
Abstract
Nickel oxide and silver oxide nanoparticles have wonderful properties that could be employed in numerous applications. Thus, synthesis of nickel silver oxide nanostructures with different characteristics is of great interest. In this review, many synthesis methods were reported such as: electrodeposition, electrochemical method, simple immersion process and subsequent RF-sputtering deposition, chemical oxidative polymerization, followed by acidic sol–gel process, flame-based process, liquid-phase reduction technique, sol–gel, hydrothermal method, co-precipitation method, simple precipitation method, thermal decomposition, chemical wet synthesis, low and high-temperature reduction, high-pressure autoclave, thermal treatment method, and laser-liquid–solid interaction technique. Reporting all methods employed for the fabrication of NiO and Ag2O nanostructures is useful to produce and develop novel nanomaterials with enhanced properties and applications. Studying the factors that tuned their properties: particle size, shape, and capping agents as well as solution pH is highly recommended in future works. Also, further research studies should be conducted for finding another/other facile and effective synthesis method/methods.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Nanoscience and nanotechnology are research scientific topics employed to examine materials at nanoscale in various sciences. In fact, nanoscience is the analysis of structures and molecules, whereas nanotechnology is the research of practical applications [1]. Richard P. Feynman is a physicist, who was known as the father of nanotechnology, has been made a revolution in nanotechnology during his famous lecture in 1959 “There’s Plenty of Room at the Bottom” [2,3,4]. Materials with nanoscale range can be found in range between 1 and 100 nm [4, 5]. Nanoparticles (NPs) can be formed in many dimensions based on their shape such as: zero-dimensional (0D), one-dimensional (1D), two-dimensional (2D) or three-dimensional (3D) nanostructures [4,5,6]. Based on physical and chemical characteristics, NPs are classified into the following types: carbon-based, metal, ceramics, semiconductor, polymeric, and lipid-based NPs [5].
Metal oxide nanoparticles earn consideration because of their various assets and broad applications [7,8,9,10,11,12,13]. Gold and silver are noble metals that were employed in the manufacture of metal and metal oxide nanoparticles [14]. Many researchers synthesized silver NPs and determined its characterizations [15,16,17,18,19,20,21]. Because of the distinctive assets of silver nanoparticles, it was broadly utilized in different applications [22, 23]. Conversely, other researchers synthesized nickel NPs and determined their characterizations [14, 24,25,26,27,28]. Recently, nickel and copper are widely used because of their economic value compared with gold and silver. Also, these NPs have antibacterial activities that can be utilized in many applications [14]. Besides, nickel NPs have good electrocatalytic properties which were investigated by Chemchoub et al. [29] for the first time.
Metal–metal oxide composite materials also have been synthesized because of their powerful properties and wide applications [30]. Nickel oxide (NiO) and silver oxide (Ag2O) NPs are widely studied in the current research studies. Many researchers synthesized NiO and Ag2O NPs through different synthesis routes. Nickel oxide NPs were synthesized by sol–gel [31,32,33,34,35,36], hydrothermal method [37,38,39,40], thermal decomposition [41, 42], co-precipitation method [43, 44], microwave combustion method [45], aqueous chemical growth procedure [46], chemical reduction method [47], wet chemical technique [48], electrochemical technique [49], calcination process [50], thermal oxidation of nickel sulfide NPs [51], plasma chemical technique in a plasma of a low-pressure arc release [52], combination of precipitation and reduction/calcination techniques [53], and after all by green production techniques using Agathosma betulina plant extract [54], medicinal plant Prunus persica [55], Calotropis gigantea leaves extract [25], Ananas comosus leaf extract [56], Avicennia Marina leaf extract [57], Solanum trilobatum leaf extract [58], Acalypha Indica leaf extract [59], silk of Zea mays lea plant extract [60], Cydonia oblonga extract [61], E. heterophylla (L.) leaves extract [62], and Gymnema sylvestre extracts [63]. Conversely, silver oxide NPs were synthesized by different methods: thermal decomposition [23], co-precipitation [64], simple chemical method [65], simple solution method [66], capping method [67], combustion method by Gomutra (cow urine) [68], and finally, green synthesis using different extracts: C. edulis extract [69], Daphne alpina [70], Lippia citriodora plant powder [71], Zephyranthes rosea flower extract [72], Ficus benghalensis prop root extract [73], Centella asiatica and tridax plant powder [74], Lactobacillus mindensis [75], Artocarpus Heterophyllus plant extract [76], and medicinal plant Cyathea nilgirensis Holttum [77].
Metal oxide nanostructured materials have developed significant research interest for the last few years to uses in several application. Nickel oxide NPs are known as a magnetic material that have certain magnetic properties. Novel and interesting magnetic properties that range from nanosized to bulk-like behavior can be achieved by varying particle size, crystal structure, and morphology [31]. Different particle sizes (average or range) of NiO NPs have been reported in numerous previous published studies: 1.42 ± 1.76 nm [56], 3 nm [47], 5 nm [31, 34], 7 nm [49], 12 nm [52], 14 and 18 nm [63], 22.7 nm [46], 25 nm [41], 28 nm [36, 55], 30 nm [42], 74.5 nm [61], 10–20 nm [60], 12–15 nm [62], 13–18 nm [53], 8–20 nm [44], 19–30 nm [45], 15–35 nm [39], 20–30 nm [51], 25–30 nm [58], 20–40 nm [25], 50 nm [32], ~ 10–100 nm [33], 30–100 nm [57], and several hundred nanometers [37]. Similarly, properties of Ag2O NPs can be controlled by varying particle size. The average particle sizes (or ranges) of Ag2O NPs were also registered in many published studies: 1.5–2 nm [69], 2–10 nm [67], 2–20 nm [75], 11–12 nm [74], 14 nm [76], 20 nm [68, 71], 10–30 nm [72], 27 nm [70], 51.4 nm [73], and 62.0 ± 5.0 nm [66], respectively. Different studies reported that the shape of NiO NPs was spherical or nearly spherical [31, 32, 38,39,40,41,42, 44, 45, 49, 52, 55, 60, 63]. Also, other shapes were reported in the literature such as: nanorods [34, 37, 47, 58], nanosheets [48], nanoplates [78], linear flakes shape [36], and thin films [33]. Additionally, spherical shape was reported for Ag2O NPs [64, 66,67,68, 71,72,73,74,75,76,77]. Other studies found that the shape of Ag2O NPs was cube, and ellipsoid [69].
Recently, the use of NiO and Ag2O NPs in various applications has been gradually increased. These NPs are classified as p-type semiconductors materials, which have wide band gaps and good optical properties. Common applications have been reported in the literature for these NPs: used as sensors, catalysts, optoelectronics, photocatalytic, antibacterial agent, solar cells devices, energy storage devices, and for water treatment applications. Although extensive research studies have been found in the literature investigating nickel and silver or their oxides nanostructures, there is no review paper found for reviewing the synthesis methods, characterizations and applications for these NPs.
In summary, brief descriptions for synthesis methods, properties, and applications of nickel oxide, silver oxide, and nickel silver oxide nanomaterials (NMs) will be introduced. Different methods were widely used to prepare these NMs with tunable characterizations. In this review, comprehensive descriptions of all methods are introduced and discussed in detail to help researchers find the required information easily for enhancing or improving methods, characterizations and applications.
2 Syntheses methods
Several techniques can be followed for manufacturing nickel silver oxide NPs. These techniques are classified into two main classes: top-down and bottom-up syntheses. Top-down approaches are known as destructive approaches, which convert larger molecule into smaller NPs such as: electrodeposition, electrochemical method, and laser-liquid–solid interaction technique [79,80,81]. Conversely, bottom-up approaches are fabricated from relatively simpler substances, which are known as building up approaches such as simple bathing procedure and subsequent RF-sputtering deposition, chemical oxidative polymerization tracked by acidic sol–gel process, flame-based process, liquid-phase reduction technique, sol–gel, hydrothermal method, co-precipitation method, simple precipitation method, thermal decomposition, chemical wet synthesis, low-temperature reduction, high-temperature reduction, high-pressure autoclave, thermal treatment method [5, 82]. In this review, syntheses methods of nickel silver oxide NPs will be introduced:
2.1 Electrodeposition
Electrochemical deposition was widely employed for the production of metal NPs such as: silver [83] and nickel [84]. It could be occurred at the interface of an electrolyte solution which consists of metal substrate (electrically conducive) and another metal (to be deposited). Regarding counter and working electrodes, the glassy carbon rod and metallic silver were used, respectively. Silver NPs were fabricated by an electrolyte that composed of AgNO3 (0.01 mM). This electrolyte was formed using deionized water (DW), at where these two electrodes were immersed parallel to each other at a distance of 2 cm. A certain voltage was applied through the electrodes at ambient temperature to initiate Ag ions reduction. But it was noticed that as the deposition process proceeded, the pH of the electrolyte was decreased. Also, Ag ions was found to be aggregated and consequently a dendritic shape was formed [83].
Wang and Zheng also prepared Ag NPs [85]. They used this synthesis route for preparing the hydrogen peroxide (HP) sensor. Electrodeposition of Ag NPs was done on a modified glassy carbon electrode with ZnO film. A sequential polish with different thicknesses of alumina powder was created on the glassy carbon electrode, followed by the sonication for 5 min in ethanol and then in water. ZnO films were electrodeposited on carbon electrode using zinc nitrate electrolyte by applying a voltage of −0.7 V for 20 min at 62 °C. Then, it was washed with water and was dried. After that, Ag was electrodeposited on ZnO/glassy carbon electrode using AgNO3 to produce Ag NPs/ZnO/glassy carbon electrode, which was rinsed with water and then was dried before use [85]. In another work, Ag NPs-decorated degraded graphene oxide (dGO) has been electrodeposited on indium tin oxide (ITO) using a cyclic voltammetry process. GO was fabricated using a streamlined Hummers’ route [86]. The electrodeposition of Ag NPs-dGO was carried out in a three-electrode electrochemical cell. Ammonia was added to AgNO3 solution to produce silver ammonia solution which was then mixed with GO solution to produce another homogeneous solution. Three-electrodes: ITO, platinum foil and drenched calomel have been utilized as working, counter and reference electrodes, respectively, for performing cyclic voltammetry. This scan was operated at a rate of 25 mV s−1 from −1.5 to 0 V. Finally, working electrode was rinsed with double distilled water after deposition [87]. Additionally, ternary nanocomposite (NC) (Ag NPs-CDs-dGO) consisting of Ag NPs, carbon dots (CDs) and dGO were fabricated through one-step electrodeposition method [88]. Like Moradi lsheikh et al. study, silver-ammonia solution was prepared from mixing of ammonia with AgNO3 solutions until no precipitates were seen. Also, aqueous solutions of CDs and GO were prepared and then were kept in different containers. A polished glass carbon electrode with mirror-like surface was submerged in deoxygenated phosphate buffered saline solution. A mixture of CDs-GO solution was added, and then, silver ammonia solution was also added. A scan was performed at a rate of 25 mV s−1 in a potential ranged from 0 to 1.0 V. Finally, the NCs of Ag NPs-CDs-GO were constructed and electrodeposited on the glass carbon electrode interface, which confirmed the production of Ag NPs-CDs-dGO/GCE [88].
Conversely, NiO NPs were prepared using this method [84]. The rose-type structure NiO NPs were prepared on ITO glass substrates through electrodeposition in nickel sulfate hexahydrate solution. Both ITO and nickel sulfate hexahydrate were employed as substrates, which were immersed in the solution for electrodeposition. The electrolyte was prepared from dissolving nickel sulfate hexahydrate into DW using a magnetic stirrer. Before deposition, ITO substrates were washed and were cleaned in different solvents. Nickel NPs were deposited on ITO substrates by using a current of −0.5 mA for 200 s. NiO nanorods were produced in a uniform size and morphology which could be used in sensing applications [84].
Nickel silver oxide nanostructures were synthesized in different studies. Usman et al. prepared carbon nanocoils-nickel foam decorated with Ag NPs/sheets by stirring assisted electrodeposition route for non-enzymatic glucose sensor. Fe/Sn (the mole ratio was ~10:1) solution was chosen as a precursor catalyst for the growth of carbon nanocoils (CNCs), synthesized by sol–gel method. Next, Ni foam (NF) was coated with the aforementioned catalyst through a dipping method. CNCs were grown on catalyst-coated NF by chemical vapors deposition. Before electrodeposition, the electrode CNC@NF hydrophilic was produced using 0.03 M cetyltrimethylammonium bromide (CTAB) solution, which was stirred for 12 h. Then, an ordinary two-electrode system was applied since CNC@NF substrate and NF were employed as cathode and anode, respectively. The precursor solution was made using 1 mM AgNO3. Then, the prepared cell was put on a stirrer, a certain current (0.01 mA) was applied for 1 h, and stirred continuously. After that, the electrode (Ag@CNC@NF) was rinsed with DW and then was dried at 60 °C for 1 d in an oven [89]. It was found that the 3D CNC-based unique structure has been used as a glucose sensor because of its good performance in detecting glucose. The Ag@CNC@NF's, which has a unique 3D helical shape, showed an enormous surface area and a short diffusion of the electrolyte ions. That new design of the electrode demonstrated wonderful features which was useful in sensitive glucose sensing applications [89].
In another study, a nanostructured Ag-catalyzed Ni foam cathode for an aluminum–hydrogen peroxide fuel cell was synthesized by electrodeposition [90]. Ni foam was used as a metal substrate, which has a 3D grid structure, high porosity and specific surface area. The metal surface was cleaned and etched using acetone and hydrochloric acid. Acetone was also used to remove grease from Ni foam substrates surfaces. After that, they were immersed in 3 M HCl at room temperature for 20 min to eliminate the oxide layer and etch the foam surface. Finally, Ni foam substrates were washed with DW to eliminate any unwanted substances. Silver catalyst was prepared using direct current electrodeposition onto the foamed Ni substrate, by adjusting the composition of the electrolyte system and deposition parameters. The solution which was utilized in the deposition included KAg(CN)2 (50 g l−1). Electrodeposition was employed via a current density of 5 mA cm−2 at an ambient temperature. Time was used to adjust the thickness of coated material which was chosen as 1 μm in Yang et al. [90] study. After deposition, a gray metallic Ag film was created on the fiber wall of porous Ni foam substrate. It was found that nanostructured Ag-catalyzed Ni foam cathode revealed high catalytic activity and produce stable electrode performance. That electrode could be used in Al–H2O2 fuel cell since it provided a high surface area in the catalyst layer and a lower mass transport resistance [90].
In another study, Ag nanowires (NWs) were synthesized, purified by dispersing them in acetone (in a ratio of 1:5), and then centrifuged (8000 rpm for 20 min). Centrifugation process was repeated using ethanol. The produced solid content was taken to fresh ethanol for spray-coating. Also, Ag NWs were deposited onto polyethylene terephthalate (PET) and glass substrates by spray-coating process. The solvent was removed directly through deposition process by heating substrates to Ni(OH)2 shell, which was potentiostatically deposited onto the Ag NW network electrodes. The deposition of Ni(OH)2 was confirmed by using the previously prepared nickel acetate (Ni(CH3COO)2.4H2O) solution (0.1 M). The working electrode, counter and reference electrodes were Ag NW network film (on glass or PET substrate), platinum foil, and Ag/AgCl (saturated KCl), respectively. A constant potential (−0.9 V) was applied for different time intervals (200, 400, 600 and 800 s). Synthesized electrodes were rinsed with DW, dried with N2 gas and then kept at 100 °C on a hot plate [91]. In their study, the synergy of highly conductive Ag NWs and high capacitive Ni(OH)2 encouraged the transportation of both ion and electron. As a result, the electrochemical properties were enhanced under a specific capacitance of 1165.2 F g−1 at a current density of 3 A g−1. It was also reported that this coaxial network structure was simply inserted to other conducting polymer or metal oxides. So, this geometry has an advantage about the possibility of using this material in other applications [91].
2.2 Electrochemical method
A simple electrochemical technique was employed for preparing a highly ordered NiOx NPs on Ni foam by the subsequent heat treatment of Ni(OH)2. The Ni foam was initially treated using 1 M aqueous HCl for 30 min and then dried in air. Aqueous solutions of Ni(NO3)2.6H2O (0.05 M) and KNO3 (0.50 M) were used as electrolytes in the electrolysis. In this method, the pretreated Ni foam, Pt wire, and Ag/AgCl were utilized as working, counter, and reference electrodes, respectively. A fixed potential (−0.90 V) was applied to fabricate NiOx NPs versus Ag/AgCl to working Ni foam electrode for 10 min. Then, Ni foam was put into a furnace at 300 °C for 4 h. Many advantages were reported for this approach: low cost, free binder, and relatively stable NPs [49].
In another study, two electrodes were employed in the galvanostatic regime to prepare NiO nanoplates [78]. A solution of Ni(NO3)2.6H2O (0.005 M) was prepared for electrodeposition. A cathodic stainless-steel substrate was inserted between two parallel graphite anodes to construct the electrochemical cell. Deposition processes were operated for 1 h at a constant current density (0.1 mA cm−2) in a bath with a temperature of 60 °C. Next, the electrode was removed from the electrolyte, rinsed with DW for many times, and then dried for 48 h. NiO was obtained after scraping the hydroxide deposit from the steel electrode and heat treatment for 3 h at 400 °C [78].
2.3 Facile immersion method followed by RF-sputtering deposition
Ag-NiO core–shell nanoflower arrays were fabricated on Cu substrate using a simple immersion method, followed by RF-sputtering technique [92], and Ag-NiO core–shell NP arrays were prepared using the same procedures [93]. Copper foil with a thickness of 0.1 mm was washed repeatedly with water and ethanol. The cleaned Cu foil was vertically submerged into a beaker containing AgNO3 aqueous solution (5 mM, 100 mL) for 30 s, and at 30 °C. When immersing was completed, the Cu foil was directly removed from the solution, washed with DW and ethanol, and then was dried by Ar gas. The RF-sputtering was adopted for depositing NiO layers onto the as-prepared Ag NP arrays using NiO target. The deposition was operated at certain conditions (25 °C, 1.0 Pa, and 100 W). The sputtering technique was continued for 40 min [92, 93]. In fact, the preparation of Ag NP arrays on Cu substrate using solution immersion method has many benefits such as: facile method, timesaving, and it occurred under ordinary conditions without adding extra substances. Ag-NiO core–shell NP arrays would be produced after the deposition of the NiO layer. Once applied in lithium-ion batteries (LIBs), the conductivity and adhesion of the NiO layer to the current collector could be enhanced using Ag NPs. The in situ prepared Ag NPs were improved the interfacial strength between the active substance and substrate, and also improved the electrical conductivity of an electrode, which enhanced the electrochemical performance [93]. Additionally, the highly conductive Ag core provided a support to NiO layer, and therefore, the conductivity was enhanced during charging and discharging. When they were used as anode materials for LIBs, the hybrid nanostructured electrodes delivered a long cycle life and the power performance was improved in comparison with a planar electrode [92].
2.4 Chemical oxidative polymerization and subsequent acidic sol–gel technique
Successive steps are needed for preparing complex NCs by this method. First, cellulose nano-whisker (CNW) were fabricated using a bleached bagasse pulp. A solution of cellulose and sulfuric acid, in a liquor ratio of cellulose to diluted H2SO4 solution (1:10 g mL−1), was heated at 45 °C under continuous stirring for 30 min. Cold water was added to the prepared solution, which was centrifuged at 10,000 rpm for 10 min. To remove the excess amounts of H2SO4, distilled water was used and then a sonicator was operated for 10 min at 1000 W. The suspended CNW was kept in refrigerator at 4 °C. Then, polypyrrole/silver NPs (PPy/AgNPs) NCs were fabricated by oxidizing of pyrrole using AgNO3. Sodium dodecyl sulfate (SDS) was dissolved completely in distilled water for 30 min under continuous stirring. Pyrrole and AgNO3 solution were also added progressively. A polymerization reaction occurred on a stirrer overnight at ambient temperature. The resulted black precipitate (PPy/AgNPs) was filtered off, was rinsed with water, and then was dried in an oven. Additionally, CNW/PPy/AgNPs NCs were fabricated by adding AgNO3 solution to the suspended CNW under continuous stirring. Then, pyrrole/SDS solution was added drop by drop to the suspended CNW, stirred overnight to complete the reaction. A precipitate of CNW/PPy/AgNPs was formed, filtered off, was washed using distilled water, and then was dried at 40 °C. As a result, sol–gel Ni2O3 NPs were formed employing citric acid as a coordinating agent, distilled water or ethylene glycol as a solvent, and nickel nitrate (Ni(NO3)2.6H2O) as a solute. The solution of Ni2O3 NPs was aged for 3 d, and then, it was mixed with CNW/AgNPs and CNW/PPy/AgNPs composites to form CNW/PPy/AgNPs-Ni2O3NPs NCs. Finally, the fabricated NCs were dried at 50 °C to form zero gel NCs [94]. In this method, a combination of CNW (hosting polymer), PPy (conductive polymer), and Ni2O3 NPs participated in the fabrication of polymeric NCs with structural, morphological and electroconductive characteristics [94].
2.5 Flame-based method
Mohammadi et al. demonstrated a synthesis method of bimetallic Ni-Ag nanopowders (NPs) by a flame-driven high-temperature reducing jet (HTRJ) method. This procedure enables the quick creation of aerosol (gas stage) of multicomponent metal NPs from low-cost metal precursors in relatively high production rates. The combustion products of fuel flame (H2, O2, and N2) were flowed through a diverging nozzle. A solution of metal precursors (Ni, Ag, and/or Cu) was inserted to the throat section of the nozzle. A hot high-velocity gas stream was used for atomization, producing droplets which were evaporated in H2 atmosphere at high temperature (~550 °C). Next, the products were cooled through the application of a nitrogen quench flow, and then, they were gathered on a filter membrane. Since the common solvents could not disperse the produced NPs, capping for these NPs was found to be required for creating nano-inks in one step. For in situ surface functionalization of NPs, octylamine was used as a ligand for that purpose. A redesigned constant output atomizer was injecting micron-sized octylamine aerosol droplets in the quench zone of the HTRJ process. The temperature of the quench zone was kept in the range of 150–200 °C to vaporize all octylamine droplets. Then, the functionalized nanopowders (FNPs) were gathered on a filter membrane. Lastly, nano-inks were created via the dispersion of FNPs in toluene and a subsequent sonication [95]. Since the flame-based processes could be used for printing structures using electrical conductivity at ambient temperature, it also might be accomplished for conductive nano-ink fabrication. This method was reported for the fabrication of different NMs such as: titania, fumed silica, and carbon in huge amounts per year. Due to the simple steps required for production of conductive inks using this approach compared with other solution phase approaches, it was adopted by many researchers. These steps included the addition of precursor to a solvent, injection of reducing and capping agent, reaction, rinsing, and removal of additional capping agent. Unlike other approaches, it would minimize the use of solvents in preparation [95].
Additionally, one-step continuous flame-based method was employed for the preparation of multi-component metal-decorated crumpled reduced graphene oxide NCs. Crumpled reduced graphene oxide balls (CGBs) were fabricated by injecting an aqueous dispersion of graphene oxide (GO) into the throat part of the nozzle using a syringe pump in HTRJ reactor at a rate of 250 mL h−1. Then, they were decorated by transition metal NPs such as: Ni, Fe, Co, and Pd for the production of metal CGB (M-CGBs) by adding metal nitrate precursors to the GO aqueous dispersion. The mass ratio of GO to metal was selected at 2 in Mohammadi et al. study [96]. A hot high-velocity gas stream was used for atomization of the aqueous precursor, producing droplets evaporated in the reaction chamber containing excess H2 at high temperature (550 or 600 °C). Then, the products were cooled, were diluted directly by blending them with N2 (100 SLM) and then were gathered on a filter membrane [96]. The main benefit of the HTRJ compared with standard flame-base aerosol techniques was the isolation between the zones of flame and product creation, which permitted the generation and/or reduction of NMs. They could be reduced by H2 when H2O was existed. Generally, various NMs could be fabricated by aqueous precursors of inexpensive metal salts and dispersed GO [95]. The combustion products from a hydrogen-rich flame (H2, O2, and N2) were transferred through a converging–diverging nozzle in 11, 3.2, and 5 standard liters per minute (SLM), respectively.
In another study, bimetallic Cu-Ni nanostructured coatings were synthesized by HTRJ method through deposition and sintering of metal NPs producing an aerosol. A gas phase (aerosol) of metal NPs could be produced easily using inexpensive and water soluble metal precursors. Copper nitrate hemipentahydrate and nickel nitrate hexahydrate (Ni(NO3)2.6H2O) were used as precursors in Sharma et al. study. The total metal concentration of the aqueous precursor solution was 10 mM, and the flow rate was adjusted at 180 mL/h. As an advantage of this method, the fabricated material was produced in powder forms which could be used in different applications such as conductive inks. The oxygen flow rate was used to control the flame temperature of the reactor system. For high temperatures, the flow rate of O2 was set at ~2.3 SLM and totally at ~14 SLM for H2 and N2. Similarly, the hot combustion products were accelerated using a converging–diverging nozzle. The aqueous precursor solution was emerged into the throat of the nozzle using four small identical inlets. The precursor solution was atomized using a hot and accelerated gas stream which caused it to be evaporated, decomposed, and finally, metal or metal oxide particles were produced. Nanostructured coatings were deposited on glass windows. A single deposition substrate was positioned perpendicular to the gas flow above the reactor exit. The substrate temperature was determined as 200 °C, and the total time for coating was 3 h [97].
Similarly, metal NPs were fabricated by a flame-based aerosol reactor, thermal decomposition, and hydrogen reduction in Scharmach et al. [98] study. Carbon-coated copper NPs were synthesized using a copper formate precursor solution. Carbon-coated metals were known for their stability against oxidation and degradation. Also, Cu NPs with a surface layer consisted of carbon were employed for stabilization of particles, keeping their electrical and thermal conductivity near from the values reported for substances that were used in conductive inks. Because of their low cost, they were used instead of Au and Ag in conductive inks and other different applications [98]. In this method, a fuel-rich hydrogen flame was used as an energy saving source. Hot combustion products was moved through a nozzle, producing high-temperature reducing jet. Then, the liquid precursor solution was directly atomized, followed by evaporation, and then, it was decomposed to create the particles. For the high-temperature-reducing jet reactor, a flow with different gases was allowed: O2 (~2 SLM) in small amounts was entered into the combustion region, but H2 and N2 (~10 SLM total) in larger amounts were entered through the surrounding concentric annular region. The hot combustion products were then accelerated, and the precursor was inserted to the diverging portion of the nozzle. The time required for nucleation and growth was controlled through the variations in dimensions of the area found between nozzle and quench zones. The residence time was set as 0.05 s in this study. At the exit, the products were quenched by blending with a plenty of cold N2 (~100 SLM), temperature was lowered directly, and particle growth was stopped. Fast quenching was useful in minimizing the formation of hard agglomeration. Additional dilution for N2 (up to 100 SLM) could be also used at the filter inlet for extra dilution and cooling. Particles were then collected efficiently using a 0.2 micron pore size filter. Particles were recovered by centrifuging, and they were rinsed by acetone and acetic acid [98].
2.6 Liquid-phase reduction technique
Ni-Ag core–shell NPs were synthesized through the deposition of Ag on Ni nanocores by employing the liquid-phase reduction method in aqueous solution [99]. Successive steps were followed for the production of Ni-Ag core–shell NPs: fabrication of Ni NPs, Ag NPs, Ni-Ag core–shell NPs, and preparation of the conductive paste. Ni NPs were prepared by dissolving nickel acetate tetrahydrate (10 mmol) in DW (50 mL), followed by the addition of sodium dodecyl sulfate (SDS) (0.5 g) to the prepared solution under stirring. Then, NaOH solution (1.0 mol/L, 30 mL) was added, followed by the addition of hydrazine hydrate (10 mL), stirred, was heated to 70 °C, and then was preserved at that temperature for 1 h. Next, the suspension was cooled to room temperature. A precipitate was produced after centrifuging, was rinsed repeatedly with water and ethanol, and then was dried in vacuum at 40 °C for 10 h [99]. Next, Ag NPs were synthesized by dissolving silver nitrate (AgNO3) (10 mmol) in DW (50 mL). An aqueous reductant solution was prepared from glucose (12.5 mmol) and tartaric acid (1.25 mmol), which was added as drops to initiate the reaction at room temperature for ~2 h. Next, the sample was produced after it was centrifuged, was washed many times using water and ethanol, and then was dried under vacuum at 40 °C for 10 h [99].
After that, Ni-Ag core–shell NPs were synthesized by following these steps: Ni powder was dissolved in HCl solution (0.01 mol/L) till saturation, and then, a treatment was applied for the removal of the oxidized Ni layer. Next, the powder was rinsed with DW till no precipitate was observed, because of the addition of AgNO3 solution. Then, it was saturated and stirred in DW (80 mL) and AgNO3 solution (10 mL). The reductant solution was added at a rate of 10 drops/min. After 2 h, it was centrifuged, was rinsed repeatedly with water and ethanol, and then was dried under vacuum at 40 °C for 10 h. Finally, a conductive paste was formed from Ni, Ag, and Ni-Ag core–shell NPs by following these ratios: 60% of metal NPs, 35% of organic vehicle, and 5% of glass powder. Three containers were used for mixing these pastes at 90 °C for 1 h. Then, they were cooled to room temperature. The pastes were screen-printed as a thick film onto a polycrystalline silicon substrate. Then, these films were dried at 150 °C under N2 atmosphere for 2 h to eliminate the organic solvent. Lastly, the films were sintered at (500–800) °C under N2 atmosphere for 10 min. The thickness of the sintered films was determined as 11 ± 1.5 mm in Jing et al. study [99]. The characteristics of the prepared NPs were determined. Crystallinity was improved, and the thicknesses of Ag nanoshells could be easily controlled. It could be also used in conductive paste since Ag surface had an oxidation resistance, and Ni core had electroconductive properties. Therefore, the use of as-prepared NPs as an electrode material was preferred than pure Ag NPs that were used in conductive pastes [99].
In another study, Ni NP-containing polyimide composite films were synthesized by liquid-phase reduction of Ni2+ ions with potassium borohydride (KBH4) as a reducing agent. The procedure followed for fabrication of Ni NP-containing polymer was initiated through the insertion of carboxyl groups into the polyimide resin for cation exchange through surface enhancement by employing KOH. The precursor resin was prepared provided that the cation exchange groups should be replaced by Ni ions. Then, Ni was fabricated by liquid-phase reduction using KBH4 as an aqueous solution. For details, the polyimide films of the pyromellitic dianhydride oxydianiline (PMDA-ODA)-type were utilized as the polymer matrix. The first step was the enhancement of polyimide films (2 × 2 cm2, 125 mm thick) by immersing them into KOH solution at 30 °C for 5 min. Then, they were washed with distilled water for 5 min. Thereafter, these films were also dipped in NiSO4 solution at 30 °C for 5 min and then were washed with distilled water for 5 min. Next, the Ni2+ ion-doped films were submerged in KBH4 solution at 30 °C for 20 min to reduce the Ni2+ ions to Ni NPs. Other extra reduction procedures were carried out at 40 °C or 50 °C. Also, KBH4 concentration was changed from 0.02 to 0.20 mol dm−3. An aqueous solution of CH3COOH (0.25 mol dm−3) was processed in polyimide NC films incorporated with Ni NPs at 1 min to eliminate any remaining Ni2+ ions that might be adsorbed. All films were washed with distilled water for 5 min, dried and kept under usual conditions. The produced films were heated at 300 °C for 2 h under N2 atmosphere [100].
2.7 Sol–gel
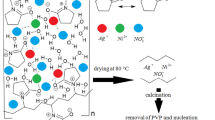
A series of Ag/NiO NC (AgxNi1−x O) was synthesized by the sol–gel method. Precursors including AgNO3 and (Ni(NO3)2.6H2O) were dissolved in DW and stirred. Four different concentrations were prepared as follows: 0:1, 0.2:0.8, 0.4:0.6, and 0.6:0.4 mol, respectively. Then, an aqueous solution of citric acid (2.0 M) was added to the prepared solutions. The pH was set at ~7 by adding ammonia. The solution was stirred and heated to 70 °C to form the gel, which was dried overnight at 120 °C in an oven to produce xerogel. Finally, the xerogel was crushed to produce a homogeneous powder which was calcined at 400 °C for 5 h [101]. Another study used sol–gel to fabricate Ag-doped NiO NPs (AgxNi1−x O). For preparation, Ni(NO3)2.6H2O was dissolved in DW at room temperature, AgNO3 was used as the dopant source in different ratios (1, 3, and 5%), and Cydonia oblonga plant extract was employed as a reducing and stabilizing agent. The AgNO3 solution was added into Ni(NO3)2.6H2O solution, and then, Cydonia oblonga extract solution (20 mL) was added drop by drop. The mixture was stirred for 5 min and then put on an oil bath at 80 °C for 12 h. The produced green gel was dried at 100 °C for 6 h and then was calcined at 400 °C for 2 h to produce a black-colored powder of Ag-doped NiO NPs [102]. Many advantages were known for this method such as: simple, eco-friendly, need inexpensive precursors, highly pure products, and produce considerable quantities [102]. The XRD results revealed that the diameter of the AgxNi1−x O NPs was increased as the concentration of Ag was varied from 1 to 5%. This growth in Ag doping size was related to an increase in internal structural weakens, which caused the growth rate to increase. Based on the results of toxicity studies, high cytotoxicity effect upon PC12 cell line was found due to the use of Ag-doped NiO NPs which inhibited cancer cells [102].
Conversely, NiO NPs were prepared by sol–gel method in different research studies [31,32,33,34,35,36]. Different precursor materials were used to synthesize NiO NPs such as: Ni(NO3)2.6H2O [31, 35, 36], Ni(NO3)2 [34], NiCl2.6H2O [32], and nickel acetate tetrahydrate [33]. The calcined temperatures used to prepare NiO NPs were reported as: 300 °C [31], 450 °C [34], 500 °C [32, 36], 800 °C [35], and from 300 to 1100 °C [33]. In fact, no reported study was found for preparing Ag2O NPs by sol–gel method. But Ag NPs were prepared by using AgNO3 as a precursor material in sol–gel method [103, 104].
In Adiba et al. study, NiO NPs were synthesized using Ni(NO3)2.6H2O as a precursor via sol–gel method. DW was used as solvent for preparation of two solutions: Ni(NO3)2.6H2O (0.01 mol) and C6H8O7 (0.02 mol), respectively. These two solutions were mixed together, stirred via a magnetic hotplate at 500 rpm and 80 °C, producing a green colored gel. The produced gel was dried overnight and then was calcined at 800 °C in muffle furnace for 8 h, producing green-colored NiO powder [35]. In another study, pure NiO NPs were fabricated using sol–gel by adjusting the pH at 11, and the calcined temperature at 450 °C. Many parameters should be adjusted to produce pure NiO NPs including: structure, calcined temperature, and pH value of the solution. The produced shape was dependent on the correct choice of the calcined temperature, which was chosen as 450 °C to fabricate a spherical shape in Zorkipli, Kaus, and Mohamad study. Additionally, it was found that sol–gel is the appropriate route to fabricate NiO NPs since it showed homogeneous mixture, improved crystallinity, uniform particle distribution, and smaller particle size [105]. Ni(NO3)2·6H2O was dissolved separately in 20 ml of both isopropanol alcohol and polyethylene glycol. The solutions were stirred for 1 d until all chemicals completely dissolved. Ammonium hydroxide (NH4OH) was added to adjust the pH of the solutions at 11. Triton X-100 [C14H22O (C2H4O)n] was also added to prevent particle agglomeration. Then, these solutions were heated at 80 °C until gel was created, was dried at 200 °C, and then was grounded [105].
NiO NPs were synthesized simply by a sol–gel method using agarose polysaccharide without any surfactant and reducing agent. Agarose, extracted from red seaweed, was known as an outstanding gel forming. The procedure followed by Alagiri, Ponnusamy, and Muthamizhchelvan study was known as multifaceted, low cost, eco-friendly and could be employed for fabrication of other magnetic metal oxides. Agarose powder (2 g) was added slowly to the distilled water (100 mL), and stirred at 50 °C for 20–30 min until the solution turned transparent. Thereafter, 1 wt% of Ni(NO3)2.6H2O solution was added to the agarose gel and stirred at 80 °C until a homogeneous gel was formed. The produced gel was heated at 100 °C for 4 h; a dried precursor was produced which was calcined at 400 °C for 3 h to produce NiO NPs [106]. TEM images revealed that the prepared NiO NPs were spherical in shape with a size of 3 nm. Also, magnetic measurements demonstrated that NiO NPs were superparamagnetic character at 300 K and ferromagnetic character at 4.2 K [106].
The Ag NPs were prepared by sol–gel method using CH3COONa and hydrazine as reducing agent in water at ambient temperature. Specified amounts of metal nitrate, citric acid, and sodium hydroxide solution were created keeping the molar ratio of nitrates to citric acid 1:1. Ammonia was added to the solution to set the pH value at 7. Ag NPs were fabricated by chemical reduction route employing N2H4.H2O as a reducing catalyst. Three concentrations of AgNO3 aqueous solution (6 mM, 7 mM, and 8 mM in 100 ml) were prepared, citric acid (0.1 M), and NaOH (0.1 M), which were mixed and stirred to produce three mixture solutions with different concentrations (12 mM, 16 mM, and 20 mM). In each solution, N2H4.H2O was added and stirred which formed a black solution due to the reduction of Ag+ ions. Next, the produced solutions were stirred for 3 h at room temperature till they became clear, and Ag particles were shining in the containers. The particles were gathered using a filter paper, were rinsed with DW, and then were dried in air [104]. In fact, sol–gel is considered as a simple, convenient, clean, nontoxic, time-saving, economical, and eco-friendly route for the fabrication of Ag NPs. Also, the particle size could be varied at room temperature conditions without using any additive [104]. High pressure, energy, temperature, dangerous substances, downstream processing were all avoided in each stage of synthesis (Shahjahan 2017).
2.8 Hydrothermal method
The basic principle of this method is the ability of solvents (water and aqueous solutions) to dissolve substances at high temperatures. The synthesis is usually occurred in autoclaves that are sealed steel cylinders capable of withstanding at high temperatures and pressure [107, 108]. This method was widely used for preparing different silver or nickel nanostructures. Metallic Ag/NiO honeycomb nanoarrays were prepared by the hydrothermal method using AgNO3, Ni(NO3)2.H2O, hexamethylenetetramine (HMT), and urea as precursors [109]. Two different solutions were prepared separately by dissolving AgNO3 and Ni(NO3)2.H2O in a double distilled water. Next, these two solutions were mixed under stirring for 15 min, followed by the addition of 100 mM of HMT (10 ml) to the precursor solution. Then, urea solution (0.6 M, 20 ml) was added under continuous stirring for 30 min. The prepared solution was kept in a covered glass bottle, and left in an oven at 100 °C for 1 d. The produced precipitate was centrifuged, was rinsed repeatedly with water and ethanol, was dried at 100 °C for 1 d and then was calcined at 400 °C for 2 h [109]. It was found that Ag enhanced the electronic conductivity of the NiO nanoarrays, which would be more suitable for the electrochemical redox reaction at the electrode/electrolyte interface. Based on electrochemical studies, Ag/NiO nanoarray was approved as good electrode material that could be used for supercapacitor applications. In their study, asymmetric device was constructed and investigated, which produced a high specific cell capacity, and a maximum energy density. These findings were better than those of lead-acid batteries and analogous to those of metal hydride batteries [109].
Additionally, Ag NPs were successfully synthesized by combined green and hydrothermal methods using red cabbage extract [110]. This combination of these two methods was used to prevent aggregation of metallic NPs synthesized in plant extracts. Two main reasons were reported for minimizing aggregation problem of metallic NPs: the use of red cabbage extract that mainly have anthocyanins which changed the pH of the extract solution to be near from neutral condition, and the availability of supercritical conditions that almost increased the solubility of metal ion and anthocyanins at high temperatures and pressure [110]. So, this combination would be effectively adopted for fabrication of many biosynthesized metallic NPs [110]. The major advantage of this method compared with low-temperature routes, i.e., co-precipitation or sol–gel, was the fabrication of size controllable NPs at large scales [110]. Also, microwave hydrothermal synthesis was used for preparing Ag NPs in the 6-O-carboxymethyl-chitin (CMC) matrix. The AgNO3 solution (10 mM, 5 mL) and D-glucose solution (10%, 0.5 mL) were added to CMC solution (1.0%, 5 mL) in water. The produced mixture that consisted of CMC, AgNO3, and D-glucose, was treated in a microwave at 130 °C and 600 W for 4 min to obtain the products [107].
NiO NPs were fabricated simply via one-step hydrothermal route using Ni(NO3)2·6H2O as a precursor. First, Ni(NO3)2·6H2O was dissolved in DW (50 mL). Next, urea (50 mM) was added drop by drop to the prepared solution and stirred at ambient temperature till it became clear. The produced solution was then heated at 150 °C in autoclave for 10 h. Then, it was cooled, was centrifuged, was rinsed with DW for many times, and then was dried in an oven at 50 °C for 5 h. Finally, the product was annealed at 450 °C in air for 5 h [40].
In Y. F. Li et al. [111] study, Ag NPs were synthesized via hydrothermal method using Arabic gum (AG) as both reductant and steric stabilizer without any other surfactant. Well-dispersed and quasi-spherical Ag NPs were synthesized under the following optimal conditions: 10 mmol/L of AgNO3, mass ratio of AG/AgNO3 = 1:1, 160 °C, and 3 h. Typically, AgNO3 (0.5 mmol/L) and AG (0.1 g) were dissolved into 50 mL of DW in a suitable vessel. The produced mixture was sealed in autoclave and was kept at 160 °C for 3 h. Then, it was turned off to minimize the temperature to the room temperature, was centrifuged, and then was washed with water and ethanol many times to gather the produced samples [111].
Conversely, high-purity Ni NPs in the powder form were fabricated by hydrothermal route using Ni(II) chloride hexahydrate (NiCl2.6H2O) and NaOH with different concentrations (from 5 to 25 mol/L). The fabrication of these NPs happened via a chemical reduction at relatively low temperature (140 °C). Typically, 4 g of NaOH (5 mol/L) was dissolved in 20 mL of DW, followed by adding 1 mL of NiCl2.6H2O (0.5 mol/L) and 0.35 mL of ethylenediamine (EDA) under continuous stirring for 60 min. Next, 0.25 mL of hydrazine hydrate (N2H4.H2O 30%) was also added and stirred for 30 min. The mixed solution was sealed and kept at 140 °C for 4 h, and then, it was cooled down to room temperature. A black fluffy product floating on the solution was removed using a magnetic bar, and then, it was washed with distilled water and ethanol. That procedure was repeated for many times to eliminate unwanted impurities. The produced sample was dried in air at 40 °C for 4 h. The results showed that the concentration of NaOH and the type of the precursor could be adjusted to fabricate Ni NPs with different sizes, shapes, and morphologies [112].
2.9 Co-precipitation method
Silver oxide (Ag2O) NPs were simply and directly prepared via co-precipitation method using AgNO3 and DW [64]. AgNO3 (1.70 g) was mixed with DW (10 mL) and stirred to produce a homogeneous solution. Triethylamine (10 mL) was added to the solution and stirred for 200 min. The resulted precipitates were centrifuged at 6500 rpm for 7–10 min, and rinsed with distilled water or ethanol. Then, they were dried at 30 °C and located in an incubator for 1 d. After that, these precipitates were dried in an oven at 200 °C for 4 h, and then in a furnace at 800 °C for 1 d. The shape and size of Ag2O NPs could be varied according to many parameters such as: morality ratio of solution, temperature, pH, time, and addition of seed solution. Also, the size of the prepared NPs should be smaller than the pore size of a biological model (i.e., in the range of 100 nm) to be successfully used for cancer cell toxicity [64].
In another work, NiO NPs were fabricated by a chemical co-precipitation route. A mixture of Ni(NO3)2.6H2O (0.5 M) and aqueous solution of NaOH (1 M) was prepared in DW and stirred for 2 h at ambient temperature. The produced precipitate was rinsed to eliminate Na ions and dried in air at 80 °C, which was crushed and after all calcined at 700 °C for 2 h. It was reported that NiO NPs had utilized as a photocatalyst for degradation of methylene blue dye from aqueous mediums [113].
Silver nickel oxide (AgNiO2) was synthesized by co-precipitation method using AgNO3 and Ni(NO3)2.6H2O as precursors in alkaline medium (NaOH) and in the existence of Na2S2O8. After reaction, a black precipitate was created, aged at 90 °C for 1 h, was filtered and washed with distilled water. Next, the precipitate was dried at 90 °C for 4 h [114]. The unique electromagnetic properties of mixed AgNiO2 indicated that the lattice properties of mixed AgNiO2 were modified compared with individual silver and nickel oxides. Also, the mixed AgNiO2 was interacted efficiently with CO at ambient temperature. The existence of O2 in the reaction environment caused the amount of carbonate-like species to minimize compared with the reaction with pure CO. When the temperatures raised above 150 °C, the structure of AgNiO2 was completely destructed and Ag0/AgOx and NiOx particles would be resulted [114].
In another study, bimetallic silver nickel (Ag Ni) NPs were prepared via co-precipitation through the sequential reduction of the metal ions in diethylene glycol, ethylene glycol, glycerol, and pentaerythritol solutions, with concomitant precipitation of Ag/Ni bimetal solutions. Additionally, AgNO3, nickel (II) acetate, polyvinylpyrrolidone (PVP), methanol, and ethanol were used in preparation procedure [115]. A mixture solution of glycerol (15 mL) and PVP (0.03–0.08 mmol) was stirred and heated to 150 °C. Then, Ni(CH3CO2)2.4H2O (0.41–2.01 mmol) was added to the prepared solution, caused the color to change to Prussian green. After the growth of Ni NPs, AgNO3 (0.35–1.24 mmol) was added to the mixture, and was stirred for 4 h. As a result, Ag/Ni solution with pale brown color was formed, was rinsed with methanol for many times, and was centrifuged at 4400 rpm for 10–15 min to eliminate excess unreacted stabilizer. The Ag Ni solution was re-dispersed in ethanol. Similarly, the aqueous co-precipitation of Ag Ni solution was fabricated by employing the following substances: PVP (0.02–0.13 mmol), PET (24.01–37.25 mmol) used as capping agent, AgNO3 (0.12–0.82 mmol), and Ni(CH3CO2)2.4H2O (0.66–1.48 mmol) in DW (100 mL) at 90 °C. These steps were repeated at different conditions: 160 °C, 2 h, and 175 °C, 2 h; and then for ethylene glycol at 160 °C, 3 h, and 175 °C, 2 h; diethylene glycol at 190 °C, 2 h, and at 200 °C, 2 h. The change in color was considered as an evidence for the successful production of Ag Ni NPs [115]. In Adekoya et al. study, co-precipitation method was employed to prepare stable Ag Ni NPs up to 6 months. The preparation by organic phase was usually designed to control anisotropic nucleation and particle growth processes [115].
Ashokkumar and Muthukumaran fabricated Zn0.96−xCu0.04NixO (0 ≤ x ≤ 0.04) NPs by co-precipitation route by employing the following precursors: zinc nitrate hexahydrate (Zn(NO3)2.6H2O), copper (II) nitrate trihydrate (Cu(NO3)2.3H2O), Ni(NO3)2.6H2O, and NaOH. Certain amounts of Zn(NO3)2.6H2O, Cu(NO3)2.3H2O, and Ni(NO3)2.6H2O were dissolved in double distilled water and stirred till the solution became transparent. Alkaline solution was prepared by dissolving NaOH in double distilled water and then added as drops to the mixture solution. The product was stirred for 2 h at ambient temperature. The precipitates were filtered and washed many times to eliminate any impurities. Then, they were dried by an oven at 80 °C for 2 h. The dried samples were grinded, and then was annealed at 500 °C in air for 2 h. Similarly, all steps were repeated to other Ni concentrations (x = 0, 0.01, 0.02, 0.03 and 0.04) [116]. Based on the results, the mean crystallite size was decreased from 27 to 22.7 nm as the Ni concentration was increased from 0 to 2% because of the suppression of nucleation and subsequent growth of ZnO by Ni-doping, whereas the crystallite size was increased from 22.7 to 25.8 nm as a result of Ni-doping from 2 to 4% because of the creation of distortion centers and Zn/Ni interstitials. Also, the noticed red shift of energy gap (Eg) was decreased from 3.65 to 3.59 eV as the Ni concentration was increased from 0 to 2%, due to sp-d exchange interactions between the band electrons and the localized d-electrons of the Ni2+ ions, while the blue shift of Eg was increased from 3.59 to 3.67 eV as the Ni concentration was increased from 2 to 4% due to Burstein–Moss effect [116].
In another study, nickel ferrite (NiFe2O4) NPs were fabricated via co-precipitation route using nickel nitrate, ferric nitrate, and NaOH as precursors [117]. Oleic acid was used as surfactant. A mixture solution was prepared by adding ferric nitrate and nickel nitrate into DW and stirred, and then, NaOH solution was added dropwise till the pH value became 10–11. Next, it was heated at 80 °C for 1 h, followed by the addition of oleic acid (5 ml), and was heated again at 90 °C for 30 min. The produced mixture was cooled at ambient temperature, and then, a few drops of HNO3 were added and a precipitate was collected. This precipitate was rinsed with water and acetone to remove impurities. Finally, it was dried to obtain powder which was calcined different temperatures 250, 350, 450, and 550 °C for 2 h, respectively [117].
A simple co-precipitation method was employed to fabricate pristine ZnO NPs and NiO-ZnO NCs with many concentrations of Ni2+ ions. This approach was reported in the literature as simple and suitable method for preparing semiconductor NCs with controlled physical, chemical and magnetic properties. Therefore, it could be subjected for biological applications such as bactericidal agents. Pristine ZnO NPs and NiO-ZnO NCs were synthesized using Zn(NO3)2.6H2O, Ni(NO3)2.6H2O and NaOH as starting substances. For the synthesis of pristine ZnO NPs, Zn(NO3)2.6H2O (0.5 M) was dissolved in DW (100 mL), followed by adding NaOH solution as drop by drop under continuous stirring, leading to the creation of white precipitate (ZnO). Then, the precipitate was rinsed many times with DW and ethanol to eliminate any impurities. ZnO powder was dried in an oven at 70 °C for 6 h and then was calcined in a furnace at 400 °C for 2 h to improve crystallinity. For the synthesis of NiO-ZnO NCs, a solution was prepared by dissolving Zn(NO3)2.6H2O (0.5X M) and Ni(NO3)2.6H2O (predetermined amount; X = 0.03, 0.06, 0.09 and 0.12 M) in deionized distilled water. Next, NaOH solution was added dropwise into the prepared mixture solution, and stirred. Finally, the upcoming steps were followed like those mentioned for the fabrication of pristine ZnO NPs [118].
2.10 Simple precipitation method
In Liu et al. [119] study, a NC consisting of Ag NPs on NiO porous thin-film nanostructures was synthesized using simple chemical precipitation technique and silver mirror reaction. These thin films have well-distributed pores and high surface areas to enhance levofloxacin (LEV) oxidation and signal amplification. In fact, “LEV is used in antibacterial infections caused by gram-negative and gram-positive bacteria, especially in the skin, soft tissues, urinary and respiratory system.” A mixture solution was made by dissolving Ni(NO3)2·6H2O (0.02 mol) and hexamethylenetetramine (HMT) (0.2 mol) in DW. The prepared solution was heated at 100 °C under stirring for 2 h. Then, the solution was cooled down to ambient temperature. Next, blue-green precipitates were produced, were rinsed for many times with DW and absolute ethanol, and followed by drying at 60 °C overnight. Green powders (Ni(OH)2) were calcined at 300 °C in air for 2 h to produce NiO porous thin films. The produced thin film (0.4 g) was dispersed in the mixture solution which consisted of AgNO3 (0.01 mol.dm−3) and glucose (0.05 mol.dm−3), was heated at 75 °C, and then was stirred for 3 h. A silvery-white product was rinsed with DW and absolute ethanol repeatedly, followed by drying overnight at 60 °C to get porous NiO-Ag thin film NC [119]. In this study, Ag NPs were successfully prepared using inexpensive substances compared with other noble metals, and also they were utilized in electrochemical sensing applications. Also, many advantages and benefits were found from the fabrication of such NCs. The addition of Ag NPs via a simplified silver mirror reaction was enhanced the electrical conductivity and catalyst activity of NiO porous thin film. Actually, porous NiO-Ag thin film NC was synthesized for LEV determination that showed a low detection limit and wide linear detection ranges. In addition, this fabricated sensor could be used effectively to detect serum human samples and it showed excellent results. Thus, easy and cheap electrochemical technique was used in medicine and clinics for the quantitative measurement of LEV [119].
Conversely, NiO NPs were synthesized by chemical precipitation method by employing Ni(NO3)2·6H2O and NaOH aqueous solution at 60 °C. Typically, two aqueous solutions were made by dissolving Ni(NO3)2·6H2O (0.25 M) and NaOH (0.5 M) in distilled water separately. The NaOH solution was heated at 60 °C, followed by the addition of Ni(NO3)2·6H2O solution drop by drop, and stirred for 2 h. The resulted light green solution was left for 2 h, was filtered, then was rinsed twice with DW and ethanol, and was dried in an oven at 80 °C for 3 h. The dried product (Ni(OH)2) was grounded and then was calcined at 400 °C for 4 h. A black powder was obtained, which was crushed once more to produce pure NiO NPs [120].
2.11 Simple wet chemical microemulsion method
In Senapati et al. study, prickly Ni NWs were fabricated, and then, the surfaces of these NWs were modified with Ag by a well-known Tollens’ reagent to synthesize Ni/Ag core–shell nanostructures. First, NiCl2.6H2O aqueous solution (4 M) was located in a flask containing a mixture of cetyltrimethylammonium bromide (CTAB) (2 g) and hexane (20 mL). After that, 1-butanol (co-surfactant) was added under continuous stirring for 20 min. As a result, a green homogeneous and transparent microemulsion (known as type 1) was created. Similarly, another microemulsion (known as type 2) was also formed, but the only difference that NaOH (8 M) and N2H4.H2O (3 mL) were utilized instead of aqueous NiCl2 and the pH of the prepared solution was set at 12. Next, type 1 microemulsion was added dropwise to microemulsion type 2, and then, the produced homogeneous solution was refluxed at 70 °C. A black precipitate was formed after 30 min which was affected by a magnetic field that can be used in separation processes from the reaction mixture. After 90 min, the final product was created, was rinsed with ethanol and then by water, followed by drying in vacuum. Next, Ni/Ag core–shell nanostructures were fabricated via a redox-transmetalation process through the addition of the freshly prepared Tollens’ reagent (10 mL) to the previously prepared black powder, dispersed in water. The resulted solution was sonicated for 30 min and then was refluxed at 60 °C for 3 h. The final product was rinsed repeatedly with water, and was dried in vacuum at 40 °C [121]. Based on the results, Ni/Ag core–shell nanostructures were more efficient catalyst compared to Ni NWs for the reduction of p-nitrophenol by sodium borohydride (NaBH4) at room temperature. Also, the addition of Ag shell in Ni/Ag nanostructures was successfully enhanced the antibacterial activity in comparison with other commercially antibacterial agents. Moreover, Ni in the core simplified the magnetic separation of core–shell Ni/Ag from the reaction mixture [121].
Conversely, NiO NPs were fabricated in a water-in-oil (w/o) microemulsion [122]. In this study, four nickel oxides with several particles size were fabricated by changing the experimental parameters. Typically, nickel chloride hexahydrate (0.666 g) and aqueous ammonia (2.2 mL) were separately dissolved in DW to form two solutions (solution I and II), respectively. N-hexanol (40.4 mL), Triton X-100 (67.0 mL), and cyclohexane (112.5 mL) were mixed together to form solution III. The mobile phase was 44% oil (cyclohexane), 33% surfactant (Triton X-100), 16% co-surfactant (cyclohexane), and 7% water (inorganic precursor) (w/w). Solutions I and II were subsequently added into two solutions from III to form microemulsion. Then, these two reverse microemulsions were mixed and stirred. The green precipitates were filtered, were rinsed, and were calcined at 500 °C for 2 h to synthesize NiO NPs. By following similar procedure, the other three sensing materials were prepared by varying the parameters used in the above procedure. Gas sensors made from NiO NPs had a better sensing performance to hydrogen sulfide, ethanol and nitrogen dioxide, compared with those based on normal bulk NiO. The reasons were summarized as: small NiO particles with large surface area, porous nanostructures, and high ratio of charge accumulation layer to the radius of a particle [122].
Additionally, NiO NPs were fabricated using microemulsion technique in another study [123]. Certain amounts of many substances (methylcyclohexane, water, surfactant AEO9, and octanol) were blended in oil phase and stirred (1500 rpm) at 30 °C, producing microemulsion system. Then, nickel nitrate aqueous solution (0.1 mol/L) was added to the prepared system and stirred till completely dissolved producing microemulsion solution consisting of nickel ions. Also, lithium borohydride was added to that system and stirred at low-temperature conditions. After stirring the microemulsion system for 1 h, NiO NPs were obtained. The solution turned dark due to the continuity of reduction reaction. Next, the particles were regained, were rinsed with distilled water and ethanol, and followed by drying for 4 h at 50 °C in an oven. The product was calcined in a furnace for 3 h at 550 °C, and then, it was crushed using pestle and mortar to obtain a powder sample [123].
The inverse microemulsions approach was also employed for the fabrication of Ag NPs via CTAB, Tergitol, and Triton X-100 as the surfactants to adjust the particle size and morphology of the prepared NPs. Initially, CTAB was utilized as a surfactant and the weight ratios the constituents used in the microemulsions were determined as (16.76% of CTAB, 13.90% of 1-butanol, 59.29% of isooctane, and the remaining was 10.05% of aqueous phase). Actually, CTAB (59.5 g) was put into a flask to which iso-octane (300 ml) and 1-butanol (60.5 ml) were added. Then, aqueous solution of AgNO3 (0.1 M) was added in drops to the system under stirring till it became optically transparent and clear microemulsion was formed. Also, similar amount of NaBH4 (0.1 M) solution was added as drops till a brownish green turbidity appeared. After stirring for 6 h, the microemulsion system turned brownish black. The formation of light gray precipitate was approved the creation of pure Ag. Next, it was centrifuged to produce a silvery white precipitate, which was rinsed with 1:1 chloroform/methanol mixture, was centrifuged, and then was dried in oven at 60 °C for 2 h to obtain Ag NP. After that, Tergitol was utilized as a surfactant in the microemulsion systems. Two microemulsions (I and II) were prepared by using cyclohexane (150 ml) as an organic solvent, Tergitol (17.5 ml), and 1-octanol (13.0 ml) as a co-surfactant. Aqueous AgNO3 (0.1 M) was added to microemulsion (I) to obtain a clear solution, while NaBH4 solution (0.1 M) was added to microemulsion (II). Thus, two different transparent w/o microemulsions were formed. A dark-colored solution resulted directly after slow mixing of the two microemulsions using a magnetic stirrer. The color changed from greenish brown to silvery white as the solution was evaporated to 60 °C, producing tiny particles in the bottom of the flask. This precipitate was rinsed with acetone for many times, was centrifuged and then was dried in oven at 60 °C. Like microemulsions (I and II), the third set of reaction was prepared except Triton X-100 (22.5 ml) as a surfactant, and 1-hexanol (15 ml) as a co-surfactant. Again, these two microemulsions were blended to produce a dark brown precipitate. This precipitate was washed with a mixture of methanol and chloroform (1:1), then was centrifuged, and finally was dried in an oven at 65 °C for 2 h [124].
Also, Ag2O NPs were fabricated in microemulsions using two different surfactants, namely Tergitol and Triton X-100. Using Tergitol as a surfactant, two microemulsion systems (I and II) were made; microemulsion (I) composed of Tergitol (17 ml) as a surfactant, 1-octanol (13 ml) as a co-surfactant, cyclohexane (150 ml) as an organic solvent, and aqueous AgNO3 solution (7.5 ml of 0.1 M). Similarly, microemulsion (II) had the same composition of (I) but aqueous sodium hydroxide solution was used instead of aqueous AgNO3. Then, these two microemulsions were combined to produce a light brown system, which was turned into dark brown color after stirring for 12 h. The microemulsion system was heated at 60 °C caused the solvent to be evaporated, then the produced solution was centrifuged. The precipitate was rinsed with acetone and then dried in an oven at 60 °C. Next, two microemulsions (I and II) were again fabricated using Triton X-100 as a surfactant. The first one (I) composed of Triton X-100 (22.5 ml) as a surfactant, 1-hexanol (15 ml) as the co-surfactant, cyclohexane (150 ml) as an organic solvent, and aqueous AgNO3 (7.5 ml of 0.1 M), while in microemulsion (II) the aqueous phase composed of aqueous NaOH solution (0.1 M). The size of the Triton X-100-based reverse micelles or droplets ranged from 5 to 18 nm according to the aqueous phase concentration that was related to stable reverse micelles. Optimum concentrations were selected for the formation of monodisperse and small size micelles of different components of reverse micelles. No change in color was detected when the two microemulsions were combined. A light yellow was seen after stirring for 2 h. A spectrum of colors (yellow, orange brown, and maroon) was observed due to the stirring for extra 10 h. Next, the microemulsion solution was centrifuged, a precipitate was formed, and then, it was rinsed with a mixture of methanol and chloroform (1:1), was centrifuged, and then was dried in oven at 60 °C for 2 h. In the reaction system, NaOH represented the hydroxide ion donor, thus Ag+ ions came from AgNO3 and reacted with hydroxide ions to create AgOH, which was heated to produce Ag2O. The formed Ag2O nuclei were subjected to Ostwald ripening and growth to produce NPs in their final size [125]. Hexagonal Ag2O NPs had been fabricated with a size of 10 nm by Tergitol, whereas nonspherical Ag2O NPs with a larger size of 40 nm was synthesized via Triton X-100. The UV–visible analysis confirmed the existence of two surface plasmon resonance peaks at 420 and 650 nm for the Ag2O NPs prepared by Tergitol-based microemulsion. Also, the surface areas for Ag2O NPs prepared using Tergitol and Triton X100 surfactants were 29.5 m2/g and 13.3 m2/g, respectively [125].
In another study, Ag/Cu bimetallic nanocatalysts supported on reticulate-like γ-alumina were fabricated by microemulsion using N2H4.H2O as a reducing agent. Triton X-100, n-butanol, cyclohexane, and water system were the catalysts of Ag–Cu NPs supported on γ-alumina that were used for fabrication. Basically, AgNO3 and Cu(NO3)2 microemulsion with various Ag/Cu atomic ratios and the hydrazine hydrate microemulsion were produced with the same ingredients ratio like Al(NO3)3 microemulsion. Ammonia microemulsion was added as drops to Al(NO3)3 microemulsion, stirred and the reaction was left for 40 min. Then, different atomic ratios AgNO3 and Cu(NO3)2 microemulsion were added under stirring for 1 h at 20 °C. After that, hydrazine hydrate microemulsion was added to the mixture producing Ag–Cu bimetallic NPs supported on Al(OH)3. The prepared mixture was stirred for 2.5 h at 20 °C, the precipitates were obtained and centrifuged once at 12,000 rpm for 40 s, and then were calcined in air at 620 °C for 4 h. Lastly, the solids were reduced at 600 °C in H2 (0.15 mL/min) for 1 h, followed by cooling to ambient temperature under N2 flow (0.15 mL/min) to get AgCu/Al2O3 catalyst [126].
Additionally, Mn–Co–Ni–O ultrafine ceramic NPs were fabricated using the reverse microemulsion in a triton X-100, n-hexanol, and cyclohexane system. Typically, aqueous solutions were prepared by dissolving these starting substances in DW: manganese dinitrate (Mn(NO3)2), cobalt (II) nitrate hexahydrate (Co(NO3)2·6H2O), and Ni(NO3)2·6H2O with a mole ratio of Mn:Co:Ni = 4:5:1. The reverse microemulsion was formed by mixing: 40 ml of emulsifier, surfactant, Triton X-100 (polyethylene glycol octylphenol ether), 60 ml of co-emulsifier, n-hexanol, and 200 ml of the oil phase, cyclohexane, and 30 ml of the previously prepared solutions. The prepared reverse microemulsion (w/o) was stirred until it became homogeneous and stable. Next, 22 ml (25 wt%) of NH4OH (precipitating agent) with a mole ratio of 1.5:1 for NH4OH:Mn–Co–Ni–O was added drop by drop into w/o microemulsion under continues stirring for 4 h at ambient temperature to produce a precursor after reaction completion. The precursor was then centrifuged and rinsed with ethanol and DW for many times to eliminate unwanted substances or impurities. Lastly, this precursor was dried at 80 °C for 1 d, and then calcined for 1 h at various temperatures [127].
2.12 Thermal decomposition
Silver-doped nickel oxide (Ag:NiOx) NPs were fabricated by thermal decomposition approach [128]. Two samples were prepared; 0.52 g of nickel acetylacetonate was used in sample A, while in sample B the mass used was 1.04 g. Then, silver acetate (0.17 g) and nickel acetylacetonate (0.26 g) were added to diphenyl ether (10 ml). The prepared solution was refluxed for 2 h at 200 °C, and later cooled to ambient temperature. Methanol (30 ml) was added to that solution, centrifuged at 2500 rpm for 20 min, rinsed with methanol and centrifuged again. The produced NPs were dried in an oven overnight at 90 °C, annealed in a furnace at 350 °C for 3 h. In sample A, a product with a greenish color was formed after heating for 3 h. In addition, a black colored powder was formed after heating for 3 h in sample B [128].
Based on Thaver, Oseni, and Tessema study, Ag-doped NiOx was identified as an active layer dopant in order to enhance charge collection and power conversion efficiency in organic solar cells (OSCs). Silver is usually known for its efficiency in OSC, because of its excellent local surface plasmonic resonance (LSPR) properties in dielectric medium. Actually, the combined NiOx and Ag NC in the OSC photoactive layer was designed to modify the device performance substantially because of their optoelectronic characteristics. The results showed that the synthesized Ag-doped NiOx NCs had improved device performance and lower cost compared with the reference solar cells [128].
In another study, solid-state thermal decomposition method was employed for the fabrication of Ag and Ag2O NPs using sodium salicylate (Ag(Hsal)) as a new precursor [23]. A solution was formed by dissolving AgNO3 (2 mmol) in distilled water (40 mL). Also, Ag(Hsal) was dissolved in distilled water (40 mL) and was added to the previously prepared solution drop by drop under continuous stirring. A white precipitate was formed after stirring the prepared solution for 30 min, rinsed with distilled water and ethanol for many times to eliminate any impurities, and then dried at 70 °C in vacuum. This economical method was reported as a free solvent and surfactant in preparation steps which was the main advantage of this approach [23].
Conversely, NiO NPs were fabricated by the decomposition of nickel octanoate Ni(octa)2 as a new precursor. The fabrication was carried out in moderate conditions using oleylamine (C18H37N) and triphenylphosphine (C18H15P) [41]. Actually, Fereshteh et al. modified the old method for the fabrication of metals and oxides NPs which utilized the thermal decomposition of metal–surfactant complexes in hot surfactant solution. In that study, a reaction was occurred between Ni(octa)2 (0.4 g) and oleylamine (5 ml) to produce Ni(octa)2-oleylamine complex solution, which was heated to 145 °C for 1 h. The produced solution was introduced into triphenylphosphine (TPP) (5 g) at 240 °C for 45 min, and then was cooled to room temperature. The black powders were precipitated due to the addition of ethanol to that solution. The products were rinsed with ethanol repeatedly to eliminate unwanted substances, and then were dried at 100 °C. Then, that product was re-dispersed in nonpolar organic solvents, such as hexane or toluene [41].
Also, NiO NPs were fabricated by nickel acetate tetrahydrate (Ni(CH3COO)2.4H2O) and bis(acetylacetonato) nickel(II) complex (Ni(acac)2∙2H2O). Two solutions A and B were prepared; solution A by dissolving Ni(CH3COO)2.4H2O (0.025 mol, 6.22 g) in DW (25 mL), whereas, solution B by dissolving acetylacetone (0.05 mol) in DW (10 mL). An alkaline solution was prepared by dissolving sodium hydroxide or ammonium hydroxide (0.05 mol) in DW (15 mL), which was then added to the prepared mixture solution. Next, that mixture was refluxed for 5 h, cooled, and then kept in a refrigerator for many hours. The blue-green shiny solid was filtered off, was rinsed with water, and was dried at 80 °C overnight. The resulted complex was dried in a desiccator overnight at room temperature in the existence of P2O5. The dried complexes which were constructed previously utilizing NaOH and NH4OH were kept for next work as samples 2 and 3, respectively. Then, Ni(CH3COO)2.4H2O and the two prepared complexes (1.0 g sample) were placed in a crucible for calcination. After that, all three samples were heated in a furnace to 500 °C with a heating rate of 5 °C/min under air and kept at 500 °C for 3 h, and then cooled to ambient temperature to produce gray-green products [129].
In another study, NiO NPs were synthesized simply by calcination of a nickel precursor. Initially, NiCl2.6H2O (0.5 g) was dissolved in an aqueous solution of KOH (1 M, 5 ml), followed by the addition of oxalic acid (0.5 g) under continues stirring for 30 min. The mixture was dried completely at 100 °C for 3 h, then annealed at 600 °C in air atmosphere for 3 h, producing black products which were rinsed for many times with DW and ethanol, and then were dried overnight at 65 °C in oven [42].
In another research study, two organic sources, namely pomegranate peel extract and cochineal dye, were used for the fabrication of Ag NPs [130]. Ag nano-sized structures were successfully produced by using pomegranate peel extract, but they were produced with high agglomeration using cochineal dye. So, another simple solid-state method was used as for both natural precursors, which were produced well-dispersed NPs with narrow size distribution [130]. Three beakers were used to dissolve dried and powdered pomegranate peel (1, 2, and 3 g) in water (30 ml) under continuous stirring for 30 min, and then they were filtered off. another solution was prepared from dissolving AgNO3 (0.3 g) in water (10 ml), which was added separately to the three filtered solutions and stirred for 1 min at 80 °C. All solutions were heated for 3 h and cooled to room temperature producing three precipitates. Next, they were rinsed with distilled water and ethanol for many times and were dried in vacuum at 80 °C for 10 h [130]. This study showed a successful method for the fabrication of Ag NPs by a facile, inexpensive, and green approach [130].
In Adner et al. study, Ag NPs were also fabricated by thermal decomposition of Ag (I) 2-phenyl-3,6,9-trioxadecanoate in solutions of varied concentrations (1–10 mM) and temperatures (100–165 °C). Initially, Ag (I) 2-phenyl-3,6,9-trioxadecanoate (3) was synthesized through successive steps. Initially, a mixture of triethylamine (4.15 mL) and rac-2-phenyl-3,6,9-trioxadecanoic acid (7.62 g, 30 mmol) was gradually added to a solution of silver (I) nitrate (5.10 g, 30 mmol) in a mixture of acetonitrile (3.5 mL) and ethanol (50 mL). The mixture was stirred for 30 min at room temperature and then was heated to 75 °C, and last by the addition of acetonitrile and ethanol mixture till the solution became clear. It was cooled to −25 °C producing a solid precipitate which was filtered off, rinsed with ethanol and then dried. Next, Ag NPs were prepared through the following steps. A solution of prepared silver(I) 2-phenyl3,6,9-trioxadecanoate (3) (10 mL) in n hexadecylamine (1–10 mM) was heated to 75 °C, followed by heating to the reaction temperature (100–165 °C) with a pre-heated oil bath. The reaction time was determined (10 min to 24 h), and then, the solution mixture was cooled by air. The particles were separated by centrifugation after the addition of ethanol (10 mL), and cleaned by repeating dispersion in ethanol and centrifugal isolation [131]. Based on the results, aspherical and spherical Ag NPs were successfully synthesized by this method. Monodisperse aspherical particles with hexagonal contours and sizes of 7–10 nm were produced after short or long reaction times (i.e., 10 min or 1 day, respectively). Actually, spherical NPs were resulted after long reaction times [131].
In another research work, clean Ag NPs were produced through the thermal decomposition of aqueous Ag2O colloids at ambient temperature without any external reducing agent [132]. Ag NPs were fabricated via simple, clean, low-cost, safe and green chemistry route. The steps of preparation were summarized as: preparation of aqueous Ag2O colloids by mixing different aliquots of silver salts (AgNO3, AgClO4) and strong alkali (NaOH, KOH) solutions to produce silver ions. After successive steps Ag NPs were obtained [132].
Conversely, NiO NPs were fabricated by the formation of nickel linoleate and the subsequent thermal decomposition of the precursor. Metal complexes were prepared by dissolving Ni(NO3)2.6H2O (0.01 mol, 0.75 g) and linolenic acid (0.02 mol, 1.5 ml) in ethanol (25 ml) at 80 °C. After that, triethylamine (0.02 mol, 0.7 ml) was gradually added drop by drop and stirred till a green precipitate was formed. Then, it was filtered, was rinsed with ethanol, and was dried at 100 °C for 12 h. Lastly, nickel linoleate was produced and then was calcined in a furnace at 400 °C for 2 h in air. In fact, many advantages were reported for this green chemistry solvent method such as: simple, cost effective, time saving, and produce smaller NPs [133].
Also, NiO NPs were synthesized by a thermal decomposition of [Ni(HNA)2] as a precursor in the existence of C18H37N. There was no need for providing two surfactants for size control since the precursor used in that study had steric hindrance. This precursor was heated during preparation without using any surfactant in spite of the key role of surfactant in lowering the reaction temperature. Initially, the precursor complex (Ni(HNA)2) was prepared by following this procedure. A homogeneous solution was prepared by dissolving Ni(OOCCH3)2.H2O (2 mmol) into methanol (10 mL). Then, 2-hydroxo-1-naphthaldehyde was dissolved in methanol, which was added as drops to the previously prepared solution under continuous stirring. After that, it was refluxed in 60 °C for 2 h, and then, a green precipitate was centrifuged, was washed with ethanol for many times, and finally was dried at 70 °C [134].
Nickel NPs were also prepared by this facile method [135]. Typically, nickel acetylacetonate (0.256 g, 1 mmol) was dissolved in alkylamine (11.2 mmols) and then heated to 100 °C under the existence of N2. In a different container, trioctylphosphine (2.5 mL, 5.6 mmol) was degassed briefly under N2 at 100 °C and then was heated to 220 °C for 10 min. A combination of nickel acetylacetonate and alkylamine was introduced into the hot trioctylphosphine and was heated at 220 °C for 30 min under N2. A change in the mixture color from blue to black was an evidence about the formation of Ni NPs. Then, the mixture was left for cooling to the ambient temperature. To precipitate Ni NPs, anhydrous ethanol (30 mL) was added to the mixture solution. Then, this solution was centrifuged at 8000 rpm for 30 min, the black solid was eliminated from solution, and was re-dispersed in hexane. Again, ethanol was added to precipitate NPs and then it was centrifuged. Finally, a black solid was produced due to centrifugation and was re-dispersed in hexane [135].
2.13 Chemical wet synthesis
The chemical wet synthesis has been used to prepare free NPs. Low-temperature borohydride reduction, high-temperature oleylamine, and high-pressure method in an autoclave are classified as chemical wet approaches. The first two methods were produced free NPs included in a stabilizing layer of organic molecules, which prevents sintering during heat treatment of NPs [136].
In the chemical wet approaches, a reduction occurred for metal precursors in the appropriate solvents. Nickel sulfide NPs (Ni9S8) were prepared by chemical precipitation route using tri ethanol ammine (TEA) as a capping agent. Nickel acetate (1 M, 10 ml) and tri ethanol ammine (0.1 M, 2 ml) were added into a beaker to create a solution. Na2S (1 M, 10 ml) was added in drops to the prepared solution and was stirred continuously. A precipitate was obtained, centrifuged, washed for many times with water and acetone and then was dried in air to produce nickel sulfide in nano-powder form. The samples were annealed at 700 °C to produce NiO NPs [51].
Also, NiO nanosheets were synthesized by Ahmad et al. study. Two solutions were formed by dissolving Ni(NO3)2·6H2O (0.6 mM) and NaOH (1.2 mM) in DW (60 mL). These solutions were mixed and were stirred for 30 min at 600 rpm. Next, the prepared mixture solution was poured into a Teflon-lined (100 mL) autoclave and was heated at 200 °C for 10 h. As the reaction completed, the resulted material was washed for many times with DW and centrifuged. Finally, the produced NiO nanosheets was dried at 60 °C in an oven and then was calcined at 450 °C in air for 1 h. Extra step for the surface modification of NiO nanosheets with Au NPs was done successfully which was required for sensing application such as hydrazine sensing [48].
In Raza et al. study, Ag NPs with different shapes and sizes were synthesized by solution-based chemical reduction approaches. In this method, AgNO3 was used as a precursor, tri-sodium citrate (TSC, Na3C6H5O7) and NaBH4 were used as reducing agents, and PVP was utilized as a stabilizing agent. In Raza et al. study, five different samples of Ag NPs were prepared by wet chemical reduction methods following the same procedure but with some modifications. All solutions were prepared in different volumes and concentrations. Typically, AgNO3 was utilized as a precursor material, Na3C6H5O7 was utilized as a reducing agent in one sample and NaBH4 was used for the other four samples. A solution of AgNO3 (1 mM, 50 mL) was prepared by dissolving AgNO3 into water, was heated to the boiling temperature, and stirred till fully dissolved. Then, Na3C6H5O7 (1%, 5 mL) aqueous solution was added in drops to the boiling AgNO3 solution till the reaction was completed under continuous stirring and refluxing condition. It was observed that the color was changed from transparent, to pale yellow, to bright yellow, and at the completion of the reaction it appeared in greenish yellow. Then, the solution was cooled at ambient temperature under stirring. The product could be known as citrate-stabilized Ag NPs due to the role of Na3C6H5O7 as a stabilizer [137].
The wet-chemical precipitation is widely studied because it is appropriate for the fabrication of metal oxide NPs. Many properties were reported for the NPs, which were fabricated by this approach such as: high specific surface area, controlled micro structure, narrow pore size, homogeneous and pure products, and uniform particle distribution. Additionally, many advantages were introduced in the literature such as: low-temperature processing, high yield, and cost effective. This technique was found to be the appropriate method for the fabrication of NPs since it allowed a molecular-level mixing, and worked on raw substances and precursors at relatively low temperature to obtain nano-structured bulk, powder, and thin films [138]. In Ponnusamy et al. study, NiO NPs were fabricated by wet-chemical approach. Initially, Ni(NO3)2·6H2O (0.1 M, 100 mL) aqueous solution was formed, and then, NaOH (0.1 M, 100 mL) was added as drops to the prepared solution above under continuous stirring for 2 h at ambient temperature till a precipitate was formed. This precipitate was centrifuged (3000 rpm) for 5 min and rinsed with distilled water for many times for removing Na ions. Next, it was dried for 12 h and then annealed at 350 °C for 2 h. The obtained dried sample was crushed to produce the final product of undoped NiO NPs [138].
In another study, wet chemical route was used for the fabrication of Ag2O NPs [65]. Two different procedures were followed to determine the effect of pH value on the synthesized NPs. First, the pH value of the solution mixture was varied according to the reaction. Second, the pH value of the solution mixture was set between 9.8 and 10 by adding NaOH solution to keep the pH value constant till the reaction was completed. In route 1, polyethylene glycol (PEG) (20 g) was dissolved in reverse osmosis (RO) water (1 L), was heated to 50 °C under continuous stirring for 1 h till dissolving all PEG, and then forming a homogeneous solution. The obtained aqueous PEG solution was filtered to eliminate any impurities. After that, another solution was prepared using AgNO3 (0.5 g), which was added to the prepared PEG solution at 50 °C under stirring for 1 h till the reaction was completed. Then, the solution was filtered, was rinsed with RO water and then with ethanol for many times, and then was dried in oven at 60 °C overnight. Similarly, all steps were repeated in route 2 except the adjustment of pH value after the addition of AgNO3 solution. The pH of the solution mixture was controlled by the slow addition of NaOH solution (0.1 M) till the pH value became in range of 9.8–10. Based on the results, adjustment of pH was found to be necessary to synthesize Ag2O NPs with high purity and crystallinity. Otherwise, some impurities such as silver carbonate would be produced with NPs if the pH was not adjusted during preparation. Also, the average particle size was smaller in route 2 compared with that reported in route 1, which indicated that adjustment of the pH value is highly required to synthesize Ag2O NPs with smaller particle size [65].
2.13.1 Low-temperature reduction
In this process, NaBH4 or KBH4 could be used as a reducing agent at low temperatures below 0 °C. The solvents were chosen to be polar molecules such as: water, alcohols, polyglycols, whereas both inorganic and organic salts could be utilized as metal precursors. For example, Ag2SO4 could be employed as a precursor for Ag and KBH4 as a reducing agent at 0 °C in water–methanol mixture for the synthesis of Ag–Cu. Also, CuSO4 and PEG with L-(+)-ascorbic acid could be used as a capping agent and KBH4 as a reducing agent in water under Ar atmosphere for the fabrication of Cu NPs. Then, these samples had been rinsed with methanol and separated by evaporating the solvent under vacuum [136].
In another research work, pure metallic Ni NPs were fabricated via the chemical reduction of nickel chloride hexahydrate (NiCl2.6H2O) using hydrazine at ambient temperature without any prevention against surfactant and inert gas [28]. Initially, NiCl2.6H2O was dissolved in absolute ethanol to form solution A. Another mixture was prepared via mixing potassium hydroxide and hydrazine monohydrate to form mixture B. Next, solution A was poured in solution mixture B under continuous stirring at room temperature till the reaction was completed after 2 h. The resulted sample was rinsed with DW and then with acetone to remove any impurities. After that, the black particles were soaked in acetone in a closed container for further characterizations [28].
2.13.2 High-temperature reduction
In this method, oleylamine could be used as a solvent, reduction agent, and stabilizing ligand. The conditions required for this approach were reported as: The availability of a vacuum/inert gas, and reactions must be operated at high temperatures (200–240 °C). In fact, oleylamine is known as a hazardous substance which causes critical skin burns and eye damage. The metal precursor should be chosen correctly due to the variations in metal electrochemical potentials. Initially, a precursor solution was injected into a hot solvent mixture. The oleylamine fabrication had been utilized for the preparation of Ag–Cu NPs. Silver acetate and copper acetylacetonate could be used as precursors which were dissolved in oleylamine, and directly injected into a mixture of oleylamine and l-octadecene at 230 °C under N2 atmosphere. Then, the resulted sample was precipitated with acetone, separated by centrifugation, and purified. A colloidal solution of the Ag–Cu NPs in hexane was produced. This procedure could be developed by choosing another Ag precursor such as: [Ag(NH2C12H25)2]NO3, silver oleate Ag(Olea), or silver malonate Ag2(Mal). The Cu precursor also could be replaced by copper oleate Cu(Olea)2. Therefore, the use of a suitable precursor can affect the structure of the core of Ag–Cu NPs (core/shell, random alloy) [136].
For the fabrication of Au-Ni, the chosen precursors were tetrachloroauric acid (HAuCl4) and nickel acetylacetonate that were dissolved in oleylamine to produce a mixture solution. This prepared solution was inserted into another mixture of oleylamine and l-octadecene under N2 atmosphere. Conversely, nickel acetylacetonate and copper acetylacetonate were the chosen precursors for the synthesis of Cu-Ni, which were dissolved in oleylamine. A solution was produced which was inserted into a hot mixture of solvents l-octadecene and oleylamine at 230 °C. The product was a colloidal solution of the Cu-Ni NPs in hexane. Additionally, Ag–Ni NPs could be fabricated using AgNO3 and nickel acetylacetonate. Generally, the product from the oleylamine fabrication is usually a colloidal solution of the alloy NPs in a non-polar solvent to prevent any oxidation for the metal NPs. Also, the produced NPs are generally spherical in shape [136].
2.13.3 High-pressure autoclave method
High-pressure autoclave is not mostly employed for the fabrication of metal NPs although it has been utilized for the fabrication of pure metals NPs, especially Au and Ag [136]. The selected metal precursors were dissolved in N,N-dimethylformamide solvent (DMF) having a reducing ability. In a closed autoclave equipped with an inert Teflon liner, the pressure was increased with heat which derived the chemical reaction. In this method, no need to work with inert gases and vacuum which is an advantage [136].
2.14 Thermal treatment method
Silver NPs were prepared using AgNO3, PVP and DW as a metal precursor, a capping agent, and a solvent, respectively. Many aqueous solutions were formed by dissolving concentrations of PVP varied from 2 to 4% in DW (100 ml) at ambient temperature. After that, AgNO3 (50 mg) was added to all PVP solutions separately, and was stirred for 3 h till totally dissolved. Each produced solution was poured into glass Petri dishes and then dried in an oven at 80 °C for 1 d. The obtained solid was grounded to produce a powder, which was calcined at 600 °C for 3 h in O2 and N2 flow to obtain Ag NPs [139].
In another research work, NiO NPs were synthesized by simple heat treatment method. A polymer solution was formed by dissolving PVP (4 g) in DW (100 ml). Then, Ni(NO3)2.6H2O (0.2 mmol) was added to the polymer solution, was stirred, was poured in a petri dish, and then was dried in oven for 1 d at 80 °C. The dried product was grounded and divided into fifths to be calcined separately at different temperatures from 500 to 800 °C for 3 h. Organic matter was removed, and NiO NPs were successfully formed [39].
Also, NiO NPs were fabricated by heat treatment of a new precursor namely nickel octanoate Ni(octa)2. Also, NaOH (5 g) was dissolved in DW (5 mL) and absolute ethanol (10 mL), and then, this solution was injected into octanoic acid (2.5 g), and was stirred for 2 h. Then, DW (15 mL) was added to the previously produced solution, and was placed on ice bath. Another solution was formed by adding NaCl (15 g) to DW (100 mL) which was added to the solution that placed on ice bath. A white precipitate was created after 10 min, was filtrated, was rinsed by cool DW, and then was dried. Actually, the white precipitate is sodium octanoate (Na(octa)) which was dissolved in DW. Then, nickel(II) nitrate was dissolved in DW and was added to the prepared solution to produce a deep green precipitate after refluxing for 2 h. This precipitate was gathered via filtration and rinsed with DW. Next, the solid was re-precipitated due to the addition of MeOH and then was dried in an oven in 50 °C for 5 h. The as-prepared Ni(octa)2 precursor was then calcined at 300–900 °C in a furnace for 4 h to produce NiO NPs. In parallel, pure Ni NPs were also fabricated by calcining Ni(octa)2 precursor under Ar atmosphere at 350–800 °C for 4 h [140].
In another study, a facile thermal reduction route was employed for the fabrication of high-purity reduced graphene oxide/silver (rGO/Ag) NCs. Silver acetate (AgCH3O2) and GO were utilized as a silver precursor and a substrate for Ag NPs, respectively. This NCs were synthesized via grinding GO and Ag precursor and were directly heated at 1000 °C in a furnace for 20 s at usual conditions. The simplified Hummer’s method was used for preparing GO from natural graphite. A certain amount (100 mg) of both silver precursor and dry GO was grounded to produce powder, which was left in a preheated furnace at 1000 °C for 20 s at an ambient conditions to produce a black powder of rGO/Ag [141].
Additionally, a modified thermal treatment using alternate O2 and N2 flow was utilized to fabricate pure Ag NPs. Many advantages were reported for the thermal treatment method such as: simplicity, low cost, eco-friendly since no toxic and unwanted products were produced, and no extra substances were needed in the reaction. Initially, PVP solution was formed by dissolving PVP (2 g) in DW (100 mL), followed by adding AgNO3 (50 mg) under continuous stirring for 3 h to obtain a colorless and transparent solution with no precipitates. Next, it was poured into a glass Petri dish, was heated in an oven at 80 °C for 1 d producing a solid which was then grounded to produce a powder, which was calcined at 400–800 °C by 100 °C step, initially in O2 flow for 3 h to remove organic compounds, and then in N2 gas flow for other 3 h to remove oxygen from oxides to form pure Ag NPs [142].
2.15 Laser-liquid–solid interaction technique
Metastable Ag–Ni alloys were synthesized by laser irradiation of a metallic substrate immersed in a solution made by dissolving nitrate or acetate precursors of Ag and Ni in ethylene glycol, diethylene glycol, and 2-ethoxyethanol. The liquid was completely mixed and then cooled using a cooling coil to keep the temperature at 25 °C. Additionally, the reactor was usually purged with N2 to avoid oxidation of the organic and powders. The precursor solution was irradiated by a continuous wave CO2 laser with powers (150–300) W, and interaction time (1–3) minutes. The metallic disk was rotated at 5–100 rpm, and it is required to minimize the formation of vortex. The rotation of the substrate caused regular cooling of the irradiated spot, re-dispersion of the prepared particles into solution, and particle agglomeration was avoided by repeated interaction with laser beam. The particles were separated by centrifugation for 15 min, rinsed with distilled water for another 15 min, and then dispersed in ethanol using an ultrasonic bath, and finally allowed to settle overnight [143, 144].
3 Structural, compositional, and morphological properties
3.1 Structural and morphological properties
X-ray diffraction (XRD) is utilized to study the structural properties of fabricated NPs. Nagamuthu and Ryu studied the XRD pattern of Ag/NiO NPs, which was prepared by the hydrothermal method (Fig. 1a). A cubic crystal structure was confirmed for NiO and metallic Ag. The peaks of NiO were found at 37.22°, 43.254°, 62.83°, and 75.35, while the metallic Ag peaks were formed at 38.26°, 44.47°, 64.71°, 77.74°, and 81.91° [109]. Also, Iqbal and Kriek investigated the effect of Ag on NiO crystal structure by preparing AgxNi1−xO NCs (Fig. 1b). The peaks of NiO were appeared at 39.00°, 43.50°, 50.05° and 74.00° that showed a face centered cubic (fcc) crystalline structure. The peaks of Ag were found at 44.5°, 52.0° and 77.0°, which confirmed the foundation of cubic crystalline silver. Based on their study, the crystallinity of synthesized compounds was decreased due to the increase in the silver content. The average crystallite sizes were evaluated as: 9.60, 34.4, 34.3 and 28.4 nm for NiO, Ag 0.2Ni 0.8O, Ag 0.4Ni 0.6O and Ag 0.6Ni 0.4O NCs, respectively [101].

Senapati et al. studied the diffraction patterns of the Ni/Ag core–shell nanostructures. The peaks of fcc Ni (JCPDS card no 04-0850) appeared at 52.17°, 61.015°, 91.76°, 114.93° and 123.48°, respectively. No diffraction peaks of NiO or hydroxide were found, which approved the successful fabrication of pure Ni. Additional peaks of fcc Ag (JCPDS 040,783) were observed at 44.56°, 76.81°, 93.23°, and 98.62°, respectively [121]. Also, XRD patterns of Ni, Ag, and Ni-Ag core–shell NPs were studied by Jing et al. The peaks of fcc Ag and Ni were clearly observed at 38.2°, 44.5°, 51.8°, 64.6°, and 77.6°, respectively. Since no impurities were appeared in the XRD patterns, the purely synthesized Ni-Ag core–shell NPs were consisted of Ni and Ag [99].
In another study, XRD patterns were identified for Ni-Ag NPs at varied compositions started from pure Ni to 50 wt% Ni. Peaks referred to Ni and Ag were matched fcc for both Ni (PDF card No. 00-004-0850) and Ag (PDF card No. 00-001-1167), respectively. Small NiO peaks were observed in nickel-rich samples because of the slight oxidation occurred to Ni NPs when they were exposed to humid air. However, oxide peaks disappeared when Ag was added. It was found that Ag content was affected the XRD pattern because it caused a stronger scattering of X-rays compared with Ni, even though in samples with small Ag content such as 20 wt%. Also, Ag peaks were dominated in the XRD patterns especially at high concentrations of Ag. For example, the major Ni (1 1 1) peak position was overlapped Ag (2 0 0) peak position. The XRD patterns of Ni NP were found to be comparable with XRD of pure Ag. Actually, these patterns implied that the bimetallic NPs consisted of two phases since Ag and Ni were not mixed, but with significant Ni content in Ag-rich phase and Ag content in the Ni-rich phase [95].
Other researchers synthesized a coaxial NC that composed of Ag NW network core and Ni(OH)2 shell. The XRD pattern and the crystal structure of Ag NW network and Ag NW/Ni(OH)2 NCs were determined. Two diffraction peaks were found for Ag NWs at 38.1° and 44.4° (JCPDS card no. 04-0783), whereas the peaks of Ag NW/Ni(OH)2 NCs (hexagonal structure) were observed at 38.2°, 52.1° and 64.4° (JCPDS card no. 74-2075), respectively. It was found that Ag NW/Ni(OH)2 NC composed only of pure Ag and b-Ni(OH)2 [91]. In another study, Ag nanoflowers were synthesized and then the crystal phase was recognized by XRD. All diffraction peaks were identified according to the standard values of cubic Ag (JCPDS No. 65-2871). Also, they were indexed to the metallic Cu substrate. Moreover, no peaks for any other impurities were appeared [92].
In the XRD pattern of NiO NPs (Fig. 2), the peaks were appeared at 2θ values of 37.05°,43.17°, 62.44°, 75.22°, and 79.25°, which were indexed to (111), (200), (220), (311), and (222) planes, respectively, of cubic NiO structure [32,33,34,35,36]. The crystallite size of NiO NPs was reported in the literature as: 12.54 nm [36], 6.5 nm [31]. Conversely, five peaks of metallic Ag fcc structure were observed at 2θ values of 38.1, 44.2, 64.5, 77.34, and 81.38, respectively, which were indexed to (111), (200), (220), (311), and (222), respectively [104]. The crystallite sizes of Ag NPs were 6, 19 and 22 nm [104]. The XRD results in another study indicated that pure cubic Ag and hexagonal phase Ag2O were formed after annealing at 400 °C for 3 h in Ar gas and air, respectively [23].

XRD pattern of NiO NPs [35]
In several studies, high-resolution TEM (HRTEM) was employed to study the morphology and shape for synthesized silver nickel oxide nanostructures. Nagamuthu and Ryu used HRTEM to confirm the honeycomb Ag/NiO nanoarrays structures. Many advantages were reported for this nanoarray: large contact area, higher amount of active material in a unit area, and high porosity at the electrode/electrolyte interface which was good for supercapacitor applications [109]. Another TEM image from another study showed Ag NPs, which scraped from Cu substrate (Fig. 3a). Ag NPs were produced in spherical shapes with diameters range of 100 ~ 200 nm. Another HRTEM image of an individual Ag NP was with a lattice spacing of 0.236 nm (Fig. 3b). An image after the deposition of NiO layer using RF-sputtering is shown in Fig. 3c. A clear difference between the shell and core appeared in the image which indicated the formation of the core–shell structure [93].

a TEM image, b HRTEM image of Ag NP scraped from Cu substrate, and c TEM image of Ag-NiO core–shell NPs [93]
In Senapati et al. study, TEM and HRTEM analyses were done for Ni/Ag core–shell nanostructure. The TEM proved the production of a network structure of interconnecting Ni NWs of diameters 200–500 nm. Also, the surfaces of the NWs were consisted of nanocones with diameters range of 80–120 nm. Conversely, the concentric ring in the investigated area electron diffraction pattern of Ni indicated that fcc structure was formed. Additionally, the spacing of the equispaced lattice fringes was found to be 0.203 nm by HRTEM analysis, which were connected with (111) plane of fcc Ni. Finally, TEM image of the Ni/Ag nanostructures confirmed that Ag particles (diameter ~30 nm) were deposited on the surface of the prickly Ni wire [121]. In Jing et al. study, TEM analysis was performed for investigation of Ni-Ag core–shell NPs that were fabricated at varied concentrations of AgNO3. Based on the analysis, it was found that Ni-Ag core–shell NPs were spherical in shapes and Ag wholly covered the surfaces of Ni cores. HRTEM image of a Ni-Ag core–shell nanocrystal showed that the spacing of Ag fringes was 0.236 nm, which was related to {100} for fcc Ag [99]. In Mohammadi et al. study, particle sizes based on the TEM analysis ranged from 4 to 5 nm. In the TEM image of Ni-Ag 50:50 wt%, some agglomeration appeared because of the nature of gas phase methods and the residence time needed for NPs to reach the electrostatic precipitator during preparation of TEM sample [95]. In Yuksel et al. study, HRTEM showed that Ag NW networks were covered by Ni(OH)2 layer. Also, Ni(OH)2 consisted of nanocrystals with sizes smaller than 20 nm [91].
Additionally, TEM and HRTEM analyses were used to characterize core–shell nanoflower arrays samples. The TEM image of the edge section of an individual Ag nanoplate was flat and thin, whereas clear and continuous fringes were found by HRTEM. The spacing of lattice fringes was found to be 0.236 and 0.204 nm, which were assigned to the interplanar spacings of {111} and {200} planes of Ag, respectively [92]. Also, Alexandrova et al. determined the size and shape of Ag NPs by TEM analysis. It was found that Ag NPs were formed in spherical shape with a particle size of 3–20 nm, which were formed by microwave hydrothermal technique [107]. Similarly, other studies confirmed that Ag NPs were formed spherical shape [76, 145], but the average particle size was reported as: 86 nm [145], and ~14 nm [76].
In another study, NiO NPs were appeared in nearly a spherical shape [32]. Conversely, Ni nanorods with a Ni–O shell structure were obtained in Kaviyarasu et al. study. The shape of the particles was varied from nearly spherical to cylindrical, ellipsoidal, hexagonal and polyhedral [34]. The particle size of NiO was determined in different studies as: 5 nm [31, 34], 28 nm [36], 50 nm [32], ~10–100 nm [33].
SEM images were employed for studying the surface and composition of different silver nickel oxide nanostructures. Zhao et al. introduced the SEM images of Ag NPs on Cu substrate before and after the coating of NiO layer (Fig. 4a). The Ag NPs were nearly spherical in shape and were distributed uniformly on Cu substrate. Also, the size of Ag NPs was in range of 100~200 nm which were increased to 200~300 nm (Fig. 4b) due to the deposition of NiO layer [93]. Iqbal and Kriek also studied the surface morphology of AgxNi1−xO as shown in Fig. 5. SEM analysis showed that the surface of NiO was non-uniform distributed like cotton which indicated the NiO particles were aggregated (Fig. 5a). The accumulation of grains caused many small particles with various sizes to form with high surface energy that was being applicable in higher catalytic activity. The surface morphology of AgxNi1−xO (e.g., x = 0.2; Fig. 5b) indicated more porous structure as well as agglomeration of particles, mainly at lower concentrations of Ag content, with the SEM image of Ag0.2Ni0.8O being representative of all fabricated composites [101].

SEM image of the a Ag NPs arrays on Cu substrate, and b Ag-NiO NPs arrays on Cu substrate [93]

SEM images for a pure NiO, and b Ag0.2Ni0.8O [101]
SEM images were also employed in Senapati et al. study to the growth of prickly Ni NWs. It was found that spherical particles were produced at 30 min, which were transformed into wires after 1 h. If the reaction time was expanded to 1.5 h, Ni would undergo full transition to a wire-like shape. They were also studied the production of wire-like morphology at lower magnification with diameters that ranged between 300 and 600 nm and lengths between 25 and 40 μm by FESEM. Conversely, the higher-magnification image indicated the creation of spiky surfaces of Ni wires. On their surface, smaller Ag particles with sizes ranging from 16 to 30 nm were seen to deposit. [121]. On the other hand, SEM image of the Ni NPs revealed that the Ni NPs were formed in spherical shape with a diameter (mean value) of 104 ± 15 nm [99]. In the same study, different amounts of Ag(NO)3 were added to the reaction to adjust the thickness of Ag nanoshells in the Ni-Ag core–shell NPs [99]. Also, Yuksel et al. used SEM images to determine variations occurred in thickness of Ag NW/Ni(OH)2 NCs during the electrodeposition process. In their study, they were used 1600 s as the maximum deposition time. It was found that using thicker Ni(OH)2 layers induced fractures in Ag NW networks [91].
Another SEM analysis for the as-synthesized Ag nanoflower arrays on the Cu substrate was introduced by another study. They found that Ag nanoflower consisted of nanoplates that were grown vertically to the substrate. The nanoplates’ width ranged from hundreds of nanometers to many micrometers, whereas the thickness was in tens of nanometers [92]. In another study, Ag NPs were prepared by combined green and hydrothermal methods using red cabbage extract that was analyzed by SEM which showed that these NPs were of spherical shape but with different particle sizes ~30, ~45, ~65, ~80, and ~100 nm [110]. Also, spherical shape (Fig. 6a) was confirmed by SEM analysis for Ag NPs in Shahjahan study [104]. SEM images were also performed for Ag2O and Ag NPs, which indicated that the morphology of the Ag2O and Ag was particle-like with a particle size ranged between ~40 and 50 nm [23]. Conversely, the surface morphology of NiO NPs appeared in the form of linear flakes, as shown by FESEM [36], and nanorods as shown in Fig. 6b [34].

Also, EDX analysis determined the elemental composition of the synthesized NPs in different research studies. It was used for confirming the existence of impurities or not in the prepared samples. In Iqbal and Kriek study, EDX analysis was employed to find the stoichiometry of AgxNi1−xO NPs. The results revealed that Ni and Ag were distributed well in the scanned area besides a considerable amount of oxygen [101]. In another reported study, silver-nickel oxide core–shell NPs were analyzed by EDX. From the analysis, Cu and Ag elements were appeared, which originated from Ag NPs and Cu substrate, respectively. Then, EDX patterns of that sample were investigated after RF-sputtering deposition. Actually, all diffraction peaks were correlated with Ag and Ni besides Cu substrate, which were resulted from Ag NPs and NiO layer that confirmed the fabrication of Ag-NiO core–shell NPs [93]. In another study, EDX analysis revealed that the prepared silver-doped nickel oxide (Ag:NiOx) NCs which was prepared by a thermal decomposition method contained traces of carbon. The reason might be due to the contamination occurring on the samples because they were not rinsed before heating in a furnace [128].
EDX spectrum of Ni NW, in Senapati et al. study, showed Ni peaks besides a small peak of oxygen in very low intensity. Also, the spectrum of Ni/Ag core–shell nanostructures confirmed the existence of Ni and Ag. It was found that the magnetic field would not able to separate Ag from Ni, which proved the production of a combined state [121]. The EDX spectrum of the Ag NPs nanocellulose NCs is shown in Fig. 7. The results approved the presence of metallic Ag in the prepared NCs by Xuewei Zhang et al. study [145].

EDX spectrum of the NCs [145]
XPS analysis was utilized in different studies to determine the oxidation valence states and the binding energy. Nagamuthu and Ryu determined the valance state of Ni, Ag, and O in their study. The existence of the material and its oxidation valance states (Ni 2p, Ag 3d, and O 1 s), and the core level spectrum of Ni 2p (Ni2+) were confirmed as seen in Fig. 8a and b, respectively. Also, three binding energies at 854.1, 855.4, and 861.5 eV were related to Ni 2p3/2 state and satellite peaks, respectively. The binding energy for the local screening from lattice oxygen to Ni 2p core hole, the nonlocal screening from the lattice despite it has been reported to contain a partial contribution from surface states, Ni 2p1/2 state which was detected from Ni–O species, and its corresponding satellite peak, were all determined as 854.1, 855.4, 874.1, and 879.6 eV, respectively. A high-resolution spectrum (Fig. 8c) shows two peaks at 368.3 and 374.3 eV, which were corresponded with Ag 3d3/2 and Ag 3d5/2, respectively. From the results, it was confirmed that metallic Ag was existed in the Ag/NiO honeycomb nanoarrays. Also, three peaks (529.4, 531.2 and 532.5 eV) were detected at the high-resolution spectrum of oxygen (Fig. 8d), which were related to lattice oxygen (O2−), adsorbed water and hydroxyl groups (OH) [109].

a Survey scan of Ag/NiO, b spectrum of Ni 2 P, c spectrum of Ag 3d, and d spectrum of O1s for Ag/NiO nanoarrays
In Yuksel et al. study, XPS measurements were employed for Ag NW/Ni(OH)2 NC. Different signals were appeared from C, O, Ni and Ag species in the XPS spectrum. Two major peaks with binding energies at 854.0 and 871.6 eV appeared due to Ni 2p3/2 and Ni 2p1/2, respectively. Also, two satellite peaks were produced from Ni 2p3/2 and Ni 2p1/2 at core level spectrum [91]. In Kaviyarasu et al. study, XPS spectrum for NiO NPs showed Ni2P3/2 peak at 855.1 eV and Ni2P1/2 883.5 eV, which confirmed the existence of NiO for samples annealed at 450 °C. The peak at a binding energy of 531.54 eV was correlated with O1s peak of NiO, which was recorded for thin film [34].
FTIR analysis is also employed to investigate the optical characteristics of the synthesized NPs. In Nagamuthu and Ryu study, FTIR spectrum was determined for Ag/NiO nanoarrays by Nagamuthu and Ryu. It ranged from 400 to 4000 cm−1 (Fig. 9). Three peaks were found in the spectrum: Ni–O stretching vibrations (475 and 568 cm−1), N–H wagging mode (953 cm−1), and CH2 stretching modes (1422 cm−1) [109]. In Mohammadi et al. study, the results of 50:50 wt% Ni-Ag NPs were displaced for FNPs and compared with bare nanopowders (BNPs). Three additional sharp peaks were found in the FNPs compared with the BNPs: N─H bending near 1500–1600 cm−1, N═C═O stretching near 2250–2350 cm−1, and C─H alkyl stretching near 2800–2900 cm−1. The existence of octylamine was observed on the surface of the NPs, since its spectrum contained N─H and C─H-related peaks. For both prepared samples, a broad O─H stretching peak (1300–1400 cm−1) appeared which was correlated with adsorbed water, and a sharper O─H stretching peak that was featured with surface hydroxyl groups. These peaks basically appeared due to the nature of the synthesis procedure and the existence of byproduct in the reaction chamber such as water vapor [95].

FTIR spectra of Ag/NiO nanoarrays [109]
In Williams et al. study, FTIR spectra of the fabricated NiO NPs showed a broad absorption band at 3434 cm−1 which was recorded as O–H stretching vibrations and a weak band near 1632 cm−1 which was related to H–O–H bending modes of vibration. The reason for the appearance of these bands was due to the adsorption of water molecules on the surface of NiO NPs. The peak centered at 423 cm−1 was related to the stretching mode of Ni–O, which confirmed the formation of NiO NPs [36]. In another study, Ni–O phase formation was found in the area of 820–400 cm−1, which was related to Ni–O stretching vibration mode [34]. Conversely, FTIR analysis for Ag2O NPs was conducted by Archana et al. [76]. FTIR spectra showed the existence of functional group absorbance peaks at 3468 cm−1, 2842 cm−1, 1637 cm−1, 1586 cm−1, 1407 cm−1, 1355 cm−1, 766 cm−1 that were corresponded to ─OH, ─CH2, ─COOH, C─C, C═C, C─H, Ag─O groups, respectively, which was presented in phytochemicals [76]. FTIR spectra for Ag2O and Ag NPs showed that the functional group of the absorbance peaks were found at 3432 and 1638 cm−1 for (─OH) stretching and bending vibrations, and 524 cm−1 for (Ag─O) groups, respectively [23].
3.2 Optical properties
The optical properties of nickel silver oxide nanostructures were widely studied in the literature review. The UV–Vis analysis is usually operated to investigate the variation in the optical band gap of nanostructures that were exposed to heat treatment routes [146]. In Nagamuthu and Ryu study, the optical properties of Ag/NiO honeycomb nanoarrays were investigated using UV–Vis spectroscopy. Two maximum absorbance bands were found in the UV spectrum at 340 and 552 nm. So, the electronic transition from valance band to conduction band in the NiO nanoarrays semiconductor was confirmed [109]. In Jing et al. study, the UV–Vis analyses of pure Ni, pure Ag, and Ni-Ag core–shell NPs were done. The characteristic absorption peaks of the Ag colloids, Ni colloids, and Ni-Ag core–shell NPs were positioned at about 420, 302, and 318 nm, respectively (Fig. 10). They found that the intensity of absorption peak was increased as the Ag(NO)3 concentration was increased [99]. In another work, the optical measurements showed that a distinct band edge with surface plasmon resonance was ranged from 400 to 425 nm, and excited emission with maximum peak at 382 nm which was evocative for cluster-in-cluster surface enriched bimetallic silver-nickel solutions [115].

UV–visible absorption spectra of a–d Ni-Ag core–shell, e pure Ni, and f pure Ag NPs. The molar ratios of Ni to Ag were a 1.5, b 0.75, c 0.5, and d 0.375 [99]
The optical band gap of NiO was reported as 3.74 eV at 500 °C [36], 3.41 eV at 450 °C [34]. In another study, UV–Vis–NIR analysis revealed that NiO NPs (crystallite size 35 nm), which was annealed at 700 °C, had a direct band gap of 3.81 eV, whereas the black NiO (crystallite size 5 nm) that was annealed at 250 °C had the band gap of 3.41 eV [32]. The intense band gap also appeared, with some spectral tuning, giving a range of absorption energies from 2.60 to 3.41 eV [34]. It was found by another study that the band gap decreased as the NP size was increased, which was consistent with quantum confinement. The average particle size and the related band gap energy for NiO NPs were ranged from 3.9 eV for the smallest (9 nm) NPs to 3.65 eV for the largest (27.5 nm) NPs. This meant that the optical band gap was varied from 3.65 to 3.9 eV, as expected by the quantum confinement effect [33]. As a conclusion, optical properties of NiO NPs could be controlled by adjusting different parameters during synthesis of fabrication of these NPs which also tuned the average size of NiO NPs [33].
Conversely, the optical bang gap energy of Ag NPs was evaluated as: 3.5 eV [103], and 3.39 eV, which can be used in optical and optoelectronic devices [83]. In Alexandrova et al. study, the resulting colloidal solution of Ag NPs in 6-O-carboxymethyl-chitin (CMC) was investigated by UV–Vis technique. The spectrum showed a distinct intense absorption band at 410–420 nm, which was an evidence of the presence of Ag NPs [107]. In another study, the band gap of Ag2O NPs was calculated by Tauc’s equation as 2.1 eV [76]. In Iqbal et al. study, it was clearly observed from Ag2O NPs spectrum that the maximum wavelength of absorption (blue shift) was seen at 500 nm to 390 nm at a calcined temperature of 800 °C. The characteristic peak of Ag was found at 400 nm which was related to resonating oscillations by surface plasmon and interband transitions. The spherical shape of particles was confirmed by the single surface plasmon peak [64].
PL analysis was also performed for optical properties. Figure 11a and b represents the PL spectra for Ag2O and Ag NPs, respectively. The band gap of Ag2O and Ag NPs was evaluated as approximately 1.4 eV (885 nm) and 3.1 eV (400 nm), respectively [23].

PL spectra of a Ag2O and b Ag NPs at room temperature [23]
4 Applications of reviewed nanostructures
4.1 Applications of nickel oxide nanostructures
Nickel oxide NPs has various properties including optical, thermal, electrical, chemical and physical properties [45, 147]. NiO is a p-type semiconductor material, which has a band gap energy range of 3.6–4.0 eV that produces a high chemical stability [55, 56, 146, 147]. Besides, its electron transfer capability, electrocatalytic, and superconductive properties are properties that make it widely used in different applications [55, 148, 149]. Many applications of NiO NPs were recorded in different scientific publications: electrochemical performance including sensing applications [38, 46, 55, 150,151,152], Li-ion batteries [37, 42], catalysts for hydrogen evolution reaction [38], biosensors [43, 147], fabrication of hybrid perovskite solar cells [32], energy storage and memristor technologies [33, 148, 152], optoelectronics applications [34, 146, 152], photocatalytic degradation [56], antibacterial applications against a gram-positive and gram-negative pathogens [45, 55], and water treatment [55, 146].
4.2 Applications of silver oxide nanostructures
Among the NMs, Ag2O NPs are well-known materials that have been used almost as antimicrobial (antibacterial) agent [64, 68,69,70,71,72,73,74, 77, 153]. Other applications were reported in many research studies such as: dye degradation, and insecticidal activity [69], antifungal activity [70], photocatalytic agent for dye degradation [68, 71, 74], antibacterial activity against dental bacterial strains [73], anti-inflammatory, antioxidant, anti-diabetic [72], cytotoxic, insecticidal, phytotoxic, antioxidant, anthelmintic agents [153], anticancer agent [64, 77], sensors [23], methanol sensing applications [66], pharmaceutical products, cosmetic, food industry, water treatment, gas sensing, electronics, construction materials, paints industry [64], fuel cells, photovoltaic cells, all-optical switching devices, optical data storage systems and as a diagnostic biological probes [65, 75, 154].
4.3 Applications of nickel silver oxide nanostructures
Different applications and various uses of nickel silver oxide nanostructures were reported in the literature such as electrical [94, 99], electrochemical [91], super capacitor [109, 155], conductive inks [95], and antibacterial applications [121]. Also, they were used as electrocatalyst for the oxygen evolution reaction (OER) in alkaline medium [101], and utilized as anode materials for LIBs [92, 93]. Jing et al. determined the electrical properties of Ni-Ag core–shell NPs, which appeared after sintering at 650 °C. They found that the resistivity was 11 mΩ/square at that temperature. The resistivity was fixed for temperatures greater than 650 °C. Since the electrical properties of the Ni-Ag core–shell paste were similar to those of pure Ag paste, Ni-Ag NPs were partly used instead of pure Ag NPs in conductive pastes. Also, Ni-Ag core–shell NPs were employed as an electrode material in the conductive pastes [99]. Also, El-Nahrawy et al. studied the dielectric properties of CNW/PPy/AgNPs-Ni2O3 composite such as dielectric constant, AC conductivity, and dielectric loss. They found that the electrical conductivity was improved because of the insertion of PPy onto CNW composites. Also, AC conductivity was increased when the temperature was increased, which illustrated semiconducting activity [94].
Conversely, coaxial core–shell Ag NW/Ni(OH)2 NC electrodes were synthesized in Yuksel et al. study. They found that coating with Ni(OH)2 facilitated the efficient charge to move to the inner NW network. They investigated the electrochemical properties by determining the specific capacitance and current density as 1165.2 F g−1, and 3 A g−1, respectively. As a result, ion and electron were transported easily, and electrochemical properties were enhanced due to the synergy of highly conductive Ag NWs and highly capacitive Ni(OH)2. It was found that after 3000 cycles, synthesized NC electrodes showed 93% capacity retention. The fabricated geometry was reported to be useful due to the potential of NW-based coaxial energy storage devices [91]. Also, Nagamuthu and Ryu found that Ag/NiO nanoarrays were used as an electrode material for supercapacitor applications. Also, they found that the electronic conductivity of NiO nanoarrays was enhanced due to the existence of Ag. A specific capacity of 824 C g−1 at a specific current of 2.5 A g−1 was produced from these nanoarrays. In their study, an Ag/NiO positive electrode-based asymmetric device was synthesized and investigated. This device provided a high specific cell capacity of 204 C g−1 at a specific current of 2.5 A g−1, and a maximum energy density of 63.75 W h kg−1 at a power density of 2812.5 W kg−1. Actually, they reported that the values found from their study were higher than the values yielded by lead acid batteries and comparable to those of metal hydride batteries (NiMH) [109].
Other researchers studied silver nickel oxide NCs and investigated them as electrocatalyst for OER in alkaline medium. They observed that the addition of Ag had not enhanced the electrocatalytic activity. So the maximum OER activity was registered by using only pure NiO. They used an overpotential of 263 mV to deliver a current density of 10 mA cm−2 at 25 °C. Conversely, the existence of Ag was found to be a determinant agent for OER activity, while the presence of iron had a positive effect on the OER activity, which actually needs further investigations [101].
Ag-NiO core–shell was employed as anode materials for LIBs. The electrode showed improved capacity and cycling performance compared with the planar NiO electrode when it was tested as an anode [92, 93]. The in situ fabricated Ag NPs enhanced the interfacial strength between the active material and substrate. So, the electrical conductivity of that electrode was improved and hence its performance. The discharge capacity of Ag-NiO electrode was gradually decreased and then stabilized at about 500 mAh g−1 after 20 cycles. No capacity decay was noticed from 20 to 100 cycles. Comparatively, the planar NiO film suffered from continuous capacity decay, and was dropped to about 100 mAh g−1 after 50 cycles [93]. In another study, the Ag-NiO core–shell nanoflower arrays had a capacity of ~800 mAh g−1 at a current density of 215 mA g−1 after 100 cycles, and excellent rate capability (94% of the capacity retained from 0.3 to 5C), which was much better than planar electrode. The performance was improved the conductivity during charging and discharging, which was suggested to be tested to other anode materials of LIBs [92].
Mohammadi et al. introduced a good idea about the fabrication of conductive inks by employing a continuous one-step flame-based method. They worked on reducing the ink costs by using bimetallic conductive nano-inks without using an additional sintering step [95]. Since Ag NPs have been known for their antimicrobial properties [74], Ni/Ag nanostructures were also investigated by Senapati et al. [121] for their antibacterial properties and hence were used in appropriate applications. Actually, the insertion of Ag shell in Ni/Ag nanostructures considerably enhanced their antibacterial activity compared with a commercially available antibacterial agents [121]. The abbreviations meaning of this work have been mentioned in Table 1.
5 Conclusions
In this review article, we have successfully summarized various methods used for the synthesis of different nickel silver oxide nanostructures. Electrodeposition, electrochemical method, simple immersion technique and subsequent RF-sputtering deposition, chemical oxidative polymerization and subsequent acidic sol–gel, flame-based process, liquid-phase reduction technique, sol–gel, hydrothermal method, co-precipitation, simple precipitation, thermal decomposition, chemical wet synthesis, low- and high-temperature reduction, high-pressure autoclave, thermal treatment method, and laser-liquid–solid interaction technique are the methods that were reported in extensive research studies for the fabrication of nanostructures. In each study, characterization of the synthesized material was discussed in detail to determine their properties and applications. Nickel silver oxide nanostructures can be used in various applications such as: electrical, electrochemical, super capacitor, conductive inks, and antibacterial applications. Also, it can be used as electrocatalyst for OER in alkaline medium and utilized as anode materials for LIBs. Few scientific papers studied nickel silver oxide nanostructures despite their good properties and wide applications, so it is highly advised to fabricate novel binary and composite NMs containing nickel and silver oxides with enhanced properties. It is also recommended to select the suitable synthesis method for the fabrication of metal oxides NMs with these benefits: simple, fast, eco-friendly, purity of the product, and low-cost method such as thermal treatment method.
Change history
24 November 2021
A Correction to this paper has been published: https://doi.org/10.1007/s00339-021-05079-3
References
S. Bayda, M. Adeel, T. Tuccinardi, M. Cordani, F. Rizzolio, The history of nanoscience and nanotechnology: from chemical-physical applications to nanomedicine. Molecules 25, 1–15 (2020). https://doi.org/10.3390/molecules25010112
R. Landauer, Information Is Inevitably Physical, Feynman Computation (CRC Press, 2018), pp. 77–92. https://doi.org/10.1201/9780429500459
A.R. Wu, L. Yu, There’s plenty of room at the bottom of a cell. Chem. Eng. Prog. 113(10), 47–51 (2017)
P.I. Dolez, Nanomaterials Definitions, Classifications, and Applications (Elsevier, 2015). https://doi.org/10.1016/B978-0-444-62747-6.00001-4
I. Khan, K. Saeed, I. Khan, Nanoparticles: properties, applications and toxicities. Arab. J. Chem. 12, 908–931 (2019). https://doi.org/10.1016/j.arabjc.2017.05.011
J.N. Tiwari, R.N. Tiwari, K.S. Kim, Zero-dimensional, one-dimensional, two-dimensional and three-dimensional nanostructured materials for advanced electrochemical energy devices. Prog. Mater. Sci. 57, 724–803 (2012). https://doi.org/10.1016/j.pmatsci.2011.08.003
A.A. Alswata, M.B. Ahmad, N.M. Al-Hada, H.M. Kamari, M.Z.B. Hussein, N.A. Ibrahim, Preparation of zeolite/zinc oxide nanocomposites for toxic metals removal from water. Results Phys. 7, 723–731 (2017). https://doi.org/10.1016/j.rinp.2017.01.036
E. Absi, M.A. Saleh, N.M. Al-Hada, K. Hamzah, A.M. Alhawsawi, E.M. Banoqitah, Binary nickel and silver oxides by thermal route: preparation and characterization. Appl. Phys. A Mater. Sci. Process. 127, 1–11 (2021). https://doi.org/10.1007/s00339-021-04775-4
N.M. Al-Hada, A.M. Al-Ghaili, H. Kasim, M.A. Saleh, H. Baqiah, J. Liu, J. Wang, Nanofabrication of (Cr2O3)x (NiO)1–x and the impact of precursor concentrations on nanoparticles conduct. J. Mater. Res. Technol. 11, 252–263 (2021). https://doi.org/10.1016/j.jmrt.2021.01.007
A.A. Baqer, K.A. Matori, N.M. Al-Hada, A.H. Shaari, H.M. Kamari, E. Saion, J.L.Y. Chyi, C.A.C. Abdullah, Synthesis and characterization of binary (CuO)0.6(CeO2)0.4 nanoparticles via a simple heat treatment method. Results Phys. 9, 471–478 (2018). https://doi.org/10.1016/j.rinp.2018.02.079
N.M. Al-Hada, E. Saion, H.M. Kamari, M.H. Flaifel, A.H. Shaari, Z.A. Talib, N. Abdullahi, A.A. Baqer, A. Kharazmi, Structural, morphological and optical behaviour of PVP capped binary (ZnO)0.4 (CdO)0.6 nanoparticles synthesised by a facile thermal route. Mater. Sci. Semicond. Process. 53, 56–65 (2016). https://doi.org/10.1016/j.mssp.2016.06.004
N.M. Al-Hada, E.B. Saion, A.H. Shaari, M.A. Kamarudin, M.H. Flaifel, S.H. Ahmad, S.A. Gene, A facile thermal-treatment route to synthesize ZnO nanosheets and effect of calcination temperature. PLoS ONE 9, 2–10 (2014). https://doi.org/10.1371/journal.pone.0103134
N.M. Al-Hada, R.M. Kasmani, H. Kasim, A.M. Al-Ghaili, M.A. Saleh, E.M. Banoqitah, A.M. Alhawsawi, A.A. Baqer, J. Liu, S. Xu, Q. Li, A.M. Noorazlan, A.A.A. Ahmed, M.H. Flaifel, S. Paiman, N. Nazrin, B.A. Al-Asbahi, J. Wang, The effect of precursor concentration on the particle size, crystal size, and optical energy gap of CexSn1−xO2 nanofabrication. Nanomaterials 11, 2143 (2021). https://doi.org/10.3390/nano11082143
J. Chaudhary, G. Tailor, B.L. Yadav, O. Michael, Synthesis and biological function of nickel and copper nanoparticles. Heliyon 5, e01878 (2019). https://doi.org/10.1016/j.heliyon.2019.e01878
M.I. Alkhalaf, R.H. Hussein, A. Hamza, Green synthesis of silver nanoparticles by Nigella sativa extract alleviates diabetic neuropathy through anti-inflammatory and antioxidant effects, Saudi. J. Biol. Sci. 27, 2410–2419 (2020). https://doi.org/10.1016/j.sjbs.2020.05.005
R. Thiruchelvi, P. Jayashree, K. Mirunaalini, Synthesis of silver nanoparticle using marine red seaweed Gelidiella acerosa-a complete study on its biological activity and its characterisation. Mater. Today Proc. (2020). https://doi.org/10.1016/j.matpr.2020.07.242
S. Dangi, A. Gupta, D.K. Gupta, S. Singh, N. Parajuli, Green synthesis of silver nanoparticles using aqueous root extract of Berberis asiatica and evaluation of their antibacterial activity. Chem. Data Collect. (2020). https://doi.org/10.1016/j.cdc.2020.100411
D. Jini, S. Sharmila, Green synthesis of silver nanoparticles from Allium cepa and its in vitro antidiabetic activity. Mater. Today Proc. 22, 432–438 (2020). https://doi.org/10.1016/j.matpr.2019.07.672
P. Tamilarasi, P. Meena, Green synthesis of silver nanoparticles (Ag NPs) using Gomphrena globosa (Globe amaranth) leaf extract and their characterization. Mater. Today Proc. (2020). https://doi.org/10.1016/j.matpr.2020.04.025
A. Lunkov, B. Shagdarova, M. Konovalova, Y. Zhuikova, N. Drozd, A. Il’ina, V. Varlamov, Synthesis of silver nanoparticles using gallic acid-conjugated chitosan derivatives. Carbohydr. Polym. 234, 115916 (2020). https://doi.org/10.1016/j.carbpol.2020.115916
L. Sherin, A. Sohail, U.E.S. Amjad, M. Mustafa, R. Jabeen, A. Ul-Hamid, Facile green synthesis of silver nanoparticles using terminalia bellerica kernel extract for catalytic reduction of anthropogenic water pollutants. Coll. Interf. Sci. Commun. 37, 100276 (2020). https://doi.org/10.1016/j.colcom.2020.100276
S. Sawan, R. Maalouf, A. Errachid, N. Jaffrezic-Renault, Metal and metal oxide nanoparticles in the voltammetric detection of heavy metals: a review. TrAC Trends Anal. Chem. 131, 116014 (2020). https://doi.org/10.1016/j.trac.2020.116014
S.M. Hosseinpour-Mashkani, M. Ramezani, Silver and silver oxide nanoparticles: synthesis and characterization by thermal decomposition. Mater. Lett. 130, 259–262 (2014). https://doi.org/10.1016/j.matlet.2014.05.133
H. Chen, J. Wang, D. Huang, X. Chen, J. Zhu, D. Sun, J. Huang, Q. Li, Plant-mediated synthesis of size-controllable Ni nanoparticles with alfalfa extract. Mater. Lett. 122, 166–169 (2014). https://doi.org/10.1016/j.matlet.2014.02.028
M.I. Din, A.G. Nabi, A. Rani, A. Aihetasham, M. Mukhtar, Single step green synthesis of stable nickel and nickel oxide nanoparticles from calotropis gigantea: catalytic and antimicrobial potentials. Environ. Nanotechnol. Monit. Manag. 9, 29–36 (2018). https://doi.org/10.1016/j.enmm.2017.11.005
C. Zeng, C. Wang, F. Wang, Y. Zhang, L. Zhang, A novel vapor-liquid segmented flow based on solvent partial vaporization in microstructured reactor for continuous synthesis of nickel nanoparticles. Chem. Eng. J. 204–205, 48–53 (2012). https://doi.org/10.1016/j.cej.2012.07.096
L. Xu, C. Srinivasakannan, J. Peng, D. Zhang, G. Chen, Synthesis of nickel nanoparticles by aqueous reduction in continuous flow microreactor. Chem. Eng. Process. Process Intensif. 93, 44–49 (2015). https://doi.org/10.1016/j.cep.2015.04.010
Z.G. Wu, M. Munoz, O. Montero, The synthesis of nickel nanoparticles by hydrazine reduction. Adv. Powder Technol. 21, 165–168 (2010). https://doi.org/10.1016/j.apt.2009.10.012
S. Chemchoub, L. Oularbi, A. El Attar, S.A. Younssi, F. Bentiss, C. Jama, M. El Rhazi, Cost-effective non-noble metal supported on conducting polymer composite such as nickel nanoparticles/polypyrrole as efficient anode electrocatalyst for ethanol oxidation. Mater. Chem. Phys. 250, 123009 (2020). https://doi.org/10.1016/j.matchemphys.2020.123009
M.S. Alnarabiji, O. Tantawi, A. Ramli, N.A.M. Zabidi, O.B. Ghanem, B. Abdullah, Comprehensive review of structured binary Ni-NiO catalyst: synthesis, characterization and applications. Renew. Sustain. Energy Rev. 114, 109326 (2019). https://doi.org/10.1016/j.rser.2019.109326
M. Tadic, D. Nikolic, M. Panjan, G.R. Blake, Magnetic properties of NiO (nickel oxide) nanoparticles: blocking temperature and neel temperature. J. Alloys Compd. 647, 1061–1068 (2015). https://doi.org/10.1016/j.jallcom.2015.06.027
K. Deevi, V.S.R. Immareddy, Synthesis and characterization of optically transparent nickel oxide nanoparticles as a hole transport material for hybrid perovskite solar cells. J. Mater. Sci. Mater. Electron. 30, 6242–6248 (2019). https://doi.org/10.1007/s10854-019-00927-8
J.S. Fain, J.W. Mares, S.M. Weiss, Size-controlled nickel oxide nanoparticle synthesis using mesoporous silicon thin films. J. Nanoparticle Res. (2015). https://doi.org/10.1007/s11051-015-3122-2
K. Kaviyarasu, E. Manikandan, J. Kennedy, M. Jayachandran, R. Ladchumananandasiivam, U.U. De Gomes, M. Maaza, Synthesis and characterization studies of NiO nanorods for enhancing solar cell efficiency using photon upconversion materials. Ceram. Int. 42, 8385–8394 (2016). https://doi.org/10.1016/j.ceramint.2016.02.054
Adiba, V. Pandey, S. Munjal, T. Ahmad, Structural, morphological and magnetic properties of antiferromagnetic nickel oxide nanoparticles synthesized via sol-gel route. Mater. Today Proc. 26, 3116–3118 (2019). https://doi.org/10.1016/j.matpr.2020.02.643
L. Williams, A.R. Prasad, P. Sowmya, A. Joseph, Characterization and temperature dependent DC conductivity study of bio templated nickel oxide nanoparticles (NiO) and their composites using polyaniline (PANI). Mater. Chem. Phys. 242, 122469 (2020). https://doi.org/10.1016/j.matchemphys.2019.122469
J. Singh, S. Lee, S. Kim, S.P. Singh, J. Kim, A.K. Rai, Fabrication of 1D mesoporous NiO nano-rods as high capacity and long-life anode material for lithium ion batteries. J. Alloys Compd. 850, 156755 (2021). https://doi.org/10.1016/j.jallcom.2020.156755
V.E. Gurenko, V.I. Popkov, A.A. Lobinsky, Synthesis of NiO granular nanospheres as a novel material for high-performance supercapacitors. Mater. Lett. 279, 128478 (2020). https://doi.org/10.1016/j.matlet.2020.128478
M. Hashem, E. Saion, N.M. Al-Hada, H.M. Kamari, A.H. Shaari, Z.A. Talib, S.B. Paiman, M.A. Kamarudeen, Fabrication and characterization of semiconductor nickel oxide (NiO) nanoparticles manufactured using a facile thermal treatment. Results Phys. 6, 1024–1030 (2016). https://doi.org/10.1016/j.rinp.2016.11.031
M.Z. Nakhjiri, S. Asadi, A. Hasan, M.M.N. Babadaei, Y. Vahdani, B. Rasti, M. Ale-Ebrahim, N. Arsalan, S.V.M. Goorabjavari, S. Haghighat, M. Sharifi, K. Shahpasand, K. Akhtari, M. Falahati, Exploring the interaction of synthesized nickel oxide nanoparticles through hydrothermal method with hemoglobin and lymphocytes: bio-thermodynamic and cellular studies. J. Mol. Liq. (2020). https://doi.org/10.1016/j.molliq.2020.113893
Z. Fereshteh, M. Salavati-Niasari, K. Saberyan, S.M. Hosseinpour-Mashkani, F. Tavakoli, Synthesis of nickel oxide nanoparticles from thermal decomposition of a new precursor. J. Clust. Sci. 23, 577–583 (2012). https://doi.org/10.1007/s10876-012-0477-8
A.D. Khalaji, M. Jarosova, P. Machek, K. Chen, D. Xue, Facile synthesis, characterization and electrochemical performance of nickel oxide nanoparticles prepared by thermal decomposition. Scr. Mater. 181, 53–57 (2020). https://doi.org/10.1016/j.scriptamat.2020.02.015
A.M. Abdallah, H. Basma, R. Awad, Preparation, characterization, and application of nickel oxide nanoparticles in glucose and lactose biosensors. Mod. Appl. Sci. 13, 99 (2019). https://doi.org/10.5539/mas.v13n6p99
R. Goel, R. Jha, C. Ravikant, Investigating the structural, electrochemical, and optical properties of p-type spherical nickel oxide (NiO) nanoparticles. J. Phys. Chem. Solids. 144, 109488 (2020). https://doi.org/10.1016/j.jpcs.2020.109488
G.T. Anand, R. Nithiyavathi, R. Ramesh, S.J. Sundaram, K. Kaviyarasu, Structural and optical properties of nickel oxide nanoparticles: investigation of antimicrobial applications. Surf. Interf. 18, 100460 (2020). https://doi.org/10.1016/j.surfin.2020.100460
H. Baksh, J.A. Buledi, N.H. Khand, A.R. Solangi, A. Mallah, S.T. Sherazi, M.I. Abro, Ultra-selective determination of carbofuran by electrochemical sensor based on nickel oxide nanoparticles stabilized by ionic liquid. Monatsh. Chemie. 151, 1689–1696 (2020). https://doi.org/10.1007/s00706-020-02704-4
M. Maruthupandy, G.N. Rajivgandhi, F. Quero, W.J. Li, Anti-quorum sensing and anti-biofilm activity of nickel oxide nanoparticles against pseudomonas aeruginosa. J. Environ. Chem. Eng. 8, 104533 (2020). https://doi.org/10.1016/j.jece.2020.104533
R. Ahmad, T. Bedük, S.M. Majhi, K.N. Salama, One-step synthesis and decoration of nickel oxide nanosheets with gold nanoparticles by reduction method for hydrazine sensing application. Sens. Actuators B Chem. 286, 139–147 (2019). https://doi.org/10.1016/j.snb.2019.01.132
T.N.J.I. Edison, R. Atchudan, Y.R. Lee, Binder-free electro-synthesis of highly ordered nickel oxide nanoparticles and its electrochemical performance. Electrochim. Acta. 283, 1609–1617 (2018). https://doi.org/10.1016/j.electacta.2018.07.101
F.F. Bobinihi, O.E. Fayemi, D.C. Onwudiwe, Synthesis, characterization, and cyclic voltammetry of nickel sulphide and nickel oxide nanoparticles obtained from Ni(II) dithiocarbamate. Mater. Sci. Semicond. Process. 121, 105315 (2021). https://doi.org/10.1016/j.mssp.2020.105315
V.P.M. Shajudheen, M. Sivakumar, S.S. Kumar, Synthesis and characterization of NiO nanoparticles by thermal oxidation of nickel sulfide nanoparticles. Mater. Today Proc. 3, 2450–2456 (2016). https://doi.org/10.1016/j.matpr.2016.04.161
A.V. Ushakov, I.V. Karpov, L.Y. Fedorov, V.G. Demin, E.A. Goncharova, A.A. Shaihadinov, G.M. Zeer, S.M. Zharkov, The effect of microstructural features on the ferromagnetism of nickel oxide nanoparticles synthesized in a low-pressure arc plasma. Phys. E Low-Dimens. Syst. Nanostructures. 124, 114352 (2020). https://doi.org/10.1016/j.physe.2020.114352
B. Ebin, Simple preparation of Ni and NiO nanoparticles using raffinate solution originated from spent NiMH battery recycling. J. Inorg. Organomet. Polym. Mater. 28, 2554–2563 (2018). https://doi.org/10.1007/s10904-018-0926-4
F.T. Thema, E. Manikandan, A. Gurib-Fakim, M. Maaza, Single phase bunsenite NiO nanoparticles green synthesis by agathosma betulina natural extract. J. Alloys Compd. 657, 655–661 (2016). https://doi.org/10.1016/j.jallcom.2015.09.227
T. Zahra, K.S. Ahmad, Structural, optical and electrochemical studies of organo-templated wet synthesis of cubic shaped nickel oxide nanoparticles. Optik (Stuttg). 205, 164241 (2020). https://doi.org/10.1016/j.ijleo.2020.164241
A.A. Olajire, A.A. Mohammed, Green synthesis of nickel oxide nanoparticles and studies of their photocatalytic activity in degradation of polyethylene films. Adv. Powder Technol. 31, 211–218 (2020). https://doi.org/10.1016/j.apt.2019.10.012
P. Karpagavinayagam, A.E.P. Prasanna, C. Vedhi, Eco-friendly synthesis of nickel oxide nanoparticles using avicennia marina leaf extract: morphological characterization and electrochemical application. Mater. Today Proc. (2020). https://doi.org/10.1016/j.matpr.2020.04.183
A.A. Ezhilarasi, J.J. Vijaya, K. Kaviyarasu, X. Zhang, L.J. Kennedy, Green synthesis of nickel oxide nanoparticles using solanum trilobatum extract for cytotoxicity, antibacterial and photocatalytic studies. Surf. Interf. 20, 100553 (2020). https://doi.org/10.1016/j.surfin.2020.100553
A. Lawrence, C. Hariharan, A.N. Prabu, B. Janarthanan, Materials today: proceedings influence of nickel oxide nanoparticles on the absorption enhancement of solar radiation for effective distillation by single slope wick-type solar still. Mater. Today Proc. (2020). https://doi.org/10.1016/j.matpr.2020.10.704
A.C. Nwanya, M.M. Ndipingwi, C.O. Ikpo, R.M. Obodo, S.C. Nwanya, S. Botha, F.I. Ezema, E.I. Iwuoha, M. Maaza, Zea mays lea silk extract mediated synthesis of nickel oxide nanoparticles as positive electrode material for asymmetric supercabattery. J. Alloys Compd. 822, 153581 (2020). https://doi.org/10.1016/j.jallcom.2019.153581
S. Ghazal, A. Akbari, H.A. Hosseini, Z. Sabouri, F. Forouzanfar, M. Khatami, M. Darroudi, Sol-gel biosynthesis of nickel oxide nanoparticles using cydonia oblonga extract and evaluation of their cytotoxicity and photocatalytic activities. J. Mol. Struct. 1217, 128378 (2020). https://doi.org/10.1016/j.molstruc.2020.128378
K. Lingaraju, H.R. Naika, H. Nagabhushana, K. Jayanna, S. Devaraja, G. Nagaraju, Biosynthesis of nickel oxide nanoparticles from Euphorbia heterophylla (L.) and their biological application. Arab. J. Chem. 13, 4712–4719 (2020). https://doi.org/10.1016/j.arabjc.2019.11.003
R. Ramalingam, M.H.U.T. Fazil, N.K. Verma, K.D. Arunachalam, Green synthesis, characterization and antibacterial evaluation of electrospun nickel oxide nanofibers. Mater. Lett. 256, 126616 (2019). https://doi.org/10.1016/j.matlet.2019.126616
S. Iqbal, M. Fakhar-e-alam, F. Akbar, M. Sha, M. Atif, N. Amin, M. Ismail, A. Hanif, W.A. Farooq, Application of silver oxide nanoparticles for the treatment of cancer. J. Mol. Struct 1189, 203–209 (2019). https://doi.org/10.1016/j.molstruc.2019.04.041
N.L. Yong, A. Ahmad, A.W. Mohammad, Synthesis and characterization of silver oxide nanoparticles by a novel method. Int. J. Sci Eng. Res 4, 155–158 (2013)
M.M. Rahman, S.B. Khan, A. Jamal, M. Faisal, A.M. Asiri, Highly sensitive methanol chemical sensor based on undoped silver oxide nanoparticles prepared by a solution method. Microchim. Acta (2012). https://doi.org/10.1007/s00604-012-0817-2
M.R.H. Siddiqui, S.F. Adil, M.E. Assal, R. Ali, A. Al-Warthan, Synthesis and characterization of silver oxide and silver chloride nanoparticles with high thermal stability. Asian J. Chem. 25, 3405–3409 (2013). https://doi.org/10.14233/ajchem.2013.13874
S.P. Vinay, Udayabhanu, G. Nagaraju, C.P. Chandrappa, N. Chandrasekhar, Novel Gomutra (cow urine) mediated synthesis of silver oxide nanoparticles and their enhanced photocatalytic, photoluminescence and antibacterial studies. J. Sci. Adv. Mater. Devices 4, 392–399 (2019). https://doi.org/10.1016/j.jsamd.2019.08.004
J. Fowsiya, G. Madhumitha, Biomolecules derived from carissa edulis for the microwave assisted synthesis of Ag2O nanoparticles: a study against S. incertulas, C. medinalis and S. mauritia. J. Clust. Sci. 30, 1243–1252 (2019). https://doi.org/10.1007/s10876-019-01627-3
S. Haq, K.A. Yasin, W. Rehman, M. Waseem, M.N. Ahmed, M.I. Shahzad, N. Shahzad, A. Shah, M.U. Rehman, B. Khan, Green synthesis of silver oxide nanostructures and investigation of their synergistic effect with moxifloxacin against selected microorganisms. J. Inorg. Organomet. Polym. Mater. (2020). https://doi.org/10.1007/s10904-020-01763-8
R. Li, Z. Chen, N. Ren, Y. Wang, Y. Wang, F. Yu, Biosynthesis of silver oxide nanoparticles and their photocatalytic and antimicrobial activity evaluation for wound healing applications in nursing care. J. Photochem. Photobiol. B Biol. 199, 111593 (2019). https://doi.org/10.1016/j.jphotobiol.2019.111593
G. Maheshwaran, A.N. Bharathi, M.M. Selvi, M.K. Kumar, R.M. Kumar, S. Sudhahar, Green synthesis of silver oxide nanoparticles using Zephyranthes Rosea flower extract and evaluation of biological activities. J. Environ. Chem. Eng. 8, 104137 (2020). https://doi.org/10.1016/j.jece.2020.104137
V. Manikandan, P. Velmurugan, J.P.W. Chang, Green synthesis of silver oxide nanoparticles and its antibacterial activity against dental pathogens. 3 Biotech 7, 1–9 (2017). https://doi.org/10.1007/s13205-017-0670-4
B.N. Rashmi, S.F. Harlapur, B. Avinash, C.R. Ravikumar, H.P. Nagaswarupa, M.R.A. Kumar, K. Gurushantha, M.S. Santosh, Facile green synthesis of silver oxide nanoparticles and their electrochemical, photocatalytic and biological studies. Inorg. Chem. Commun. 111, 107580 (2020). https://doi.org/10.1016/j.inoche.2019.107580
Z.H. Dhoondia, H. Chakraborty, Lactobacillus mediated synthesis of silver oxide nanoparticles. Nanomater. Nanotechnol. (2012). https://doi.org/10.5772/55741
S.N. Archana, R. Sharma, Srivastava, silver oxide nanoparticles synthesized by green method from artocarpus Hetrophyllus for antibacterial and antimicrobial applications. Mater. Today Proc. 28, 332–336 (2020). https://doi.org/10.1016/j.matpr.2020.02.233
G. Pradheesh, S. Suresh, J. Suresh, V. Alexramani, Antimicrobial and anticancer activity studies on green synthesized silver oxide nanoparticles from the medicinal plant cyathea nilgiriensis holttum. Int. J. Pharm. Investig. 10, 146–150 (2020). https://doi.org/10.5530/ijpi.2020.2.27
M. Aghazadeh, Synthesis, characterization, and study of the supercapacitive performance of NiO nanoplates prepared by the cathodic electrochemical deposition-heat treatment (CED-HT) method. J. Mater. Sci. Mater. Electron. 28, 3108–3117 (2017). https://doi.org/10.1007/s10854-016-5899-x
M. Tulinski, M. Jurczyk, Nanomaterials synthesis methods. Metrol. Stand. Nanotechnol. (2017). https://doi.org/10.1002/9783527800308.ch4
L.A. Kolahalam, I.V.K. Viswanath, B.S. Diwakar, B. Govindh, V. Reddy, Y.L.N. Murthy, Review on nanomaterials: synthesis and applications. Mater. Today Proc. 18, 2182–2190 (2019). https://doi.org/10.1016/j.matpr.2019.07.371
S.M. Bhagyaraj, O.S. Oluwafemi, Nanotechnology: The Science of the Invisible (Elsevier Ltd, 2018). https://doi.org/10.1016/b978-0-08-101975-7.00001-4
S. Iravani, Green synthesis of metal nanoparticles using plants. Green Chem. 13, 2638–2650 (2011). https://doi.org/10.1039/c1gc15386b
R. Singaravelan, S.B.S. Alwar, Electrochemical synthesis, characterisation and phytogenic properties of silver nanoparticles. Appl. Nanosci. 5, 983–991 (2015). https://doi.org/10.1007/s13204-014-0396-0
Y. Zhang, L.Z. Xie, C.X. Yuan, C.L. Zhang, S. Liu, Y.Q. Peng, H.R. Li, M. Zhang, Preparation of 3D rose-like nickel oxide nanoparticles by electrodeposition method and application in gas sensors. J. Mater. Sci. Mater. Electron 27, 1817–1827 (2016). https://doi.org/10.1007/s10854-015-3959-2
Q. Wang, J. Zheng, Electrodeposition of silver nanoparticles on a zinc oxide film: improvement of amperometric sensing sensitivity and stability for hydrogen peroxide determination. Microchim. Acta. 169, 361–365 (2010). https://doi.org/10.1007/s00604-010-0356-7
N. Huang, H. Lim, C. Chia, M. Yarmo, M. Muhamad, Simple room-temperature preparation of high-yield large-area graphene oxide. Int. J. Nanomed. 6, 3443–3448 (2011). https://doi.org/10.1021/j100561a010
A.M. Golsheikh, N.M. Huang, H.N. Lim, R. Zakaria, C.Y. Yin, One-step electrodeposition synthesis of silver-nanoparticle-decorated graphene on indium-tin-oxide for enzymeless hydrogen peroxide detection. Carbon N. Y. 62, 405–412 (2013). https://doi.org/10.1016/j.carbon.2013.06.025
H. Guo, H. Jin, R. Gui, Z. Wang, J. Xia, F. Zhang, Electrodeposition one-step preparation of silver nanoparticles/carbon dots/reduced graphene oxide ternary dendritic nanocomposites for sensitive detection of doxorubicin. Sens. Actuators B Chem. 253, 50–57 (2017). https://doi.org/10.1016/j.snb.2017.06.095
M. Usman, L. Pan, A. Farid, A.S. Khan, Z. Yongpeng, M.A. Khan, M. Hashim, Carbon nanocoils-nickel foam decorated with silver nanoparticles/sheets using a novel stirring assisted electrodeposition technique for non-enzymatic glucose sensor. Carbon N. Y. 157, 761–766 (2020). https://doi.org/10.1016/j.carbon.2019.10.069
W. Yang, S. Yang, W. Sun, G. Sun, Q. Xin, Nanostructured silver catalyzed nickel foam cathode for an aluminum-hydrogen peroxide fuel cell. J. Pow. Sour. 160, 1420–1424 (2006). https://doi.org/10.1016/j.jpowsour.2006.02.015
R. Yuksel, S. Coskun, Y.E. Kalay, H.E. Unalan, Flexible, silver nanowire network nickel hydroxide core-shell electrodes for supercapacitors. J. Pow. Sour. 328, 167–173 (2016). https://doi.org/10.1016/j.jpowsour.2016.08.008
W. Zhao, N. Du, H. Zhang, D. Yang, Silver-nickel oxide core-shell nanoflower arrays as high-performance anode for lithium-ion batteries. J. Pow. Sour. 285, 131–136 (2015). https://doi.org/10.1016/j.jpowsour.2015.03.088
W. Zhao, N. Du, H. Zhang, D. Yang, Silver-nickel oxide core-shell nanoparticle array electrode with enhanced lithium-storage performance. Electrochim. Acta. 174, 893–899 (2015). https://doi.org/10.1016/j.electacta.2015.04.156
A.M. El-Nahrawy, A.B.A. Hammad, T.A. Khattab, A. Haroun, S. Kamel, Development of electrically conductive nanocomposites from cellulose nanowhiskers, polypyrrole and silver nanoparticles assisted with nickel(III) oxide nanoparticles. React. Funct. Polym. (2020). https://doi.org/10.1016/j.reactfunctpolym.2020.104533
M.M. Mohammadi, S.S. Gunturi, S. Shao, S. Konda, R.D. Buchner, M.T. Swihart, Flame-synthesized nickel-silver nanoparticle inks provide high conductivity without sintering. Chem. Eng. J. 372, 648–655 (2019). https://doi.org/10.1016/j.cej.2019.04.141
M.M. Mohammadi, S. Shao, S.S. Gunturi, A.R. Raghavan, N. Alexander, Y. Liu, C.M. Stafford, R.D. Buchner, M.T. Swihart, A general approach to multicomponent metal-decorated crumpled reduced graphene oxide nanocomposites using a flame-based process. Nanoscale 11, 19571–19578 (2019). https://doi.org/10.1039/c9nr05792g
M.K. Sharma, D. Qi, R.D. Buchner, W.J. Scharmach, V. Papavassiliou, M.T. Swihart, Flame-driven aerosol synthesis of copper-nickel nanopowders and conductive nanoparticle films. ACS Appl. Mater. Interf. 6, 13542–13551 (2014). https://doi.org/10.1021/am5026853
W.J. Scharmach, R.D. Buchner, V. Papavassiliou, P. Pacouloute, M.T. Swihart, A high-temperature reducing jet reactor for flame-based metal nanoparticle production. Aerosol Sci. Technol. 44, 1083–1088 (2010). https://doi.org/10.1080/02786826.2010.511320
J.J. Jing, J. Xie, G.Y. Chen, W.H. Li, M.M. Zhang, Preparation of nickel–silver core–shell nanoparticles by liquid-phase reduction for use in conductive paste. J. Exp. Nanosci. 10, 1347–1356 (2015). https://doi.org/10.1080/17458080.2015.1012751
D. Fujioka, S. Ikeda, K. Akamatsu, H. Nawafune, K. Kojima, Preparation of Ni nanoparticles by liquid-phase reduction to fabricate metal nanoparticle-polyimide composite films. RSC Adv. 9, 6438–6443 (2019). https://doi.org/10.1039/c9ra00182d
M.Z. Iqbal, R.J. Kriek, Silver/nickel oxide (Ag/NiO) nanocomposites produced via a citrate sol-gel route as electrocatalyst for the oxygen evolution reaction (OER) in alkaline medium. Electrocatalysis 9, 279–286 (2018). https://doi.org/10.1007/s12678-018-0455-5
S. Ghazal, A. Akbari, H.A. Hosseini, Z. Sabouri, F. Forouzanfar, M. Khatami, M. Darroudi, Biosynthesis of silver-doped nickel oxide nanoparticles and evaluation of their photocatalytic and cytotoxicity properties. Appl. Phys. A Mater. Sci. Process. 126, 1–8 (2020). https://doi.org/10.1007/s00339-020-03664-6
S. Maharjan, K.S. Liao, A.J. Wang, Z. Zhu, B.P. McElhenny, J. Bao, S.A. Curran, Sol-gel synthesis of stabilized silver nanoparticles in an organosiloxane matrix and its optical nonlinearity. Chem. Phys. 532, 110610 (2020). https://doi.org/10.1016/j.chemphys.2019.110610
M. Shahjahan, Synthesis and characterization of silver nanoparticles by sol-gel technique. Nanosci. Nanometrology 3, 34 (2017). https://doi.org/10.11648/j.nsnm.20170301.16
N.N.M. Zorkipli, N.H.M. Kaus, A.A. Mohamad, Synthesis of NiO nanoparticles through sol-gel method. Procedia Chem. 19, 626–631 (2016). https://doi.org/10.1016/j.proche.2016.03.062
M. Alagiri, S. Ponnusamy, C. Muthamizhchelvan, Synthesis and characterization of NiO nanoparticles by sol-gel method. J. Mater. Sci. Mater. Electron. 23, 728–732 (2012). https://doi.org/10.1007/s10854-011-0479-6
V.A. Alexandrova, L.N. Shirokova, V.S. Sadykova, A.E. Baranchikov, Antimicrobial activity of silver nanoparticles in a carboxymethyl chitin matrix obtained by the microwave hydrothermal method. Appl. Biochem. Microbiol. 54, 496–500 (2018). https://doi.org/10.1134/S0003683818050046
M. Bonomo, Synthesis and characterization of NiO nanostructures: a review. J. Nanoparticle Res. (2018). https://doi.org/10.1007/s11051-018-4327-y
S. Nagamuthu, K.S. Ryu, Synthesis of Ag/NiO honeycomb structured nanoarrays as the electrode material for high performance asymmetric supercapacitor devices. Sci. Rep. 9, 1–11 (2019). https://doi.org/10.1038/s41598-019-41446-0
I. Ocsoy, A. Demirbas, E.S. McLamore, B. Altinsoy, N. Ildiz, A. Baldemir, Green synthesis with incorporated hydrothermal approaches for silver nanoparticles formation and enhanced antimicrobial activity against bacterial and fungal pathogens. J. Mol. Liq. 238, 263–269 (2017). https://doi.org/10.1016/j.molliq.2017.05.012
Y.F. Li, W.P. Gan, J. Zhou, Z.Q. Lu, C. Yang, T.T. Ge, Hydrothermal synthesis of silver nanoparticles in Arabic gum aqueous solutions. Trans. Nonferrous Met. Soc. China (Engl. Ed.) 25, 2081–2086 (2015). https://doi.org/10.1016/S1003-6326(15)63818-3
A. Bouremana, A. Guittoum, M. Hemmous, D. Martínez-Blanco, P. Gorria, J.A. Blanco, N. Benrekaa, Microstructure, morphology and magnetic properties of Ni nanoparticles synthesized by hydrothermal method. Mater. Chem. Phys. 160, 435–439 (2015). https://doi.org/10.1016/j.matchemphys.2015.05.015
G. Jayakumar, A.A. Irudayaraj, A.D. Raj, Photocatalytic degradation of methylene blue by nickel oxide nanoparticles. Mater. Today Proc. 4, 11690–11695 (2017). https://doi.org/10.1016/j.matpr.2017.09.083
D.A. Svintsitskiy, M.K. Lazarev, T.Y. Kardash, E.A. Fedorova, E.M. Slavinskaya, A.I. Boronin, Mixed silver-nickel oxide AgNiO2: probing by CO during XPS study. J. Chem. Phys. (2020). https://doi.org/10.1063/1.5138237
J.A. Adekoya, E.O. Dare, M.A. Mesubi, N. Revaprasadu, Synthesis and characterization of optically active fractal seed mediated silver nickel bimetallic nanoparticles. J. Mater. 2014, 1–9 (2014). https://doi.org/10.1155/2014/184216
M. Ashokkumar, S. Muthukumaran, Microstructure, optical and FTIR studies of Ni, Cu co-doped ZnO nanoparticles by co-precipitation method. Opt. Mater. (Amst) 37, 671–678 (2014). https://doi.org/10.1016/j.optmat.2014.08.012
S. Joshi, M. Kumar, S. Chhoker, G. Srivastava, M. Jewariya, V.N. Singh, Structural, magnetic, dielectric and optical properties of nickel ferrite nanoparticles synthesized by co-precipitation method. J. Mol. Struct. 1076, 55–62 (2014). https://doi.org/10.1016/j.molstruc.2014.07.048
S. Thambidurai, P. Gowthaman, M. Venkatachalam, S. Suresh, Enhanced bactericidal performance of nickel oxide-zinc oxide nanocomposites synthesized by facile chemical co-precipitation method. J. Alloys Compd. 830, 154642 (2020). https://doi.org/10.1016/j.jallcom.2020.154642
C. Liu, D. Xie, P. Liu, S. Xie, S. Wang, F. Cheng, M. Zhang, L. Wang, Voltammetric determination of levofloxacin using silver nanoparticles deposited on a thin nickel oxide porous film. Microchim. Acta. (2019). https://doi.org/10.1007/s00604-018-3146-2
M.P. Deshpande, K.N. Patel, V.P. Gujarati, K. Patel, S.H. Chaki, Structural, thermal and optical properties of nickel oxide (NiO) nanoparticles synthesized by chemical precipitation method. Adv. Mater. Res. 1141, 65–71 (2016)
S. Senapati, S.K. Srivastava, S.B. Singh, H.N. Mishra, Magnetic Ni/Ag core-shell nanostructure from prickly Ni nanowire precursor and its catalytic and antibacterial activity. J. Mater. Chem. 22, 6899–6906 (2012). https://doi.org/10.1039/c2jm00143h
Y. Du, W. Wang, X. Li, J. Zhao, J. Ma, Y. Liu, G. Lu, Preparation of NiO nanoparticles in microemulsion and its gas sensing performance. Mater. Lett. 68, 168–170 (2012). https://doi.org/10.1016/j.matlet.2011.10.039
X. Zhang, Q. Liu, Z. Fan, Enhanced in situ combustion of heavy crude oil by nickel oxide nanoparticles. Int. J. Energy Res. 43, 3399–3412 (2019). https://doi.org/10.1002/er.4478
I.A. Wani, S. Khatoon, A. Ganguly, J. Ahmed, T. Ahmad, Structural characterization and antimicrobial properties of silver nanoparticles prepared by inverse microemulsion method. Coll. Surf. B Biointerf. 101, 243–250 (2013). https://doi.org/10.1016/j.colsurfb.2012.07.001
T. Ahmad, I.A. Wani, O.A. Al-Hartomy, A.S. Al-Shihri, A. Kalam, Low temperature chemical synthesis and comparative studies of silver oxide nanoparticles. J. Mol. Struct. 1084, 9–15 (2015). https://doi.org/10.1016/j.molstruc.2014.12.015
H.K. Wang, C.Y. Yi, L. Tian, W.J. Wang, J. Fang, J.H. Zhao, W.G. Shen, Ag-Cu bimetallic nanoparticles prepared by microemulsion method as catalyst for epoxidation of styrene. J. Nanomater. 2012, 1–8 (2012). https://doi.org/10.1155/2012/453915
L. Chen, W. Kong, J. Yao, H. Zhang, B. Gao, Y. Li, H. Bu, A. Chang, C. Jiang, Synthesis and characterization of Mn-Co-Ni-O ceramic nanoparticles by reverse microemulsion method. Ceram. Int. 41, 2847–2851 (2015). https://doi.org/10.1016/j.ceramint.2014.10.106
Y. Thaver, S.O. Oseni, G. Tessema, Silver doped nickel oxide nanocomposite and photon harvesting enhancement in bulkheterojunction organic solar cell. Sol. Energy 214, 11–18 (2021). https://doi.org/10.1016/j.solener.2020.11.044
M.Z. Bani-Fwaz, A.A. El-Zahhar, H.S.M. Abd-Rabboh, M.S. Hamdy, M. Shkir, Synthesis of NiO nanoparticles by thermal routes for adsorptive removal of crystal violet dye from aqueous solutions. Int. J. Environ. Anal. Chem. 00, 1–19 (2019). https://doi.org/10.1080/03067319.2019.1678599
M. Goudarzi, N. Mir, M. Mousavi-Kamazani, S. Bagheri, M. Salavati-Niasari, Biosynthesis and characterization of silver nanoparticles prepared from two novel natural precursors by facile thermal decomposition methods. Sci. Rep. 6, 1–13 (2016). https://doi.org/10.1038/srep32539
D. Adner, J. Noll, S. Schulze, M. Hietschold, H. Lang, Asperical silver nanoparticles by thermal decomposition of a single-source-precursor. Inorganica Chim. Acta. 446, 19–23 (2016). https://doi.org/10.1016/j.ica.2016.02.059
O.A.D. Gallardo, R. Moiraghi, M.A. MacChione, J.A. Godoy, M.A. Pérez, E.A. Coronado, V.A. MacAgno, Silver oxide particles/silver nanoparticles interconversion: susceptibility of forward/backward reactions to the chemical environment at room temperature. RSC Adv. 2, 2923–2929 (2012). https://doi.org/10.1039/c2ra01044e
A. Kalam, A.G. Al-Sehemi, A.S. Al-Shihri, G. Du, T. Ahmad, Synthesis and characterization of NiO nanoparticles by thermal decomposition of nickel linoleate and their optical properties. Mater. Charact. 68, 77–81 (2012). https://doi.org/10.1016/j.matchar.2012.03.011
M. Salavati-Niasari, N. Mir, F. Davar, A novel precursor in preparation and characterization of nickel oxide nanoparticles via thermal decomposition approach. J. Alloys Compd. 493, 163–168 (2010). https://doi.org/10.1016/j.jallcom.2009.11.153
K.P. Donegan, J.F. Godsell, D.J. Otway, M.A. Morris, S. Roy, J.D. Holmes, Size-tuneable synthesis of nickel nanoparticles. J. Nanoparticle Res. (2012). https://doi.org/10.1007/s11051-011-0670-y
J. Pinkas, J. Sopoušek, P. Brož, V. Vykoukal, J. Buršík, J. Vřešťál, Synthesis, structure, stability and phase diagrams of selected bimetallic silver- and nickel-based nanoparticles. Calphad Comput. Coupling Ph. Diagr. Thermochem. 64, 139–148 (2019). https://doi.org/10.1016/j.calphad.2018.11.013
M.A. Raza, Z. Kanwal, A. Rauf, A.N. Sabri, S. Riaz, S. Naseem, Size- and shape-dependent antibacterial studies of silver nanoparticles synthesized by wet chemical routes. Nanomaterials 6(4), 74 (2016). https://doi.org/10.3390/nano6040074
P.M. Ponnusamy, S. Agilan, N. Muthukumarasamy, T.S. Senthil, G. Rajesh, M.R. Venkatraman, D. Velauthapillai, Structural, optical and magnetic properties of undoped NiO and Fe-doped NiO nanoparticles synthesized by wet-chemical process. Mater. Charact. 114, 166–171 (2016). https://doi.org/10.1016/j.matchar.2016.02.020
L. Gharibshahi, E. Saion, E. Gharibshahi, A.H. Shaari, K.A. Matori, Influence of Poly(vinylpyrrolidone) concentration on properties of silver nanoparticles manufactured by modified thermal treatment method. PLoS ONE 12, 1–17 (2017). https://doi.org/10.1371/journal.pone.0186094
M. Salavati-Niasari, F. Davar, Z. Fereshteh, Synthesis of nickel and nickel oxide nanoparticles via heat-treatment of simple octanoate precursor. J. Alloys Compd. 494, 410–414 (2010). https://doi.org/10.1016/j.jallcom.2010.01.063
M. Zainy, N.M. Huang, S.V. Kumar, H.N. Lim, C.H. Chia, I. Harrison, Simple and scalable preparation of reduced graphene oxide-silver nanocomposites via rapid thermal treatment. Mater. Lett. 89, 180–183 (2012). https://doi.org/10.1016/j.matlet.2012.08.101
L. Gharibshahi, E. Saion, E. Gharibshahi, A.H. Shaari, K.A. Matori, Structural and optical properties of ag nanoparticles synthesized by thermal treatment method. Materials (Basel) 10, 402 (2017). https://doi.org/10.3390/ma10040402
D. Poondi, J. Singh, Synthesis of metastable silver-nickel alloys by a novel laser-liquid-solid interaction technique. J. Mater. Sci. 35, 2467–2476 (2000). https://doi.org/10.1023/A:1004765618078
D. Poondi, T. Dobbins, J. Singh, Novel laser-liquid-solid interaction technique for synthesis of silver, nickel and immiscible silver-nickel alloys from liquid precursors. J. Mater. Sci. 35, 6237–6243 (2000). https://doi.org/10.1023/A:1026701915796
X. Zhang, H. Sun, S. Tan, J. Gao, Y. Fu, Z. Liu, Hydrothermal synthesis of Ag nanoparticles on the nanocellulose and their antibacterial study. Inorg. Chem. Commun. 100, 44–50 (2019). https://doi.org/10.1016/j.inoche.2018.12.012
N.M. Al-Hada, H.M. Kamari, M.A. Saleh, M.H. Flaifel, A.M. Al-Ghaili, H. Kasim, A.A. Baqer, E. Saion, W. Jihua, Morphological, structural and optical behaviour of PVA capped binary (NiO)0.5 (Cr2O3)0.5 nanoparticles produced via single step based thermal technique. Results Phys. 17, 103059 (2020). https://doi.org/10.1016/j.rinp.2020.103059
T. Guo, M.S. Yao, Y.H. Lin, C.W. Nan, A comprehensive review on synthesis methods for transition-metal oxide nanostructures. CrystEngComm 17, 3551–3585 (2015). https://doi.org/10.1039/c5ce00034c
R. Paulose, R. Mohan, V. Parihar, Nanostructured nickel oxide and its electrochemical behaviour—A brief review. Nano-Struct. Nano-Objects 11, 102–111 (2017). https://doi.org/10.1016/j.nanoso.2017.07.003
R.S. Kate, S.A. Khalate, R.J. Deokate, Overview of nanostructured metal oxides and pure nickel oxide (NiO) electrodes for supercapacitors: a review. J. Alloys Compd. 734, 89–111 (2018). https://doi.org/10.1016/j.jallcom.2017.10.262
T.P. Mokoena, H.C. Swart, D.E. Motaung, A review on recent progress of p-type nickel oxide based gas sensors: future perspectives. J. Alloys Compd. 805, 267–294 (2019). https://doi.org/10.1016/j.jallcom.2019.06.329
Y.F. Sun, S.B. Liu, F.L. Meng, J.Y. Liu, Z. Jin, L.T. Kong, J.H. Liu, Metal oxide nanostructures and their gas sensing properties: a review. Sensors 12, 2610–2631 (2012). https://doi.org/10.3390/s120302610
M. Napari, T.N. Huq, R.L.Z. Hoye, J.L. MacManus-Driscoll, Nickel oxide thin films grown by chemical deposition techniques: potential and challenges in next-generation rigid and flexible device applications. InfoMat 3, 536–576 (2021). https://doi.org/10.1002/inf2.12146
S. Ahmad, H. Rashid, Q. Jalil, S. Munir, S. Khan, B. Barkatullah, R. Ullah, A.A. Shahat, A.A.N.A. A-Mishari, A.A. Shahat, H.M. Mahmood, A. Bari, Polymers encapsulated aspirin loaded silver oxide nanoparticles: synthesis, characterization and its bio-applications. Sains Malays. 48, 1887–1897 (2019). https://doi.org/10.17576/jsm-2019-4809-09
W.M. Shume, H.C.A. Murthy, E.A. Zereffa, A review on synthesis and characterization of Ag2O nanoparticles for photocatalytic applications. J. Chem. 2020, 1–15 (2020). https://doi.org/10.1155/2020/5039479
M.M. Sk, C.Y. Yue, K. Ghosh, R.K. Jena, Review on advances in porous nanostructured nickel oxides and their composite electrodes for high-performance supercapacitors. J. Pow. Sour. 308, 121–140 (2016). https://doi.org/10.1016/j.jpowsour.2016.01.056
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
No potential conflict of interest was reported by the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Absi, E., Saleh, M., Al-Hada, N.M. et al. A review on preparation and characterization of silver/nickel oxide nanostructures and their applications. Appl. Phys. A 127, 871 (2021). https://doi.org/10.1007/s00339-021-04979-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00339-021-04979-8