
Abstract
Polycrystalline lead-free (1 − x)(K0.5Bi0.5)TiO3-xBaTiO3, ((1 − x)KBT-xBT) (x = 0.00, 0.08, 0.12) ceramics were synthesized via solid-state reaction method. The powder X-ray diffraction (PXRD) and structural refinement results confirm that a single-phase tetragonal structure with space group P4mm. Charge density distribution inside the unit cell of (1 − x)KBT-xBT was investigated by the maximum entropy method. Charge density analysis reveals the reduction in ionic nature along K/Bi–O bond and enhancement of covalent nature along Ti–O bond with the addition of BaTiO3. The charge density distribution studies done using maximum entropy method for (1 − x)KBT-xBT have not been done so far. The surface morphology study was done using scanning electron microscopy (SEM). Energy dispersive X-rays spectra (EDS) were used to investigate the elemental compositions present in the system. The dielectric constant and loss tangent were studied as a function of frequency. The dielectric constant and loss were decreased with increase of frequency. Room temperature dielectric constant (ɛ) and loss (tan δ) were measured for x = 0.00 about 511 and 0.51, respectively, at a frequency of 10 kHz.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
In the past several decades, lead zirconate titanate (PZT) ceramics have dominated the electronic industries because of their excellent properties and superior technological applications in actuators, sensors, and other electronic devices. Most of them are lead containing materials which cause serious technological and environmental problems in the production of ceramic samples [1,2,3]. Recently, much attention on perovskite bismuth titanate compounds have been paid to the development of lead-free ceramics, which is considered as alternative to toxic PZT ceramics [4]. Potassium bismuth titanate belongs to lead-free ABO3 perovskites. The K0.5Bi0.5TiO3 (KBT) has tetragonal structure at room temperature, and it has high Curie temperature (T C = 380 °C) [5]. From the structural point of view, KBT has a tetragonal symmetry with a = b = 3.918 Å and c = 4.013 Å at room temperature. From a synthetic perspective, piezoelectric ceramics are usually prepared by solid-state reaction method, which consists of a calcination process below 1000 °C, where the raw materials such as K2CO3, Bi2O3, and TiO2 react with each other to form the KBT, in a sintering process above 1000 °C [6,7,8,9]. Himura et al. [10] and Wada et al. [11] reported that the high-density K0.5Bi0.5TiO3 samples are difficult to fabricate due to the high volatility of the bismuth component at sintering temperature. This situation can be improved by forming solid solutions of KBT with other ferroelectrics such as BaTiO3 [12], K0.5Na0.5NbO3 [13], and Bi0.5Na0TiO3 [14], as well as the use of sintering aids such as Bi2O3, La2O3, and MnCO3 [15].
This work is specifically concerned with investigating the electronic structure and chemical bonding of (1 − x)KBT-xBT ceramics. The solid solutions has been prepared by solid-state reaction method with different compositions, x = 0.00, 0.08 and 0.12. The effects of BaTiO3 on the crystal structure, microstructure, and dielectric properties have been analyzed. The crystal structure and charge density of (1 − x)KBT-xBT ceramics have been analyzed to investigate the effect of atomic substitution on the chemical bonding by means of XRD data. The experimental investigation on electron density distribution between the constituent atoms has not yet been reported in (1 − x)KBT-xBT, (x = 0.00, 0.08 and 0.12) ceramics.
2 Synthesis and characterizations
Polycrystalline powders of (1 − x)(K0.5Bi0.5)TiO3-xBaTiO3, (x = 0.0, 0.08 and 0.12) were prepared by solid-state reaction method. The stoichiometric amounts of K2CO3 (99.99% purity), Bi2O3 (99.99% purity), BaCO3 (99.99% purity), and TiO2 (99.99% purity) were used as starting materials. Before the solid-state reaction, the raw powders were taken, which satisfies the following reaction:
High-purity powders of starting chemicals were ball-milled for 2 h using agate balls to synthesize solid solutions. The mixed powders were dried and calcined at 800 °C for 2 h. The calcined powders were pressed uniaxially at a pressure of 200 MPa with 2 wt% polyvinyl alcohol as a binder. Then, the pellets were sintered using the conventional sintering method in air at 1050 °C for 3 h.
The synthesized ceramics were structurally characterized by powder X-ray diffraction (PXRD) using Bruker AXS D8 advance diffractometer. The XRD patterns were obtained using CuKα (λ = 1.54056 Å) radiation in the 2θ range from 20° to 80°, with the scanning step of 0.02°. The surface morphology and chemical composition were investigated by scanning electron microscopy (SEM) (Carl Zeiss EVO 18, 20 kV) and energy dispersive X-ray spectroscopy (EDS) (Carl Zeiss EVO 18) respectively. The dielectric properties were measured at room temperature using an impedance analyzer (PSM 1735, N4L) in the wide range of frequency from 10 Hz to 1 MHz.
3 Results and discussion
3.1 Structural analysis and Rietveld refinement analysis
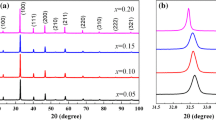
Figure 1a shows the powder XRD profiles of (1 − x)KBT-xBT, (x = 0.00, 0.08, 0.12) ceramic samples sintered at 1050 °C for 3 h. The XRD patterns reveal the tetragonal structure with space group P4mm (#99) (JCPDS PDF # 36-0339) [16]. Figure 1b shows that the enlarged diffraction peaks shift towards lower angle side of 2θ. The Ba2+ substitutions in K0.5Bi0.5TiO3 are also confirmed by peak shifting to lower angle side of 2θ indicating increase of cell parameters (Table 1). This is attributed to the fact that the ionic radius of Ba2+ (1.61 Å) cations is substituted for the smaller ionic radii of K+ (1.35 Å) and Bi3+ (0.96 Å) [17], which are expected to be primarily responsible for the increase of the unit cell dimensions. From cell parameter, tetragonality (c/a) was obtained by XRD measurement in the range from 1.001 to 1.006, which indicates that the crystal structure is tetragonal at room temperature.

a XRD patterns of (1 − x)KBT-xBT, x = 0.00, 0.08, and 0.12 ceramics at room temperature. b Enlarged (101), (111), and (200) peaks
The XRD data of all the compositions x = 0.00, 0.08, 0.12 were refined by the Rietveld method [18] using JANA 2006 software [19]. The structural refinements were carried out by taking the positional parameter of tetragonal BaTiO3 structure. The initial atomic position coordinates (x, y, z) were taken as (0, 0, 0) for potassium/bismuth, (0.5, 0.5, 0.512) for titanium, (0.5, 0.5, 0.023) for O1, and (0.5, 0, 0.486) for O2 from standard Wyckoff [20] position tables with space group P4mm. The fitted profiles of XRD data sets of (1 − x)KBT-xBT with x = 0.00, 0.08, 0.12 are shown in Fig. 2 (a)–(c). The fitting of observed and calculated profiles is quite good confirming the tetragonal structure of (1 − x)KBT-xBT (x = 0.00, 0.08, 0.12). In Fig. 2a–c, the cross symbol represents the observed XRD profile; the continuous line represents the calculated XRD profile, along with the error bar (difference (I c − I o)) and respective Bragg’s positions. The room temperature lattice constants of K0.5Bi0.5TiO3 are determined using Rietveld refinement [18] and found to be a = b = 3.9760 Å, c = 4.0018 Å and volume = 63.23 Å3 (Table 1). The fractional atomic coordinates, reliability factors, and goodness of fit were calculated from Rietveld refinement [18], and the results are listed in Table 1.

a Refined powder XRD profile for (K0.5Bi0.5)TiO3. b Refined powder XRD profile for 0.92(K0.5Bi0.5)TiO3-0.08BaTiO3. c Refined powder XRD profile for 0.88(K0.5Bi0.5)TiO3-0.12BaTiO3
3.2 Charge density by maximum entropy method
The electronic structure and bonding nature of (1 − x)KBT-xBT, (x = 0.00, 0.08, 0.12) ceramics were estimated by maximum entropy method (MEM) [21]. Charge distribution in the unit cell was carried out using the structural parameter obtained from Rietveld refinement [18]. MEM is used to reconstruct the electronic charge density structures. The MEM computations were carried out on (1 − x)KBT-xBT‚ (x = 0.00, 0.08, 0.12) unit cells, which were divided into 64 × 64 × 64 pixels along a, b, and c axes of the tetragonal lattice. Then, each pixel was assigned with a prior charge density of F 000/V, where F 000 is the total number of electrons in the unit cell and V is the volume of the unit cell. The resultant charge density is plotted using visualization software VESTA [22].
The 3D charge density distributions in the unit cell with an iso-surface level of 1.0 e/Å3 for (1 − x)KBT-xBT ceramics show the model of the structure as presented in Fig. 3a–c. Figures 4 and 5 show the 2D electron density contour in the contour range of 0–1 e/Å3 with an interval of 0.04 e/Å3. Figure 4a shows the shaded (001) plane in the 3D unit cell. Figure 4b, c demonstrates the 2D charge density contour maps corresponding to the (001) plane, which shows the K/Bi-O bonds. It can be observed that the charge accumulation between K/Bi and O ions in the undoped K0.5Bi0.5TiO3 ceramic reveals covalent nature due to the lone pair effect of Bi3+ ions. The charge density contours are further faded away at the middle portion of the K/Bi-O bond path for increasing Ba doping levels at x = 0.08 and 0.12. This indicates that the K/Bi-O bond becomes more ionic for x = 0.08 and 0.12 with Ba2+ substitution in the host lattice. Therefore, the contours of the 2D electron density distribution on (001) plane are faded away at the middle portion of the K/Bi-O bond for x = 0.12 (Fig. 4d) with respect to the x = 0.00, 0.08. Figure 5a shows the 3D unit cell with (002) plane shaded. Figure 5b, c illustrates the 2D contour maps corresponding to the (002) plane, which also show the Ti-O bonds. 2D contour maps on (002) plane show the charge overlap between Ti and O ions along the bonding region, which confirms the covalent nature of bonding between the Ti and O ions. Similar results have been reported in the previous studies [23, 24].

3D electron density distribution for (1 − x)KBT-xBT, a x = 0.00, b x = 0.08, c x = 0.12 with iso-surface level of 1 e/Å3

a 3D unit cell of (1 − x)KBT-xBT with (001) plane shaded. 2D electron density distribution on (001) plane for (1 − x)KBT-xBT, b x = 0.00, c x = 0.08, d x = 0.12

a 3D unit cell of (1 − x)KBT-xBT with (002) plane shaded. 2D electron density distribution on (002) plane for (1 − x)KBT-xBT, b x = 0.00, c x = 0.08, d x = 0.12
An analysis can be done on the 1D electron density profiles of (1 − x)KBT-xBT‚ (x = 0.00, 0.08 and 0.12) along K/Bi–O and Ti–O bonding directions and the valence contribution for the three systems is shown in Fig. 6a, b. The bond length variation and charge density values at mid-bond of K/Bi–O and Ti–O bonds are obtained from the MEM analysis and are given in Table 2. The bond length of Ba-O bond for x = 0.00 is 2.7880 Å. The bond length is increased from 2.8261 to 2.8265 Å with the increasing of Ba content for x = 0.8 and x = 0.12 respectively. In addition, the bond length for Ti-O bond for x = 1.9730 Å, which increases with the addition of Ba doping in the host lattice. The expansion of bond length with the incorporation of Ba is consistent with XRD data. The variation in bond length values with Ba doping indicates distortion in the K0.5Bi0.5TiO3 structure. From Table 2, for the concentration x = 0.00, the electron density at the bond critical point is found to be 0.4212 e/Å3. This is due to the lone pair effect of Bi3+ ions as discussed above and the electron density values are decreased with increase of Ba2+ concentration. This confirms that the K/Bi–O bond is ionic in nature for x = 0.08 and 0.12. Again, from Table 2, the mid-bond electron density values range from 0.7232 e/Å3 to 0.6265 e/Å3, which lead to the covalent behavior between Ti and O ions.

1D electron density profiles along a K/Bi-O, b Ti–O bonds in (1 − x)KBT-xBT‚ (x = 0.00, 0.08 and 0.12)
3.3 Microstructure and EDS analysis
Figure 7a–c shows the SEM micrographs of the (1 − x)KBT-xBT, (x = 0.00, 0.08, 0.12) ceramics sintered at 1050 °C. The SEM images were recorded at different magnifications (× 5000, × 10,000, ×25,000, and × 50,000) and the images with × 5000 are presented. It can be seen that there is a significant change in the surface morphology and particle size for the undoped and doped samples. Particles of irregular shape with pores heterogeneously distributed are seen for x = 0.08 sample. However, for x = 0.12 sample, very fine particles with agglomeration are noticed. For all the samples, it is observed that there is no clear grain boundary. The elemental compositional analysis of the synthesized samples was done by energy dispersive X-ray spectroscopy (EDS) and the recorded EDS spectra of (1 − x)KBT-xBT‚ (x = 0.00, 0.08 and 0.12) ceramics are shown in Fig. 8a–c. Each peak in the EDS spectra refers to particular atomic species present in the prepared sample. The atomic and weight percentages of the elements (K, Bi, Ti, Ba, and O) present in the synthesized sample with the expected proportions and are given in Table 3. It confirms that there are no impurities present in the sample.

SEM images of the (1 − x)KBT-xBT solid solutions with a x = 0.00, b x = 0.08, c x = 0.12

EDS spectra (1 − x)KBT-xBT ceramics a x = 0.00, b x = 0.08, c x = 0.12
3.4 Frequency dependence of dielectric constant measurements
Figure 9a, b shows the variation of dielectric constant and dielectric loss (tan δ) for all the samples with frequency range 10 Hz to 1 MHz at room temperature. Figure 9a shows that dielectric constant of all synthesized samples decreases with increasing frequency and becomes constant above 1 kHz. The plot at lower frequencies may be attributed to different types of polarization (electronic, atomic, interfacial, ionic, etc.). At higher frequencies, the dielectric constant decreases due to the contribution from electronic polarization [25, 26]. Variation of dielectric loss as a function of frequency at room temperature for all compositions is shown in Fig. 9b. Figure 9b shows the decreasing trend in loss values with increasing frequency. It can also be observed from Fig. 9b that the dielectric loss (tan δ) decreases with increasing Ba substitution for further doping. The decreasing value of dielectric loss at higher frequencies is expected to be due to the grain resistance [27]. The dielectric constant ε and loss (tan δ) for x = 0.00 are found to be 511 and 0.51, respectively, at 10 kHz frequency measured at room temperature. Dielectric properties of the (1 − x)KBT-xBT‚ (x = 0.00, 0.08, 0.12) ceramics at 10 kHz are given in Table 4.

a Frequency dependence of the dielectric constant and b dielectric loss (tan δ) of (1 − x)KBT-xBT‚ (x = 0.00, 0.08, 0.12) ceramics
4 Conclusion
In this study, lead-free (1 − x)(K0.5Bi0.5)TiO3-xBaTiO3, (x = 0.00, 0.08, 0.12) ceramics were synthesized by solid-state reaction technique. X-ray diffraction study confirmed that the (1 − x)KBT-xBT ceramics crystallized in a tetragonal structure. Substitution of Ba2+ at (K0.5Bi0.5)2+ site leads to increase of lattice constant and cell volumes as revealed by profile refinement. The chemical bonding nature and charge density distributions in the unit cell of (1 − x)KBT-xBT ceramics were investigated by maximum entropy method (MEM). The mid-bond electron density values reveal the covalent nature between Ti-O ions and ionic nature between K/Bi-O ions. The morphological properties of the solid solutions were analyzed from SEM images. Chemical compositions were further confirmed by EDS spectra. Frequency dependence of dielectric constant and losses was investigated at room temperature in the frequency range from 10 Hz to 1 MHz.
References
T.R. Shrout, S.J. Zhang, J. Electroceram 19, 111 (2007)
J. Rödel, W. Jo, K.T.P. Seifert, E.M. Anton, T. Granzow, D.J. Damjanovic. Am. Ceram. Soc 89, 1153 (2009)
J. Wu, D. Xiao, J. Zhu. Chem. Rev 115, 2559 (2015)
L. Hou, Y.D. Hou, X.M. Song, M.K. Zhu, H. Wang, H. Yan, Mater. Res. Bull 41, 1330 (2006)
Y.M. Chiang, G.W. Farrey, A.N. Soukhojak, Appl. Phys. Lett. 73, 3683 (1998)
V.V. Ivanova, A.G. Kapyshev, Yu..N. Venevtsev, G.S. Zhdanov, Izv. AN SSSR, Ser. fiz. 26, 354 (1962)
S. Zhao, G. Li, A. Ding, T. Wang, Q. Yin, J. Phys. D: Appl. Phys 39, 2277 (2006)
J. König, M. Spreitzer, B. Jancar, D. Suvorov, Z. Samardzija, A. Popovic, J. Eur. Ceram. Soc 29, 1695 (2009)
M. Davies, E. Aksel, J.L. Jones, J. Am. Ceram. Soc. 94, 1314 (2011)
Y. Himura, R. Aojagi, H. Nagata, T. Takenaka, Jpn. J. Appl. Phys 44, 5040 (2005)
T. Wada, A. Fukui, Y. Matsuo, Jpn. J. Appl. Phys 41, 7025 (2002)
J. Gomah-Pettry, P. Marchet, A. Simon, R. Von der Muhll, M. Maglione, J. Mercurio, Integr. Ferroelectr 61, 155 (2004)
Y. Hiruma, K. Marumo, R. Aoyagi, H. Nagata, T. Takenaka, J. Electroceram 21, 296 (2008)
T. Zaremba, J. Therm. Anal. Calorim 74, 653 (2003)
H. Du, W. Zhou, F. Luo, D. Zhu, S. Qu, Y. Li, Z. Pei., J. Phys. D: Appl. Phys 41, 11541 (2008)
G. Chang-lin, W. Yu-qin, W. Tian-Bao, Wuli Xuebao 31, 1119 (1982)
C.-F. Yang, K.-N. Chen, C.-C. Wu, Y.-Hsun Lin, Ceram. Int 42(7), 8932 (2016)
H.M. Rietveld, J. Appl. Crystallogr 2, 65 (1969)
V. Petricek, M. Dusek, J. Palatinus, The crystallographic computing system (Institute of Physics) (Czech Republic, Praha, 2006)
R.W.G. Wyckoff, C. Structures, (John Wiley, New York, 1963)
D.M. Collins, Nature 49, 298 (1982)
K. Momma, F. Izumi, J. Appl. Crystallogr 41, 653 (2008)
S. Sasikumar, R. Saravanan, S. Saravanakumar, K. Aravinth, J Mater. Sci Mater. Electron 28, 9950–9963 (2017)
J. Mangaiyarkkarasi, R. Saravanan, J Mater. Sci: Mater. Electron 28(3), 2624–2633 (2017)
C.G. Koops, Phys. Rev. B 83, 108 (1951)
D.R. Patil, S.A. Lokare, R.S. Devan, S.S. Chougule, C.M. Kanamadi, Y.D. Kolekar, B.K. Chougule, Mater. Chem. Phys 104, 254 (2007)
P. Nayak, T. Badapanda, A.K. Singh, S. Panigrahi, An approach for correlating the structural and electrical properties of Zr4+-modified SrBi4Ti4O15/SBT ceramic. RSC Adv 7, 16319–16331 (2017)
Acknowledgements
The authors S. Sasikumar and R. Saravanan thank the Naval Research Board of DRDO, India, for providing a grant in research project (No. NRB/DRDO/MAT/269). The authors S. Sasikumar and R. Saravanan would like to express their special thanks, Dr.T.Mukundan, Material Science Division and Dr.R.Ramesh, Transducers division, NPOL, Cochin. In addition, the authors thank SAIF (Sophisticated Analytical Instrument Facility), CUSAT, Cochin for the PXRD measurements. The authors thank the International Research Centre, Kalasalingam University for the SEM/EDS measurements. The authorities of The Madura College, Madurai-625 011, Tamil Nadu, India are gratefully acknowledged for their constant encouragement of the research activities of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sasikumar, S., Saravanan, R., Saravanakumar, S. et al. Preparation, electronic structure, and chemical bonding of lead-free (1 − x)(K0.5Bi0.5)TiO3-xBaTiO3 solid solution. Appl. Phys. A 124, 31 (2018). https://doi.org/10.1007/s00339-017-1444-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00339-017-1444-x