content is an indication of ROS-mediated damages in leaf due to pathogen infiltration; therefore, lipid peroxidation assay and SOD activity were performed in infiltrated leaf tissue to evaluate oxidative damage. Lipid peroxidation assay has been measured by malondialdehyde (MDA) content. Analysis showed less accumulation of MDA (0.89–1.15 µ mol g−1 FW) in each overexpression line compared to WT-infiltrated leaves (1.79 µ mol g−1 FW) (Fig. 7d). Furthermore, increased SOD activities (3.7–5.6 units/mg protein) in each infiltrated transgenic leaves suggested maximum antioxidant activity in overexpression lines compared to WT-infiltrated leaves (Fig. 7e).

Quantitative analysis of salicylic acid (SA), qRT-PCR analysis of pathogenesis-related protein 1(PR1), hydrogen peroxide accumulation, and antioxidant enzymatic assay, (a) Quantitative HPLC analysis showed SA content enhanced from the range of 2.39 to 2.80 µg/g in overexpressing lines of A. thaliana (b) PR1 gene showed significant upregulation in each overexpressing lines up to twofold from WT-infiltrated leaves (c) Reduced H2O2 formation in all overexpressing lines (d) Lipid peroxidation assay showed low MDA accumulation in each overexpressing lines (e) Enhanced SOD activity in overexpression lines. The transcript level (relative expression) was normalized to the transcript abundance of Actin genes. Data are means ± SE of three biological and two technical replicates. Mean difference of different assays was analyzed by ANOVA using SPSS software (var.16.0) and mean separation was done using Duncan’s multiple range test (p < 0.05). Alphabets (a, b, c, d) reflected and treatment means with different letters in the same column are significantly different at p ≤ 0.05 (Duncan’s Multiple Range Test)
Physiological Changes During Pathogen Stress
Chlorophyll fluorescence is a non-invasive, non-destructive, and very sensitive probe of photosynthesis that has been used for studying the localized stress responses caused by bacteria, fungus, and other environmental stress (Iqbal et al. 2012). Our result showed spatial distribution followed specific patterns for pathogen infection. P. syringae (vir) infected leaves showed water-soaked patches in WT, which eventually started necrosis after 72 h. On the contrary, avirulent infiltrated leaves showed no necrosis patches, but fluorescence was altered in WT as compared to overexpression lines (Fig. 8). Chlorophyll fluorescence image analysis after virulent infection showed that Fv/Fm of each overexpression line was slightly affected, but NPQ was higher in each overexpression line (Supplementary Fig S3a, b). On the contrary, both Fv/Fm and NPQ were severely reduced in WT plants. Hence, net photosynthetic yield [Y (II)] was greater in overexpression lines than WT (Supplementary Fig S3b). Lowering of Fv/Fm and Y (II) in infected plant leaves suggested potential photo-damage in pathogen-inoculated leaf area. However, avirulent strain interaction in each line showed Fv/Fm was slightly more than WT, but NPQ was found steady in each overexpression line. Also, during avirulent interaction, NPQ was increased in WT; consequently, the photosynthetic yield was declined (YII) in WT compared to overexpression lines.

Chlorophyll fluorescence imaging after P. syringae infiltration. False-color image showing Fv/Fm—maximum quantum yield of PSII in Arabidopsis leaves after spraying the suspension of an avirulent and virulent strain of P. syringae. Damage in WT and WsSGT-overexpressing A. thaliana leaves were visualized by chlorophyll fluorescence imaging after 72 h
Analysis of Sterol and Sterol Glycosides
It was previously reported the major sterols found in plant systems are campesterol, stigmasterol, sitosterol, and their conjugated form that participates in the growth of plant as well as provide tolerance against abiotic and biotic stresses. To establish the role of WssgtL3.1 in enhanced immunity against P. syringae, we have explored the changes of free sterol and conjugated sterol (SG + ASG) levels of pathogen-treated leaves and untreated leaves of each genotype. Phytosterol contents of each plant sample are given in Table 1, and the analyzed data are presented as mean ± SE. As deduced from the data, the WssgtL3.1 overexpression led to increased total phytosterol content in each overexpressing line with a range of 8.91% to 6.47%. However, glycosylated sterol fractions in each line revealed that the conjugated sterol content was significantly more in transgenic lines compared to WT due to overexpression of WssgtL3.1 (Table 1). Overexpression of WssgtL3.1 in A. thaliana, exemplified by one transgenic line (T1), showed significant alteration in the sterol profile (Supplementary Fig. S4). Moreover, analysis of the total phytosterol content of pathogen-infiltrated leaves of all samples revealed that total sterol content of each overexpressing line was remarkably increased in comparison to the corresponding untreated leaves (1.40 to 2.53%) as well as to the WT leaves. Besides, analysis of different sterol fractions showed that conjugated sterol contents were decreased in each overexpression line as well as WT after pathogen infiltration from respective untreated plant samples. Nevertheless, total sterol content was obviously more in all overexpression lines after pathogen infiltration due to more biosynthesis of free sterols. This enhancement was showed mainly in the free form of sitosterol and stigmasterol (Table 1). Also, the increased amount of free plant sterols in WT was not significant; therefore, total sterol content was not altered substantially. Moreover, an enhancement of free sterol in each pathogen-infiltrated leaves samples was analyzed with the relative expression of the squalene synthase (sqs) gene. Real-time PCR assay confirmed sqs gene expression enhanced up to ~ fivefold in each infiltrated overexpression lines and proposed enhanced free sterol biosynthesis during pathogen stress (Fig. 9). Hence, the quantitative analysis suggested all overexpression lines contained greater content of the free form of sterol (sitosterol and stigmasterol) and their conjugated form (SG + ASG) after pathogen infiltration due to metabolic activity.

Squalene synthase (sqs) gene expression analysis by qRT-PCR. sqs gene showed 1.5 to twofold upregulation in overexpressing infiltrated leaves. The transcript level (relative expression) was normalized to the transcript abundance of Actin genes. Data are means ± SE of three biological and two technical replicates. Student t test was used to determine the significant level at p < 0.01 for * and p ≤ 0.001 for ***
Discussion
Glycosyltransferases enzyme catalyzes the modification of plant sterol and secondary metabolite via the transfer of single- or multiple-activated sugars, resulting in the glycosylation of plant molecules (Sharma et al. 2007). Free and glycosylated sterols are the primary components of the cellular membrane that are known to be involved in the regulation of biophysical properties of the cell membrane, maintenance of the cellular homeostasis process, and likely to be participating in the defense responses against environmental challenges (Chaturvedi et al. 2011; Ferrer et al. 2017; Castillo et al. 2019). SGTs of W. somnifera belong to the GT family-1 and are defined by the presence of a C-terminal consensus sequence termed as “signature motif” and represented as UGT prosite motif/ UDP-sugar-binding domain (Sharma et al. 2007; Chaturvedi et al. 2011). Both represent binding sites for sterol and UDP sugar, respectively, and reported as active sites of SGT for substrates. These domains are principally involved in the interaction of the SGT enzyme with the activated sugar donor (Sharma et al. 2007). Sequence homology confirmed that the WsSGTL3.1 amino acid sequence has conserved PSBD domain and UGT prosite motifs which are 50–80% similar to other plant SGTs. The presence of domain characteristic of SGTs suggests the involvement of WsSGTL3.1 protein in catalyzing the binding of sterol molecule with UDP sugar and plays an important role in the phytosterol modification (Chaturvedi et al. 2012; Sharma et al. 2007). SGTs of W. somnifera have been reported to follow a “compulsory-order sequential mechanism” forming a ternary complex (Madina et al. 2007b; Sharma et al. 2007). Consequently, both the activated sugar donor UDP glucose and the sterol substrates are required to bind the enzyme at the same time to catalyze the reaction (Madina et al. 2007b; Chaturvedi et al. 2011). Moreover, SGTs of W. somnifera exhibited wide-ranging sterol specificity by glycosylating a variety of sterols and secondary metabolites (with anolides, flavonoids, etc.) that led to change in their involvement in cellular metabolism (Madina et al. 2007a, 2007b; Sharma et al. 2007; Misra et al. 2008). A previous study documented that WssgtL3.1 gene from W. somnifera has differential expression pattern in the different plant tissues (Chaturvedi et al. 2011). Also, the WssgtL3.1 gene has shown strong relative expression in W. somnifera upon treatment of salicylic acid, methyl jasmonate, and heat and cold stress (Madina et al. 2007b; Chaturvedi et al. 2011). Several studies of W. somnifera sgts suggested WssgtL3.1 gene likely to participate in plant defense and provide tolerance against abiotic stress (Mishra et al. 2021a, b). Due to the significance of WssgtL3.1 in glycosylation of sterol, the WssgtL3.1 gene was cloned in topo vector with CaMV35S promoter and transformed in A. thaliana for functional characterization of heterologous overexpression lines of A. thaliana in the presence of pathogen. Three transgenic lines were selected for further study of functional characterization. All selected transgenic lines showed strong mRNA expression compared to WT. Furthermore, enzyme activity analysis revealed WssgtL3.1 overexpression by increasing enzyme activity in all transgenic lines. Despite this, no observable phenotypic changes were observed in each transgenic line.
Upon P. syringae inoculation (both avir and vir strains), progression of bacterial growth was significantly inhibited in the leaves of WssgtL3.1 overexpression lines at different time durations. Also, overexpression lines did not show susceptibility after 3dpi, whereas WT leaves were symptomatic and necrotic. Moreover, callose deposition has shown the severity of disease during the interactions of plants with the pathogen (Singh et al. 2016). As evident, each overexpressing plant leaves following pathogen infiltration had less callose deposition which confirmed less bacterial growth in transgenics compared to WT. In addition, SA, an important factor of the signal transduction pathway, leads to plant acquired resistance and also plays a role in resistance to all microbial pathogens (Tiwari et al. 2017; Delaney et al. 1995). Analysis of SA in all pathogen-infiltrated samples revealed that the SA content did not change under normal growth conditions, whereas free SA increased in each overexpressing line compared to WT plants until 48 h of pathogen infiltration. Several studies have reported the enhanced expression of the defense marker gene PR1 due to the increment in the accumulation of salicylic acid (Tiwari et al. 2016). Hence, qRT-PCR analysis was performed, and it was observed that PR1 gene showed higher expression in transgenic infiltrated leaves (Flors et al. 2008). It has been believed that H2O2 produced during oxidative damage (ROS) could trigger hypersensitive cell death (Summermatter et al. 1995). Similarly, MDA accumulation is an important biochemical marker for predicting the oxidative damage due to increased lipid peroxidation (Mishra et al. 2013). The suppression of disease severity in each transgenic line might be due to low production of H2O2 and MDA, suggesting a maximum antioxidant activity that exhibited enhance resistance against bacterial pathogens (Martin-Rivilla et al. 2019; Stahl et al. 2019). However, it was reported that a lower dose of H2O2 might serve to activate cellular protectant genes and participates in antioxidative mechanisms (Alexieva et al. 2001; Summermatter et al. 1995). High SOD activities in each transgenic line suggested maximum antioxidant activity in all overexpression lines, which could protect plants against pathogenic bacteria (Ausubel et al. 1993; Pandey et al. 2014).
Chlorophyll fluorescence of PSII (Fv/Fm) is a useful tool for examining the spatial distribution of photosynthetic activity in the presence of different environmental stress (Busch et al. 2009). Spatial and temporal changes in chlorophyll fluorescence analysis suggested free radical compounds that generate due to pathogen stress that may also cause damage to the photosynthetic apparatus (Iqbal et al. 2012). In all the conditions, the NPQ value was comparatively higher in transgenic lines than WT plants, which might also depend on the area of tissue damage. Tissues with mild damage might have a stimulated electron flow, which could increase the NPQ as a protection mechanism. Conversely, in severely damaged tissues, inhibition of photosynthetic electron transport may result in reduced NPQ and Y (II) (Berger et al. 2007). Enhanced NPQ and decreased Y (II) adjusted with excess excitation dissipation through the zeaxanthin feedback process (Johnson et al. 2009). A high NPQ could compensate for the decrease in Y (II) or even caused a lowering of Y (NO).
Sterol conjugates are synthesized in Arabidopsis by the enzyme UDP-glucose:sterol glycosyltransferases (SGTs), i.e., UGT80A2 and UGT80B1 (DeBolt et al. 2009). A functional characterization study of double knock-out mutants of sgt gene (UGT80A2 and B1) exhibited very small amounts of sterol glycosides and acylated sterol glycosides, which correlates with increased free sterol and sterol ester levels in inflorescence and siliques, suggesting the regulatory relationship between free sterols and conjugated sterol levels (DeBolt et al. 2009; Castillo et al. 2019). Accordingly, it may be concluded that heterologous overexpression of WssgtL3.1 in Arabidopsis enhanced the sterol conjugates (SG + ASG) contents in transgenic lines. HPLC analysis demonstrated that increased level of free sterol and sterol conjugates in each overexpression lines compared to WT. This result confirmed that the all transgenic lines have maximum glycosylation activity (Pandey et al. 2014). However, after pathogen infiltration, free sterol content was increased in each overexpression line but conjugated sterol content was decreased. This metabolic interconvertible of sterols and sterol conjugates is quite rapid, suggesting a regulatory function of cell membrane via different metabolic activities (Moreau et al. 2002; Ferrer et al. 2017; Ramirez-Estrada et al. 2017). In addition, higher expression of sqs in each pathogen-infiltrated leaves suggested its positive regulation with SA during pathogen infection (Wang et al. 2012; Singh, 2015). The sqs is the main enzyme that catalyzes the first enzymatic step in sterol biosynthesis and positively regulates pathogen stress (Singh et al. 2015). By agreement with this, our result showed the greater expression of sqs in all transgenic infiltrated leaves due to maximum SA accumulation. Apart from that, the WssgtL3.1 gene was also found to be positively regulated with SA hence enzyme activity in all transgenic lines could be increased, consequently, change the glycosylated sterol level in each overexpression line after pathogen infiltration (Madina et al. 2007a, 2007b; Chaturvedi et al. 2012). Consequently, an altered ratio was established between free sterol and glycosylated sterol in each pathogen-inoculated overexpression lines due to the synergistic effect of sqs and WssgtL3.1 gene (Chaturvedi et al. 2011; Kopischke et al. 2013; Singh et al. 2015). As a result, more contents of free sterol and conjugated sterol were accumulated in all overexpression lines. An induced level of free sterol and sterol conjugate might have altered the sterol homeostasis resulting in changed plasma membrane permeability and fluidity that led to affect the functions of membrane-bound proteins such as enzymes, channels, receptors, or other signaling components (Saema et al. 2016; Ferrer et al. 2017; Ramirez-Estrada et al. 2017). It has also recently shown a reduced sterol ester levels or changes in the glycosylated sterol levels in A. thaliana enhanced the defense response against Phytophthora infestans (Kopischke et al. 2013). Similarly, the silencing of the sterol glycosyltransferases gene altered the glycosylated sterol level in W. somnifera and increased susceptibility to Alternaria alternata (Singh et al. 2016).
In conclusion, our results demonstrated that the overexpression of the WssgtL3.1 gene in A. thaliana showed enhanced activity during the pathogen treatment and altered the proportion of FS and conjugated sterol (SG + ASG) content. These additive alterations repressed the bacterial growth, resulting in reduced H2O2 and that helps in triggering the antioxidant response. Consequently, infiltrated overexpression lines showed less lipid peroxidation, high NPQ, and lowering Y (NO) resulting in high photosynthetic yields Y (II) during stress conditions. Overall observation suggested that WssgtL3.1 in A. thaliana plays a crucial role to improve the immunity of the Arabidopsis plants. However, in-depth study of the involvement of free sterols and conjugated sterols in regulating membrane function during pathogen defense is yet to be understood. A more thorough study of the complete understanding of sterol modulation and their mechanism under different biotic stress and abiotic stress conditions requires further research.
Abbreviations
- CFI:
-
Chlorophyll fluorescence imaging
- CFU:
-
Colony-forming unit
- DPI:
-
Day post-inoculation
- GTs:
-
Glycosyltransferases
- MDA:
-
Malondialdehyde
- PSBD:
-
Putative sterol-binding domain
- PSPG:
-
Plant secondary product glycosyltransferase
- ROS:
-
Reactive oxygen species
- SA:
-
Salicylic acid
- SG:
-
Sterol glycoside
- SOD:
-
Superoxide dismutase
- SQS:
-
Squalene synthase
- NPQ:
-
Non-photochemical quenching
- Y(II):
-
Yield
- WT:
-
Wild type
References
Aboobucker SI, Suza WP (2019) Why do plants convert sitosterol to stigmasterol? Front Plant Sci 10:354
Adam L, Somerville SC (1996) Genetic characterization of five powdery mildew disease resistance loci in Arabidopsis thaliana. Plant J 9:341–356
Alexieva V, Sergiev I, Mapelli S, Karanov E (2001) The effect of drought and ultraviolet radiation on growth and stress markers in pea and wheat. Plant Cell Environ 24:1337–1344
Ausubel FM, Glazebrook J, Greenberg J, Mindrinos M, Yu G-L (1993) Analysis of the Arabidopsis defense response to Pseudomonas pathogens. Advances in Molecular Genetics of Plant-Microbe Interactions, vol 2. Springer, Dordrecht, pp 393–403
Beauchamp C, Fridovich I (1971) Superoxide dismutase: improved assays and an assay applicable to acrylamide gels. Anal Biochem 44(1):276–287
Bent AF, Innes R, Ecker J, Staskawicz BJ (1992) Disease development in ethylene-insensitive Arabidopsis thaliana infected with virulent and avirulent Pseudomonas and Xanthomonas pathogens. Mol Plant Microbe Interact 5:372–372
Berger S, Benediktyova Z, Matous K, Bonfig K, Mueller MJ, Nedbal L, Roitsch T (2007) Visualization of dynamics of plant-pathogen interaction by novel combination of chlorophyll fluorescence imaging and statistical analysis: differential effects of virulent and avirulent strains of P. syringae and of oxylipins on A. thaliana. J Exp Bot 58:797–806. https://doi.org/10.1093/jxb/erl208
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. https://doi.org/10.1016/0003-2697(76)90527-3
Busch F, Huner NPA, Ensminger I (2009) Biochemical constrains limit the potential of the photochemical reflectance index as a predictor of effective quantum efficiency of photosynthesis during the winter spring transition in Jack pine seedlings. Funct Plant Biol 36:1016–1026. https://doi.org/10.1071/FP08043
Castillo N et al (2019) Inactivation of UDP-glucose sterol glucosyltransferases enhances Arabidopsis resistance to Botrytis cinerea. Front Plant Sci 10:1162
Chaturvedi P, Misra P, Tuli R (2011) Sterol glycosyltransferases–the enzymes that modify sterols. Appl Biochem Biotechnol 165:47–68. https://doi.org/10.1007/s12010-011-9232-0
Chaturvedi P, Mishra M, Akhtar N, Gupta P, Mishra P, Tuli R (2012) Sterol glycosyltransferases-identification of members of gene family and their role in stress in Withania somnifera. Mol Biol Rep 39:9755–9764. https://doi.org/10.1007/s11033-012-1841-3
Clough SJ, Bent AF (1998) Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16:735–743
DeBolt S et al (2009) Mutations in UDP-Glucose:sterol glucosyltransferase in Arabidopsis cause transparent testa phenotype and suberization defect in seeds. Plant Physiol 151:78–87. https://doi.org/10.1104/pp.109.140582
Delaney TP, Friedrich L, Ryals JA (1995) Arabidopsis signal transduction mutant defective in chemically and biologically induced disease resistance. PNAS USA 92:6602–6606
Ferrer A, Altabella T, Arró M, Boronat A (2017) Emerging roles for conjugated sterols in plants. Prog Lipid Res 67:27–37
Flors V, Ton J, Van Doorn R, Jakab G, García-Agustín P, Mauch-Mani B (2008) Interplay between JA, SA and ABA signalling during basal and induced resistance against Pseudomonas syringae and Alternaria brassicicola. Plant J 54:81–92
Griebel T, Zeier J (2010) A role for beta-sitosterol to stigmasterol conversion in plant-pathogen interactions. Plant J 63:254–268. https://doi.org/10.1111/j.1365-313X.2010.04235.x
Grille S, Zaslawski A, Thiele S, Plat J, Warnecke D (2010) The functions of steryl glycosides come to those who wait: recent advances in plants, fungi, bacteria and animals. Prog Lipid Res 49:262–288
Hartmann M-A (1998) Plant sterols and the membrane environment. Trends Plant Sci 3:170–175
Hayford MB, Medford JI, Hoffman NL, Rogers SG, Klee HJ (1988) Development of a plant transformation selection system based on expression of genes encoding gentamicin acetyltransferases. Plant Physiol 86:1216–1222
Iqbal M, Goodwin P, Leonardos E, Grodzinski B (2012) Spatial and temporal changes in chlorophyll fluorescence images of Nicotiana benthamiana leaves following inoculation with Pseudomonas syringae pv Tabaci. Plant Pathol 61:1052–1062
Johnson MP, Pérez-Bueno ML, Zia A, Horton P, Ruban AV (2009) The zeaxanthin-independent and zeaxanthin-dependent qE components of nonphotochemical quenching involve common conformational changes within the photosystem II antenna in Arabidopsis. Plant Physiol 149:1061–1075
Katagiri F, Thilmony R, He SY (2002) The Arabidopsis thaliana-pseudomonas syringae interaction. Arabidopsis Book 1:e0039. https://doi.org/10.1199/tab.0039
Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10:845–858
Keukens EA et al (1995) Molecular basis of glycoalkaloid induced membrane disruption. BBA-Biomembranes 1240:216–228
Kopischke M et al (2013) Impaired sterol ester synthesis alters the response of Arabidopsis thaliana to Phytophthora infestans. Plant J 73:456–468
Li X et al (2014) Distinct biochemical activities and heat shock responses of two UDP-glucose sterol glucosyltransferases in cotton. Plant Sci 219:1–8
Lim EK, Higgins GS, Li Y, Bowles DJ (2003) Regioselectivity of glucosylation of caffeic acid by a UDP-glucose:glucosyltransferase is maintained in planta. Biochem J 373:987–992. https://doi.org/10.1042/BJ20021453BJ20021453[pii]
Madina BR, Sharma LK, Chaturvedi P, Sangwan RS, Tuli R (2007a) Purification and characterization of a novel glucosyltransferase specific to 27beta-hydroxy steroidal lactones from Withania somnifera and its role in stress responses. BBA 1774:1199–1207
Madina BR, Sharma LK, Chaturvedi P, Sangwan RS, Tuli R (2007b) Purification and physico-kinetic characterization of 3beta-hydroxy specific sterol glucosyltransferase from Withania somnifera (L) and its stress response. BBA 1774:392–402
Marek G, Carver R, Ding Y, Sathyanarayan D, Zhang X, Mou Z (2010) A high-throughput method for isolation of salicylic acid metabolic mutants. Plant Methods 6:21. https://doi.org/10.1186/1746-4811-6-21
Martin-Rivilla H, Garcia-Villaraco A, Ramos-Solano B, Gutierrez-Mañero F, Lucas J (2019) Extracts from cultures of Pseudomonas fluorescens induce defensive patterns of gene expression and enzyme activity while depressing visible injury and reactive oxygen species in Arabidopsis thaliana challenged with pathogenic Pseudomonas syringae. AoB Plants 11:plz049
Maxwell K, Johnson GN (2000) Chlorophyll fluorescence–a practical guide. J Exp Bot 51:659–668
Mishra MK et al (2013) Overexpression of WsSGTL1 gene of Withania somnifera enhances salt tolerance, heat tolerance and cold acclimation ability in transgenic Arabidopsis plants. PLoS ONE 8:e63064. https://doi.org/10.1371/journal.pone.0063064
Mishra MK, Singh G, Tiwari S, Singh R, Kumari N, Misra P (2015) Characterization of Arabidopsis sterol glycosyltransferase TTG15/UGT80B1 role during freeze and heat stress. Plant Signal Behav 10:e1075682. https://doi.org/10.1080/15592324.2015.1075682
Mishra MK, Srivastava M, Singh G, Tiwari S, Niranjan A, Kumari N, Misra P (2017) Overexpression of Withania somnifera SGTL1 gene resists the interaction of fungus Alternaria brassicicola in Arabidopsis thaliana. Physiol Mol Plant Pathol 97:11–19
Mishra MK, Pandey S, Misra P, Niranjan A, Srivastava A (2020) An efficient protocol for clonal regeneration and excised root culture with enhanced alkaloid content in Thalictrum foliolosum DC—an endemic and important medicinal plant of temperate Himalayan region. Ind Crops Prod 152:112504
Mishra MK, Tiwari S, Misra P (2021a) Overexpression of WssgtL3.1 gene from Withania somnifera confers salt stress tolerance in Arabidopsis. Plant Cell Rep. https://doi.org/10.1007/s00299-021-02666-9
Mishra MK, Pandey S, Niranjan A, Misra P (2021b) Comparative analysis of phenolic compounds from wild and in vitro propagated plant Thalictrum foliolosum and antioxidant activity of various crude extracts. Chem Pap. https://doi.org/10.1007/s11696-021-01708-6
Misra L, Mishra P, Pandey A, Sangwan RS, Sangwan NS, Tuli R (2008) Withanolides from Withania somnifera roots. Phytochemistry 69:1000–1004. https://doi.org/10.1016/j.phytochem.2007.10.024
Moreau RA, Whitaker BD, Hicks KB (2002) Phytosterols, phytostanols, and their conjugates in foods: structural diversity, quantitative analysis, and health-promoting uses. Prog Lipid Res 41:457–500
Pandey V, Niranjan A, Atri N, Chandrashekhar K, Mishra MK, Trivedi PK, Misra P (2014) WsSGTL1 gene from Withania somnifera, modulates glycosylation profile, antioxidant system and confers biotic and salt stress tolerance in transgenic tobacco. Planta 239:1217–1231. https://doi.org/10.1007/s00425-014-2046-x
Ramirez-Estrada K, Castillo N, Lara JA, Arró M, Boronat A, Ferrer A, Altabella T (2017) Tomato UDP-glucose sterol glycosyltransferases: a family of developmental and stress regulated genes that encode cytosolic and membrane-associated forms of the enzyme. Front Plant Sci 8:984
Saema S, Rahman LU, Singh R, Niranjan A, Ahmad IZ, Misra P (2016) Ectopic overexpression of WsSGTL1, a sterol glucosyltransferase gene in Withania somnifera, promotes growth, enhances glycowithanolide and provides tolerance to abiotic and biotic stresses. Plant Cell Rep 35:195–211. https://doi.org/10.1007/s00299-015-1879-5
Schaller H (2003) The role of sterols in plant growth and development. Prog Lipid Res 42:163–175
Schreiber U (2003) Pulse amplitude (PAM) fluorometry and saturation pulse method. In: Papageorgiou G, Govindjee (eds) Chlorophyll a fluorescence a signature of photosynthesis. Advances in photosynthesis and respiration series. Kluwer Academic Publishers, Dordrecht, pp 1–41
Sharma LK, Madina BR, Chaturvedi P, Sangwan RS, Tuli R (2007) Molecular cloning and characterization of one member of 3beta-hydroxy sterol glucosyltransferase gene family in Withania somnifera. Arch Biochem Biophys 460:48–55. https://doi.org/10.1016/j.abb.2007.01.024
Singh AK et al (2015) Virus-Induced gene silencing of Withania somnifera squalene synthase negatively regulates sterol and defence-related genes resulting in reduced withanolides and biotic stress tolerance. Plant Biotechnol J 13:1287–1299
Singh G, Tiwari M, Singh SP, Singh S, Trivedi PK, Misra P (2016) Silencing of sterol glycosyltransferases modulates the withanolide biosynthesis and leads to compromised basal immunity of Withania somnifera. Sci Rep 6:25562. https://doi.org/10.1038/srep25562
Stahl E, Hartmann M, Scholten N, Zeier J (2019) A role for tocopherol biosynthesis in Arabidopsis basal immunity to bacterial infection. Plant Physiol 181:1008–1028
Summermatter K, Sticher L, Métraux J-P (1995) Systemic responses in Arabidopsis thaliana infected and challenged with Pseudomonas syringae pv syringae. Plant Physiol 108:1379–1385
Tiwari S, Lata C, Chauhan PS, Nautiyal CS (2016) Pseudomonas putida attunes morphophysiological, biochemical and molecular responses in Cicer arietinum L. during drought stress and recovery. Plant Physiol Biochem 99:108–117. https://doi.org/10.1016/j.plaphy.2015.11.001
Tiwari S, Lata C, Chauhan PS, Prasad V, Prasad M (2017) A functional genomic perspective on drought signalling and its crosstalk with phytohormone-mediated signalling pathways in plants. Curr Genom 18(6):469–482
Wang K, Senthil-Kumar M, Ryu C-M, Kang L, Mysore KS (2012) Phytosterols play a key role in plant innate immunity against bacterial pathogens by regulating nutrient efflux into the apoplast. Plant Physiol 158:1789–1802
Warnecke D et al (1999) Cloning and functional expression of UGT genes encoding sterol glucosyltransferases from Saccharomyces cerevisiae, Candida albicans, Pichia pastoris, and Dictyostelium discoideum. J Biol Chem 274:13048–13059
Acknowledgements
The authors acknowledge the support of the Department of Biotechnology, Govt. of India, for providing financial support to carry out the research work. ST acknowledges “National Post-Doctoral Fellowship (NPDF)” by the Science & Engineering Research Board (SERB), Govt. of India (File No.: PDF/2020/001377). Also, Director, CSIR National Botanical Research Institute, is gratefully acknowledged by the authors for providing the facilities.
Author information
Authors and Affiliations
Contributions
MKM and PM designed the research. MKM, ST, and MS performed the research work. AA performed HPLC, analyzed data, and revised the manuscript. MKM, ST, and PM drafted the research and revised the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Additional information
Handling Editor: Paloma Sanchez.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mishra, M.K., Tiwari, S., Srivastava, M. et al. Ectopic Expression of WsSGTL3.1 Gene in Arabidopsis thaliana Confers Enhanced Resistance to Pseudomonas syringae. J Plant Growth Regul 41, 1871–1886 (2022). https://doi.org/10.1007/s00344-021-10427-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00344-021-10427-x