
Abstract
Experimental studies of the structural, magnetic and magnetocaloric properties of the three compounds Pr0.5X0.1Sr0.4MnO3 (X = Ce, Eu and Y) are reported. Our samples were synthesized using the Pechini sol–gel method. X-ray powder diffraction at room temperature indicates that our materials crystallize in the orthorhombic structure with Pbnm space group. The compounds undergo a second-order magnetic transition from paramagnetic to ferromagnetic state around their own Curie temperatures T C ~ 310, 270 and 230 K for X = Ce, Eu and Y, respectively. A considerable magnetocaloric effect (MCE) is observed around room temperature. The maximum values of magnetic entropy change ∆S max are 3.54, 3.81 and 2.99 J/kgK for the samples with X = Ce, Eu and Y, respectively, when a magnetic field of 5 T was applied. The relative cooling power (RCP) values for the corresponding materials are 246.60, 261.66 and 298 J/kg. It is shown that for Pr0.5X0.1Sr0.4MnO3 the exponent n and the magnetic entropy change follow a master curve behavior. With the universal scaling curve, the experimental ∆S at several temperatures and fields can be extrapolated.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Recently, the invention of a magnetic cooling prototype by Zimm et al. [1] has greatly stimulated the search for new generation of refrigerators that are more efficient, economical and environmentally friendly for replacing the current refrigerators using greenhouse gases that are harmful to environment and contributing to ozone-depleting or global warming [2–4]. Practically, magnetic cooling requires the combination of a magnetic field source with high strength and a material with a sufficiently high magnetocaloric effect (MCE) at low magnetic fields, a wide temperature range and a small thermomagnetic hysteresis. In fact, the MCE is associated with temperature change of a magnetic material under the application of magnetic field (µ 0 H). Depending on the direction of µ 0 H, the material can be heated or cooled [5]. This phenomenon is originated from the coupling of a magnetic field with magnetic moments in a solid [6, 7]. For this purpose, the magnetic properties and MCE in lots of manganese oxide systems are systematically investigated, and some of them are found to possess promising MCE properties, such as Pr-based manganites [8–11]. This family of materials showing multifunctional properties has fascinated considerable scientific and technological interests due to the rich fundamental physics and their presence in useful applications [5, 12]. With prominent advantages in terms of large magnetic entropy change ΔS, Pr-based manganites ceramics has been the subject of intensive research. These systems exhibit excellent soft ferromagnetism, high magnetization, low toxicity elements and low cost. Regarding these proprieties, manganites can be considered as a candidate material for MCE application [13]. Additionally, these materials are very convenient for the preparation routes, and their Curie temperature can be justified under the various doping conditions. Fundamentally, the manganese ion exists in two different oxidation states such as Mn3+ and Mn4+ which creates two types of magnetic interactions that are ferromagnetic and antiferromagnetic interactions related to the double-exchange pair of Mn3+–Mn4+ and superexchange pairs of Mn3+–Mn3+ and Mn4+–Mn4+, respectively. Their interaction strength depends on Mn3+/Mn4+ ratio [14], the structural parameters as well as Mn–O bond and Mn–O–Mn bond angles, the average size of the A cation site r A [15] and the disorder degree of the so-called mismatch A-site effect σ 2 [16].
As the MCE properties of manganites are known only to a limited extent, the search for further compositions and preferment elaboration method likely to lead to an increased magnetocaloric effect is still desired. The present paper extends recent studies on Pr-based manganites [17–20] and aims at investigating the structural, magnetic and magnetocaloric properties of Pr0.5X0.1Sr0.4MnO3 ceramics with X = Ce, Eu and Y prepared by using the Pechini sol–gel method. In this study, our doped compounds are characterized by a fixed Mn3+/Mn4+ ratio, while the values of <r A> and \(\sigma^{2}\) depend on the nature of the substituent.
2 Experimental section
Our doped samples Pr0.5X0.1Sr0.4MnO3 were prepared through sol–gel method using nitrate–citrate route [21]. The called sol–gel ceramics were obtained by dissolving highly pure Pr6O11, SrCO3, Ce2O3, Eu2O3, Y2O3 and MnO2 in diluted nitric acid with continuous stirring and moderate heating resulting in a transparent solution. Nitric acid was added to convert the stoichiometric amounts of precursors into their respective nitrates. After total dissolution, citric acid (C6H8O7) in 1:1.5 molar ratio was added as a complexant agent and the pH of the solution was controlled by the addition of NH3. After continuous stirring for 2 h, ethylene glycol was added as a polymerization agent and the mixture is maintained at 90 °C until the formation of brown gel. The brown color of the gel indicates that there are carbonaceous species in the material, presumably due to the onset of decomposition in the polymerized network. Then, this solution was heated at 130 °C and the excess of nitric acid and water was boiled off giving a homogeneous red resin, which was then dried at 150 °C overnight to evaporate a nitrogen element and obtain a black fine powder. The Pr0.5X0.1Sr0.4MnO3 with X = Ce, Eu and Y crystallized powder was obtained by calcination at 500 °C for 6 h. The resulting powder was pressed into pellets forms for 2 min (of about 1 mm thickness and 12 mm diameter) under 4 ton/cm2 and subsequently sintered at 1000 °C for 7 h to be consolidated. The samples were characterized at room temperature by X-ray diffraction with CuKα radiation (λ = 1.5406 Å).
The structural refinement was carried out by the Rietveld analysis of the X-ray powder diffraction data with the help of FULLPROF software [22, 23]. Magnetization measurements versus temperature and versus magnetic applied field up to 5 T were carried out using BS1 and BS2 magnetometers constructed at Néel Institute. The MCE results were deduced from the isothermal magnetization data.
3 Results and discussions
3.1 Structural characterization
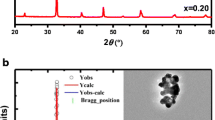
Figure 1 shows that the XRD patterns of the sol–gel made ceramic for Pr0.5X0.1Sr0.4MnO3 (X = Ce, Eu and Y) registered at 300 K including the observed and calculated profiles as well as the difference profile. The quality of the refinement is evaluated through the goodness of the fit indicator Chi-square which is lower than 1.75. The diffraction peaks can be indexed in the orthorhombic setting of the Pbnm space group like bulk Pr0.5Eu0.1Sr0.4MnO3 [24]. The atoms Pr, Ce, Eu and Y are located at 4c (x, y, 1/4) position, Mn at 4b (1/2, 0, 0) and oxygen atoms occupy two sites, specifically O1 at 4c (x, y, 1/4) and O2 at 8d (x, y, z). Regardless of the nature of elements of the substituent, the orthorhombic crystal structure with the Pbnm space group is stable. The obtained results are similar in kind to those observed in the work of Moumen et al. [23].

XRD patterns after Rietveld refinement of Pr0.5X0.1Sr0.4MnO3 (X = Ce, Eu and Y) samples. The vertical tick indicates the allowed reflections. The difference pattern between the observed data and fits is shown at the bottom
It is clear from Fig. 1 that a single phase-material is observed for X = Eu sample without any detectable impurity. A small secondary phase attributed to the presence of Pr6O11 impurity is observed in the material doped by Ce or Y marked with (*) in Fig. 1. The latter is identified with X’Pert HighScore Plus software. Given the small concentration of the impurity, the absence of manganese in it, and the negligible effect on the perovskite stoichiometry, we assume that the secondary phase does not have any significant effect on the subsequent measurements of magnetic properties [11, 25]. Detailed results of the structural parameters deduced from the refinement are summarized in Table 1. The residuals for the weighted pattern R WP, the pattern R P and the structure factor R F are given in Table 1. In Table 2, we have calculated the tolerance factor (t), the average size of A-site cations (r A) and the mismatch size (σ 2) at the A-site. The ionic radii are taken according to Shannon [26]. The lattice parameters a, b and c/\(\sqrt 2\) verified the relation c/\(\sqrt 2\) < a < b indicating that the materials are characterized by the presence of the static Jahn–Teller distortion [27]. In addition, it can be concluded that values verify the orthorhombic distortion condition (0.75 ≤ t ≤ 0.96) which is in accordance with the refinement results [28]. Refinements results of the phase Pr6O11 are reported in Table 3.
In the present samples, the substitution is achieved by replacing the X ion by a trivalent rare earth ion (Ce3+, Eu3+ and Y3+) with different sizes (\(r_{{{\text{Ce}}^{3 + } }}\) = 1.196 Å, \(r_{{{\text{Eu}}^{3 + } }}\) = 1.12 Å and \(r_{{{\text{Y}}^{3 + } }}\) = 1.075 Å). This replacement induces a modification in the chemical internal pressure that affects locally the Mn–O–Mn angle and the Mn–O distance. In our materials, the Mn3+/Mn4+ ratio is fixed and the lattice distortion is ascribed to the variation of the average ionic radius <r A> of the cation site, induced by the size of the substitution. In fact, the participation of the oxygen anion O2− in the dual exchange process allows us to conclude that the more Mn–O–Mn angle moves away from its ideal value (180°), the more the overlap between 3d orbitals of manganese ion Mn and 2p of oxygen O is weakened. Consequently, this leads to the narrowing of the band width W given by [29]:
where θ Mn–O–Mn is the Mn–O–Mn angle and d Mn–O is the Mn–O distance. The wide bandwidth W plays a significant role in the competition between ferromagnetic and antiferromagnetic interactions. A small change in W has a profound effect on magnetic properties of manganites [30]. The values of W are given in Table 1. The W decreases with decreasing the average ionic radius <r A>. Thus, an overall increase in the bandwidth W, thereby leading to an enhancement of T C. It is obvious that, in the Pr0.5Y0.1Sr0.4MnO3 sample, the distortion compared with the ideal cubic perovskite structure (θ Mn–O–Mn = 180° et t = 1) is the highest among our manganites and this leads to obtain the smallest volume (V X = Y = 227.84 (Å)3).
3.2 Magnetic properties
Magnetization versus temperature has been undertaken under an applied magnetic field of 0.05 T in the field-cooled (FC) mode for Pr0.5X0.1Sr0.4MnO3 materials and shown in Fig. 2a. All materials exhibit a pointed transition from high-temperature paramagnetic state to low-temperature ferromagnetic state. The M(T) curves do not reveal secondary magnetic phases, which indicates that the magnetic behavior is not affected by the presence of small amount of secondary phase [11]. We see clearly by examining the M(T) curve of the Pr0.5Ce0.1Sr0.4MnO3 sample shows a steplike decrease at 170 K. A similar behavior has been observed in previous works on the Pr0.6Sr0.4MnO3 [31, 32]. The authors suggest that the steplike decrease is not due to AFM transition. Using neutron diffraction study, the mentioned anomaly is attributed to orthorhombic to monoclinic structural phase transition upon cooling [28]. Besides, we find, from Fig. 2, that the transitions of our samples are sharp. This allows us to predict in advance that these samples are able to have a strong magnetocaloric behavior around Curie temperature T C [33]. T C defined as the temperature at which the (dM/dT − T) curve reaches a minimum (see Fig. 2). The Curie temperatures are equal to 310, 270 and 230 K for X = Ce, Eu and Y, respectively, and confirm the decrease in bandwidth W for the three compounds. This allows us to conclude that these materials are able to be exploited in a wide temperature range containing room temperature. So we just should associate them together to obtain a composite material usable in the magnetic refrigeration technology [34, 35].

a Temperature dependence of magnetization under 0.05 T for Pr0.5X0.1Sr0.4MnO3 (X = Ce, Eu and Y). b The dM/dT curves for Pr0.5X0.1Sr0.4MnO3
In order to confirm the ferromagnetic behavior observed in our materials, the magnetization M (T, μ 0 H) is measured for all samples. Figure 3 shows the isothermal magnetization curves for the typical Pr0.5Y0.1Sr0.4MnO3. The M–H curves reveal a strong variation of magnetization around the Curie temperature. It indicates that there is a possible large magnetic entropy change associated with the ferromagnetic–paramagnetic transition temperature occurring at T C. It can be seen that the M (T, μ 0 H) at T < T C confirmed the ferromagnetic behavior of our sample. The magnetization increases sharply with magnetic applied field for µ 0 H < 1 T but does not reach saturations up to 5 T indicating that the magnetic moment of the rare earth ions is polarized and pinned by the Mn sublattice [36]. At low temperature, the spontaneous magnetization M sp(exp) determined by the extrapolation of the M versus µ 0 H data in the high field to zero field is found to be 80.72 emu/g (~3.05 μ B/Mn), 82.58 emu/g (~3.13 μ B/Mn) and 78.86 emu/g (~2.91 μ B/Mn) for The Pr0.5X0.1Sr0.4MnO3 with X = Ce, Eu and Y, respectively (see Fig. 3b). The magnitude of the M sp(exp) is smaller than the theoretical value of 3.5 μ B/Mn calculated for full spin alignment, leading to small canted spins. This confirms the presence of the antiferomagnetic (AFM) interactions, indicating that the magnetic moment of the rare earth ions is polarized and pinned by the Mn sublattice [31].

a Isothermal magnetization curves at various temperatures for Pr0.5Y0.1Sr0.4MnO3 sample. b Magnetization versus magnetic applied field at 20 K for Pr0.5X0.1Sr0.4MnO3 (X = Ce, Eu and Y) samples
3.3 Magnetocaloric effect
To see the effect of (Ce, Eu and Y) doping on magnetic entropy change, we have calculated the total magnetic entropy change ΔS(T, μ 0 H) for all materials. Based on the classical thermodynamical theory, the magnetic entropy change ∆S (T, μ 0 H) associated with a magnetic field variation is given by:
Experimentally, there are two ways to estimate the ∆S: The first one is measurement of the magnetization versus temperature under different applied magnetic fields, and the second one is measurement of the M–H curve at several temperatures. In this work, we use the second way to estimate the magnetic entropy change. For magnetization measured at discrete field and temperature intervals, ∆S can be approximated to be:
where M i and M i+1 are the experimental magnetization values measured in µ 0 H i , at temperature T i and T i+1, respectively. The entropy change for Pr0.5X0.1Sr0.4MnO3 (X = Ce, Eu and Y) compounds calculated as a function of temperature at several magnetic applied fields is given in Fig. 4a, b and c (the symbols represent experimental data). The magnetic entropy changes reach its maximum around T C and increase with the magnetic field change. −ΔS observed for X = Ce, Eu and Y is depicted in Fig. 4d under a magnetic field of 2 and 5 T. At 5 T applied field, the maximum magnetic entropy change ∆S max is obtained to be 3.54 J kg−1 K−1 for X = Ce and 3.81 J kg−1 K−1 for X = Eu and 2.99 for X = Y. The magnetic entropy change of the Pr0.5X0.1Sr0.4MnO3 samples as a function of temperature exhibits a broad variation with temperature around T C. Such a variation covering a broad temperature range is advantageous for magnetic refrigeration [37]. This is a very interesting result, because we are able to change MCE and width of peak temperature range of the material by changing the nature of substitution and may select optimization, which is desirable for Ericsson-cycle magnetic cooling and heat exchange technology.

Experimental (open symbols) magnetic entropy change and predictions using the universal curve (continuous lines) for a Pr0.5Ce0.1Sr0.4MnO3, b Pr0.5Eu0.1Sr0.4MnO3 and c Pr0.5Y0.1Sr0.4MnO3. d Magnetic entropy change as a function of temperature at µ 0 H = 2 and 5 T for all samples
In addition to the magnitude of the ∆S, another important parameter used to characterize the cooling efficiency of the material is the relative cooling power (RCP). RCP gives an estimate of quantity of the heat transfer between the hot (T hot) and cold (T cold) end during one refrigeration cycle and is the area under the ∆S (T) curve between two temperatures (∆T = T hot–T cold) of the full width at half maximum (FWHM) of the curve. RCP values under an applied field of 5 T are about 246.6, 261.66, 298 J/kg for Pr0.5X0.1Sr0.4MnO3 (X = Ce, Eu and Y), respectively, which is ~60.1, 63.82 and 72.70 % of that of Gd metal at the same field (for Gd metal, RCP = 410 J/kg at 5 T. In Table 4, we compare the entropy changes of Pr0.5X0.1Sr0.4MnO3 with other A-site multi-element doping effect like Sr and Ca in several manganites system. The obtained values were comparable with those mentioned in Table 4. Phan et al. [13] studied the MCE in perovskite manganites, and they have explained that the large MCE in the perovskite manganites can originate from the spin–lattice coupling related to the magnetic ordering process. The results are obtained in the temperature range that covers the room temperature which is beneficial and strongly requested for magnetic cooling applications [38]. These materials are thus considered as an alternative option for refrigeration applications because the expensiveness of Gd (a prototype magnetic-refrigerant material) is limited its applicability in commercial cooling devices.
A phenomenological universal curve, for the field dependence of the magnetic entropy, has been proposed being the theoretical justification for its existence based on scaling relations [39, 40]. This universal curve can be particularly helpful for studying order of phase transition and refrigerant capacity of similar materials, such as manganite series with same universality class. The method is also used to compare the properties of several compounds and to make extrapolations to temperatures and/or fields outside the available experimental range [41].
The universal curve is made by using normalized entropy change:
where ∆S peak is the peak maximum magnetic entropy change at several magnetic fields. And the temperature axis is rescaled by a new parameter θ, defined by the expression [35]:
where T r1 and T r2 are the temperatures where ∆S equals ∆S/2. Figure 5a, b and c shows the θ dependence of ∆S(T) for typical field changes for Pr0.5X0.1Sr0.4MnO3 and confirms the predictions of a master curve behavior for several materials. The transformed curves ∆S peak/∆S versus θ of X = Ce, Eu and Y under magnetic fields are depicted in Fig. 5d. It can be clearly seen that the experimental points of the samples collapse into a single master curve of the magnetic entropy change, demonstrating the predictions of master curve behavior for Pr0.5X0.1Sr0.4MnO3 system. The existence of the universal curve of ∆S around T C confirms the nature of second-order phase transition for X = Ce, Eu and Y. This implies that the present materials have the same nature of phase transitions and belong to the same universality class. The inset of Fig. 5a, b and c shows the magnetic field dependences of the two reference temperatures (T r1 and T r2). The average curve of ∆S peak/∆S versus θ is plotted in Fig. 5 and gives a smoother depiction of the curve. This average curve, once the temperature axis is back-transformed from the reduced temperature to the unnormalized one, allows making extrapolations to lower (higher) temperatures for the high-field data and obtaining a finer description of the peak for the low-field curves [42, 43]. The inverse transformation to the experimental temperature and magnetic entropy change axes can be done by combining all data in the average universal curve. This way permits to provide a prediction about the behavior of the ∆S(T) curve. Figure 4a, b and c shows the good agreement between the predicted curves (red solid curves) and experimental points (symbols).

Shows the collapse of the experimental data onto the universal curve and shows the dependences of the two reference temperatures (T r1 and T r2) at various magnetic fields for a Pr0.5Ce0.1Sr0.4MnO3, b Pr0.5Eu0.1Sr0.4MnO3, and c Pr0.5Y0.1Sr0.4MnO3. d Master curve behavior for the magnetic entropy change of all samples
Numerous works have been done concerning the field dependence of the magnetic entropy change (ΔS) of manganites at the FM–PM transition T C. According to Oesterreicher et al. [44], µ 0 H dependences of ΔS max can be described by a power function ΔS max ≈ (μ 0 H)n [34] where the exponent ‘n’ depends up on the magnetic state of the sample. For a magnetic substance undergoing the second-order magnetic transition, the interactions existed near T C obey to the mean field theory (MFT). According MFT, the direct integration of the Curie–Weiss law indicates that n = 2 at T ≫ T C. At temperatures T ≪ T C, n tends to 1 and is magnetic field independent. At the Curie temperature, the field dependence of the magnetic entropy change corresponds to n = 2/3 [35].
Figure 6a, b and c shows the temperature dependence of n for different values of the maximum applied field for Pr0.5X0.1Sr0.4MnO3. However, n(T, µ 0 H) curves show the minima around their Curie temperature. Its peak position shifts toward high temperatures when the magnetic applied field increases. The exponent n exhibits a moderate decrease with increasing temperature. The n value reaches 1 below the T C and 2 above the T C. At T C, the exponent n for the three samples is larger than the predicted value of 2/3. The disagreements from theoretical value are due to the local inhomogeneities or superparamagnetic clusters in the vicinity of T C existed in the Pr0.5X0.1Sr0.4MnO3 with X = Ce, Eu and Y [45]. A similar result has been observed in other manganites system [46]. The universal curve is made by using exponent n as a function of the rescaled temperature (θ) for the magnetic ordering transitions of our materials. The insets in Fig. 6 show the θ dependence of the local exponent n. The procedure consists in identifying the reference temperatures as those which have a certain value of n. This value has been arbitrarily selected as n(Tr) = 1.5. The present samples (X = Ce, Eu and Y) show a collapse of n into a master curve confirming the second-order nature of phase transition (see Fig. 6d). In general, deviations from the collapse might indicate either the influence of the demagnetizing field associated with the shape of the sample [47] or to the presence of additional magnetic phases [48, 49]. The divergence of the curves is clear in the Pr0.5Y0.1Sr0.4MnO3 sample, particularly above the T C, and suggests an increased sensitivity of n to applied magnetic field.

Local exponent n at several magnetic fields for a Pr0.5Ce0.1Sr0.4MnO3, b Pr0.5Eu0.1Sr0.4MnO3 and c Pr0.5Y0.1Sr0.4MnO3. In the insets: local exponent n as a function of the rescaled temperature θ for all magnetic fields. d Local exponent n as a function of the rescaled temperature θ for all materials
4 Conclusion
In summary, the Pr0.5X0.1Sr0.4MnO3 samples were successfully prepared via the modified nitrate–citrate route method. X-ray diffraction measurements revealed that all crystallize in the orthorhombic structure with the space group Pbnm and also demonstrated the presence of a secondary phase Pr6O11 in the two samples Pr0.5Ce0.1Sr0.4MnO3 and Pr0.5Y0.1Sr0.4MnO3. The samples exhibit a typical second-order magnetic phase transition at T C, which is obtained to be 230 and 310 K for X = Y and X = Ce samples, respectively. The substitution of Pr affected the magnetization curves, but M(T) of the doped materials remains of the same order of magnitude as values obtained for Pr0.6Sr0.4MnO3. The Pr0.5X0.1Sr0.4MnO3 manganites are particularly pronounced with wide operative temperature range, as well as inexpensiveness and easy fabrication, and therefore could be considered as a potential magnetic refrigerant. The X-sample displays better magnetic properties as far as the magnetic cooling applications are considered. Consequently, it is plausible to conclude that X3+ plays an important role in modifying physical properties of these materials in spite of the fact that the Mn3+/Mn4+ ratio remains fixed by partially substituting Pr by a fixed amount of X in all samples. Furthermore, the experimental ∆S collapses onto a universal curve based on the scaling relation, where an average curve is obtained. With the universal scaling curve, the experimental ∆S at several temperatures and fields can be extrapolated.
References
C.B. Zimm, A. Jastrab, A. Sternberg, V.K. Pecharsky, K.A. Gschneidner Jr, M. Osborne et al., Description and performance of a near-room temperature magnetic refrigerator. Adv. Cryog. Eng. N. Y. 43, 1759 (1998)
K.A. Gschneidner Jr, V.K. Pecharsky, Magnetocaloric materials. Ann. Rev. Mater. Sci. 30, 387 (2000)
J.A. Barclay, Active and passive magnetic regenerators in gas/magnetic refrigerators. J. Alloys Compd. 207–208, 355 (1994)
R. M’nassri, A. Cheikhrouhou, Magnetocaloric properties in ordered double-perovskite Ba2Fe1−x Cr x MoO6 (0 ≤ x ≤ 1). J. Korean Phys. Soc. 64, 879 (2014)
A.M. Tishin, Y.I. Spichkin, The Magnetocaloric Effect and its Applications (Institute of Physics Publishing, Bristol, 2003)
K.A. Gschneidner Jr, V.K. Pecharsky, A.O. Tsokol, Recent developments in magnetocaloric materials. Rep. Prog. Phys. 68, 1479 (2005)
S.M. Benford, G.V. Brown, T–S diagram for gadolinium near the Curie temperature. J. Appl. Phys. 52, 2110 (1981)
Y. Regaieg, M. Koubaa, W. Cheikhrouhou Koubaa, A. Cheikhrouhoua, T. Mhiri, Magnetocaloric effect above room temperature in the K-doped La0.8Na0.2−x K x MnO3 manganites. J. Alloys Compd. 502, 270 (2010)
A. Selmi, R. M’nassri, W. Cheikhrouhou-Koubaa, N. Chniba Boudjada, A. Cheikhrouhou, Influence of transition metal doping (Fe Co, Ni and Cr) on magnetic and magnetocaloric properties of Pr0.7Ca0.3MnO3 manganites. Ceram. Int. 41, 10177 (2015)
R. M’nassri, W. Cheikhrouhou-Koubaa, M. Koubaa, N. Boudjada, A. Cheikhrouhou, Magnetic and magnetocaloric properties of Pr0.6−x Eu x Sr0.4MnO3 manganese oxides. Solid State Commun. 151, 1579 (2011)
S. Mahjoub, M. Baazaoui, R. M’nassri, H. Rahmouni, N. Chniba Boudjada, M. Oumezzine, Effect of iron substitution on the structural, magnetic and magnetocaloric properties of Pr0.6Ca0.1Sr0.3Mn1−x Fe x O3 (0 ≤ x≤ 0.075) manganites. J. Alloys Compd. 608, 191–196 (2014)
C.N.R. Rao, B. Raveau (eds.), Colossal Magnetoresistance, Charge Ordering and Related Properties of Manganese Oxides (Singapore, World Scientific, 1998)
M.H. Phan, S.C. Yu, Review of the magnetocaloric effect in manganite materials. J. Magn. Magn. Mater. 308, 325 (2007)
A.M. Aliev, A.G. Gamzatov, A.B. Batdalov, A.S. Mankevich, I.E. Korsakov, Structure and magnetocaloric properties of La1−x K x MnO3 manganites. Phys. B 406, 885 (2011)
V.S. Kolat, T. Izgi, A.O. Kaya, N. Bayri, H. Gencer, S. Atalay, Metamagnetic transition and magnetocaloric effect in charge-ordered Pr0.68Ca0.32−x Sr x MnO3 (x = 0, 0.1, 0.18, 0.26 and 0.32) compounds. J. Magn. Magn. Mater. 322, 427 (2010)
C. Hao, B. Zhao, Y. Huang, G. Kuang, Y. Sun, A-site-disorder-dependent magnetocaloric properties in the mono-valent-metal doped La0.7Ca0.3MnO3 manganites. J. Alloys Compd. 509, 5877 (2011)
A. Selmi, R. M’nassri, W. Cheikhrouhou-Koubaa, N. Chniba Boudjada, A. Cheikhrouhou, Effects of partial Mn-substitution on magnetic and magnetocaloric properties in Pr0.7Ca0.3Mn0.95X0.05O3 (Cr, Ni, Co and Fe) manganites. J. Alloys Compd. 619, 627 (2015)
A. Bettaibi, R. M’nassri, A. Selmi, H. Rahmouni, N. Chniba-Boudjada, A. Chiekhrouhou, K. Khirouni, Effect of chromium concentration on the structural, magnetic and electrical properties of praseodymium-calcium manganite. J. Alloys Compd. 650, 268 (2015)
A. Selmi, R. M’nassri, W. Cheikhrouhou-Koubaa, N. Chniba Boudjada, A. Cheikhrouhou, The effect of Co doping on the magnetic and magnetocaloric properties of Pr0.7Ca0.3Mn1−x Co x O3 manganites. J. Ceram. Int. 41, 7723 (2015)
R. M’nassri, N. Chniba Boudjada, A. Cheikhrouhou, Impact of sintering temperature on the magnetic and magnetocaloric properties in Pr0.5Eu0.1Sr0.4MnO3 manganites. J. Alloys Compd. 626, 20 (2015)
Huili Li, Xuejian Liu, Liping Huang, Synthesis of lutetium aluminum garnet powders by nitrate–citrate sol–gel combustion process. Ceram. Int. 33, 1141–1143 (2007)
H.M. Rietveld, A profile refinement method for nuclear and magnetic structures. J. Appl. Cryst. 2, 65 (1969)
T. Roisnel, J. Rodriguez-Carvajal, Computer programfullprof. LLB-LCSIM (2003)
M. Moumen, A. Mehri, W. Cheikhrouhou-Koubaa, M. Koubaa, A. Cheikhrouhou, Structural, magnetic and magnetocaloric properties in Pr0.5M0.1Sr0.4MnO3 (M = Eu, Gd and Dy) polycristalline manganites. J. Alloys Compd. 509, 9084 (2011)
S. Zouari, L. Ranno, A. Cheikh-Rouhou, P. Strobel, Effect of mixed alkali-element substitution on structural and magnetic properties of praseodymium manganites Pr0.9(Na1−x K x )0.1MnO3. J. Alloys Compd. 452, 249 (2008)
R.D. Shannon, Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. A 32, 751 (1976)
K. Knizek, Z. Jira’k, E. Pollert, F. Zounova, S. Vratislav, Structure and magnetic properties of Pr1−x Sr x MnO3 perovskites. J. Solid State Chem. 100, 292 (1992)
J. Cibert, J.F. Bobo, U. Lüders, C. R. development of new materials for spintronics. C. R. Phys. 6, 977 (2005)
M. Medarde, J. Mesot, P. Lacorre, S. Rosenkranz, P. Fischer, K. Gobrecht, High-pressure neutron-diffraction study of the metallization process in PrNiO3. Phys. Rev. B 52, 9248 (1995)
Y. Moritomo, H. Kuwahara, Y. Tomioka, Y. Tokura, Pressure effects on charge-ordering transitions in Perovskite manganites. Phys. Rev. B 55, 7549 (1997)
D.V. MaheswarRepaka, T.S. Tripathi, M. Aparnadevi, R. Mahendiran, Magnetocaloric effect and magnetothermopower in the room temperature ferromagnet Pr0.6Sr0.4MnO3. J. Appl. Phys. 112, 123915 (2012)
W. Boujelben, M. Ellouze, A. Cheih-Rouhou, J. Pierre, Q. Cai, W.B. Yelon, K. Shimizu, C. Dubourdieu, Neutron diffraction, NMR and magneto-transport properties in the Pr0.6Sr0.4MnO3 perovskite manganite. J. Alloys Compd. 334, 1 (2002)
R. M’nassri, W. Cheikhrouhou-Koubaa, N. Boudjada, A. Cheikhrouhou, Magnetocaloric effects in Pr0.6−x Er x Sr0.4MnO3 (0.0 ≤ x ≤0.2) manganese oxides. J. Supercond. Nov. Magn. 26, 1429 (2013)
R. M’nassri, Enhancement of refrigeration capacity and table-like magnetocaloric effect in LaFe10.7Co0.8Si1.5/La0.6Pr0.4Fe10.7Co0.8Si1.5 composite. J. Supercond. Nov. Magn. 29, 207–213 (2016)
R. M’nassri, Enhanced refrigerant capacity and magnetic entropy nearly flattening in (La2/3Ba1/3MnO3)1−x/(La2/3Ba1/3MnO2.98)x composite. J. Supercond. Nov. Magn. (2016). doi:10.1007/s10948-016-3480-8
L. Ling, J. Fan, L. Pi, S. Tan, Y. Zhan, Effect of magnetism and average radius at A-site on in (Pr, Gd, Dy) system. Solid State Commun. 145, 11 (2008)
R. M’nassri, Field dependence of magnetocaloric properties in La0.6Pr0.4Fe10.7Co0.8Si1.5. J. Supercond. Nov. Magn. 27, 1787 (2014)
S. Choura Maatar, R. M’nassri, W. Cheikhrouhou Koubaa, M. Koubaa, A. Cheikhrouhou, Structural, magnetic and magnetocaloric properties of La0.8Ca0.2−x Na x MnO3 manganites (0 ≤ x ≤0.2). J. Solid State Chem. 225, 83 (2015)
V. Franco, J.S. Blázquez, A. Conde, Field dependence of the magnetocaloric effect in materials with a second order phase transition: a master curve for the magnetic entropy change. Appl. Phys. Lett. 89, 222512 (2006)
V. Franco, A. Conde, Scaling laws for the magnetocaloric effect in second order phase transitions: from physics to applications for the characterization of materials. Int. J. Refrig. 33, 465 (2010)
R. M’nassri, N. Chniba Boudjada, A. Cheikhrouhou, 3D-Ising ferromagnetic characteristics and magnetocaloric study in Pr0.4Eu0.2Sr0.4MnO3 manganite. J. Alloys Compd. 640, 183 (2015)
L. Zhang, J. Fan, W. Tong, L. Ling, L. Pi, Y. Zhang, Scaling of the magnetic entropy change in spinel selenide CuCr2Se4. Phys. B 407, 3543 (2012)
V.M. Prida, V. Franco, V. Vega, J.L. Sanchez-Llamazares, J.J. Sunol, A. Conde, B. Hernando, Magnetocaloric effect in melt-spun FePd ribbon alloy with second order phase transition. J. Alloys Compd. 509, 190 (2011)
H. Oesterreicher, F.T. Parker, Magnetic cooling near Curie temperatures above 300 K. J. Appl. Phys. 55, 4334 (1984)
V. Franco, J.S. Blázquez, A. Conde, The influence of Co addition on the magnetocaloric effect of Nanoperm-type amorphous alloys. J. Appl. Phys. 100, 064307 (2006)
The-Long Phan, P. Zhang, T.D. Thanh, S.C. Yu, Crossover from first-order to second-order phase transitions and magnetocaloric effect in La0.7Ca0.3Mn0.91Ni0.09O3. J. Appl. Phys. 115, 17A912 (2014)
R. Caballero-Flores, V. Franco, A. Conde, L.F. Kiss, Influence of the demagnetizing field on the determination of the magnetocaloric effect from magnetization curves. J. Appl. Phys. 105, 07A919 (2009)
V. Franco, A. Conde, V. Provenzano, R.D. Shull, Scaling analysis of the magnetocaloric effect in Gd5Si2Ge1.9X0.1 (X = Al, Cu, Ga, Mn, Fe, Co). J. Magn. Magn. Mater. 322, 218 (2010)
V. Franco, R. Caballero-Flores, A. Conde, Q.Y. Dong, H.W. Zhang, The influence of a minority magnetic phase on the field dependence of the magnetocaloric effect. J. Magn. Magn. Mater. 321, 1115 (2009)
V.K. Pecharsky Jr, K.A. Gschneidner, Giant magnetocaloric effect in Gd5(Si2Ge2). Phys. Rev. Lett. 78, 4494 (1997)
M.H. Phan, H.X. Peng, S.C. Yu, Large magnetocaloric effect in single crystal Pr0.63Sr0.37MnO3. J. Appl. Phys. 97, 10M306 (2005)
M.H. Phan, S.C. Yu, N.H. Hur, Excellent magnetocaloric properties of La0.7Ca0.3−x Sr x MnO3 (0.05 ≤ x ≤0.25) single crystals. Appl. Phys. Lett. 86, 072504 (2005)
A. Rostamnejadi, M. Venkatesan, P. Kameli, H. Salamati, J.M.D. Coey, Magnetocaloric effect in La0.67Sr0.33MnO3 manganite above room temperature. J. Magn. Magn. Mater. 323, 2214 (2011)
N.S. Bingham, M.H. Phan, H. Srikanth, M.A. Torija, C. Leighton, Magnetocaloric effect and refrigerant capacity in charge-ordered manganites. J. Appl. Phys. 106, 023909 (2009)
Acknowledgments
This study is supported by the Tunisian Ministry of Higher Education and Scientific Research and the Neel Institute.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sakka, A., M’nassri, R., Chniba-Boudjada, N. et al. Effect of trivalent rare earth doping on magnetic and magnetocaloric properties of Pr0.5(Ce,Eu,Y)0.1Sr0.4MnO3 manganites. Appl. Phys. A 122, 603 (2016). https://doi.org/10.1007/s00339-016-0125-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00339-016-0125-5