
Abstract
Genetic population patterns in coral reefs are important for understanding biodiversity and forecasting recovery following disturbance. Studying gene flow between small and disrupted marine ecosystems, such as the reefs along the Singapore Strait, is especially crucial due to their fragile nature. Here we sampled the ubiquitous coral species complex Pachyseris speciosa from seven sites in Singapore and applied genotyping-by-sequencing (nextRAD) for a fine-scale population genomic assessment. Our results confirm the existence of two clearly distinct, possibly cryptic, lineages that occur sympatrically at each of the seven sampling sites and are related to the “blue” and “green” lineages found in other parts of the Indo-Pacific. The closely related lineages show contrasting patterns characterised by panmixia and substantial (but unexplained) substructuring, respectively. Analysis of barriers and corridors to gene flow reveals limited connectivity between the two largest clusters of reefs at the eastern and western sectors of Singapore’s southern coastal waters. Our results extend the geographic range of the two recently uncovered P. speciosa species, further confirming their status as distinct and widespread species. More broadly, our findings highlight how conservation and management strategies may maximise connectivity among Singapore’s urbanised reef ecosystems.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Describing all the species on Earth is a task that may never be completed (Scheffers et al. 2012). Yet, estimation of species diversity has been and continues to be of great interest, motivated mainly by the need to provide a reference point for current and future losses of biodiversity (Mora et al. 2011). Assessments on the number of marine species range widely, between 226,000 species at the conservative end (Appeltans et al. 2012) and up to 10 million and more (Frederick Grassle and Maciolek 1992; Costello et al. 2012). A significant factor for this broad range is the presence of taxonomically cryptic species (Bickford et al. 2007; Pfenninger and Schwenk 2007; Nygren 2014), and methods for delimiting them are one of the most pertinent issues in biological science (Sites and Marshall 2003). Taxonomically cryptic species are broadly defined as two or more genetically distinct taxa that are erroneously classified as a single species (Richards et al. 2016; Sheets et al. 2018; Bongaerts et al. 2021). This problem could be due to morphological resemblance despite the genetic divergence (Pfenninger and Schwenk 2007; Fišer et al. 2018). Studies on cryptic species have increased exponentially over the past three decades since genetic techniques started to be applied (Bickford et al. 2007; Nygren 2014). They are evenly distributed among biogeographical regions (Pfenninger and Schwenk 2007) and could be found in many major metazoan and plant groups (Grundt et al. 2006; Pfenninger and Schwenk 2007). In particular, marine environments are known to host numerous such cryptic species groups likely due to their species-rich habitats (Willig et al. 2003). Identification of cryptic species is crucial for biodiversity conservation, management and understanding the evolutionary processes which drive speciation (Chadè et al. 2008; Baird et al. 2011; Andrews et al. 2014). Given that most species remain undescribed, efforts to catalogue and explain biodiversity need to be prioritised (Bickford et al. 2007).
Scleractinian corals (Cnidaria: Anthozoa: Scleractinia) are difficult to classify due to their morphological plasticity (Todd 2008; Paz-García et al. 2015) and pervasive convergent evolution (e.g. Fukami et al. 2004, 2008; Huang et al. 2009). Taxonomically cryptic diversity has been documented in several coral genera, for example, in Acropora Oken, 1815 (Ladner and Palumbi 2012; Richards et al. 2016), Stylophora Schweigger, 1820 (Stefani et al. 2011), Pocillopora Lamarck, 1816 (Souter 2010; Schmidt-Roach et al. 2013; Gélin et al. 2017), and Seriatopora Lamarck, 1816 (Bongaerts et al. 2010; Warner et al. 2015). For centuries, morphology was the sole means to delimit one species from another (e.g. Lamarck 1815; Wells 1956; Chevalier 1975; Lang 1984; Veron 2002). However, integrative studies that rely on combining morphological and multi-locus molecular approaches for phylogenetic reconstruction have proved to be furthermost accurate (Benzoni et al. 2007, 2011, 2014; Kitahara et al. 2012, 2013; Huang et al. 2014; Arrigoni et al. 2014, 2015). For example, Arrigoni et al. (2019) recently described a new cryptic genus and species Paraechinophyllia variabilis, Arrigoni, Benzoni and Stolarski, 2019, which could not be distinguished from other lobophylliids based on morphological characters alone, and delimited it from other species from the family using six molecular markers (COI, 12S, ATP6-NAD4, NAD3-NAD5, histone H3 and ITS). Notably, species boundaries within the genus Leptastrea Milne-Edwards & Haime, 1849, have been evaluated by applying restriction site-associated DNA sequencing (RADseq) to sample single nucleotide polymorphisms (SNPs) across the genome, complementing detailed morphological diagnoses (Arrigoni et al. 2020). Such reduced-representation genomic approaches provide high coverage of homologous portions of the genome from multiple individuals for comparatively low cost and effort by sequencing only certain regions of DNA adjacent to restriction endonuclease sites (Toonen et al. 2013).
The zooxanthellate genus Pachyseris Milne-Edwards & Haime, 1849, is widely distributed throughout the Indo-Pacific and can be found in all reef habitats but most commonly on lower reef slopes (Veron 1980, 2000; Scheer and Pillai 1983; Sheppard and Sheppard 1991). In particular, Pachyseris speciosa Dana, 1846, the most widespread nominal species in the genus, is found in the Indo-Pacific from the Red Sea to the Philippines and Tahiti (Veron 2000; Hughes et al. 2013). There are currently six valid species in the genus (Hoeksema and Cairns 2020), which has been well represented in systematic and phylogenetic studies (Fukami et al. 2008; Kitahara et al. 2010; Terraneo et al. 2014). However, the relationships among Pachyseris species and within even the most widespread P. speciosa are unclear. Terraneo et al. (2014) reconstructed Pachyseris phylogeny inferred from Bayesian inference analysis of mitochondrial intergenic spacer between COI and 16S-rRNA. Three lineages were analysed: P. rugosa and P. speciosa were recovered as sister species and both were sister to P. inattesa. Despite the ecological prominence of P. speciosa on Indo-Pacific reefs, much of its evolution and population genetic structure remain obscure. Recently, Bongaerts et al. (2021) used reduced-representation sequencing to show that P. speciosa actually represents a species complex, with three sister species occurring sympatrically throughout Australasia. The genomic differentiation was accompanied by ecological, physiological and reproductive differences, yet there was a lack of morphological characters distinguishing the three lineages.
Members of the P. speciosa species complex are characterised as gonochores with a broadcasting reproductive mode (Kerr et al. 2011; Bongaerts et al. 2021). Their ecologically opportunistic nature is reflected in it being one of the most common species throughout Singapore’s reefs (Guest et al. 2016; Wong et al. 2018) and across depths (Chow et al. 2019). Ship movement rousing the shallow seafloor of the Singapore Strait (Browne et al. 2014, 2015) and the extensive urban coastal development projects are the main causes for the turbid water in the reefs here (Dikou and van Woesik 2006; Sin et al. 2016). Due to this high turbidity and the consequent high light attenuation (i.e. < 1% of surface PAR at ~ 9 m depth; Todd et al. 2004), foliose corals with large areal extents for light capture, such as P. speciosa, are most common at 3–8 m depth (Dikou and van Woesik, 2006; Guest et al. 2016; Chow et al. 2019).
In this study, we used a genotyping-by-sequencing approach to characterise the P. speciosa species complex and population genomic variation across seven sites on Singapore reefs (~ 2–15 km apart) situated along the Singapore Strait. We confirm the presence of two distinct and possibly cryptic species with further genetic substructuring uncovered that cannot be related to the studied geographic locations.
Materials and methods
Sample collection and DNA extraction
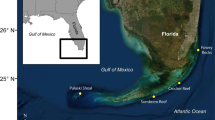
We focused on the highly urbanised reef system in Singapore where we targeted seven sampling sites—Raffles, Semakau, Hantu, TPT, Kusu, St. John and Sisters Islands (Fig. 1). Sites were chosen depending on the abundance and availability of Pachyseris speciosa. Colonies were identified to species based on the original description by Dana, 1846, supported by subsequent descriptions as well as images of the holotype and live colonies (Veron and Pichon 1980; Veron 2000; Terraneo et al. 2014). From each site, between 18 and 23 samples were taken from 4 to 6 m depth using a cutter (Table S1). We kept at least 5 m apart from sample to sample in order to avoid sampling genetically identical colonies. All samples were preserved in 100% molecular grade ethanol and stored at − 80 °C immediately after collection. Genomic DNA was extracted using Qiagen DNeasy Blood and Tissue Kit. Post-extraction, gel electrophoresis test was carried out in order to ensure that samples were clean from RNA or proteins. DNA quantification was performed using a Qubit 3 Fluorometer.

A map of all seven sampling sites at the offshore islands of Singapore
NextRAD library preparation and sequencing
All 144 samples were genotyped via nextRAD genotyping-by-sequencing (SNPsaurus, LLC), following library preparation as detailed in Russello et al. (2015). Briefly, DNA (~ 12 ng) was first fragmented and adapter-ligated with the Nextera DNA Library Prep Kit (Illumina, Inc). Fragmented DNA was then amplified with one of the primers matching the adapter and extending nine nucleotides into the genomic DNA with the selective sequence 5′–GTGTAGAGG–3′. Thus, only fragments starting with a sequence that can be hybridised by the selective sequence of the primer would be efficiently amplified. The nextRAD libraries were then sequenced on three HiSeq 4000 lanes for 150-bp single-end reads (University of Oregon).
Genotyping and quality control
The genotyping analysis used custom scripts (SNPsaurus, LLC) that trimmed the reads using bbduk (BBTools package, Brian Bushnell, Walnut Creek, CA, USA) (see Supplementary File 1). Next, all remaining reads were mapped to the Pachyseris speciosa genome (Bongaerts et al. 2021; downloaded from http://reefgenomics.org) (see Supplementary File 1). Genotype calling was performed using Samtools and bcftools (Li et al. 2009) and compiled in Variant Call Format (VCF) files using custom parameters (see Supplementary File 1). The VCF files were filtered to remove alleles with a population frequency of less than 3%. SNPs that were heterozygous in all samples, or had more than two alleles in a sample were also removed. PGDSpider (version 2.1.1.5) (Lischer and Excoffier 2012) was used to reformat the VCF files for downstream analyses. The remaining SNPs were evaluated for significant deviations from Hardy–Weinberg equilibrium and linkage using arlecore (version 3.5.2.2), with SNPs that deviated in more than five a priori populations removed as in Bongaerts et al. (2017). The clonecorrect function in R package poppr (version 2.8.1) (Kamvar et al. 2014, 2015) was used to remove potential clones from the dataset, with clonal groups reduced to a single representative per population. Finally, only SNPs with 1% missing data and samples with < 15% missing data were retained to ensure high-quality downstream analyses (see Afiq-Rosli et al. 2021). Three datasets with varying filtering parameters were assembled for analysis: overall dataset (all SNPs), neutral dataset (SNPs under selection removed) and outlier dataset (only SNPs identified as under selection). BayeScan (version 2.1) (Foll and Gaggiotti 2008) using default parameters (see Supplementary File 1) and Bayes factor cut-off of 0.05 were used to identify SNPs under possible selection.
Genetic structuring and connectivity analyses
To assess genetic structure for each dataset, Bayesian clustering analysis was performed in STRUCTURE version 2.3.4 for up to seven possible genetic clusters (K) according to the total number of collection sites. We considered correlated allele frequencies in the admixture model, using sampling locations as priors, and ran 10 iterations of 100,000 MCMC repetitions with the first 10,000 as burn-in (Gilbert et al. 2012; Janes et al. 2017). MCMC convergence, where α values reached equilibrium, was examined using the Data plot option in STRUCTURE (Porras-Hurtado et al. 2013). Variation of K values was then summarised and plotted in CLUMPAK (Kopelman et al. 2015). The optimal K was determined by examining the Ln Pr(X|K) and ΔK plots (Pritchard and Wen 2003; Evanno et al. 2005; Janes et al. 2017). Principal component analysis (PCA) was performed in the R package SNPRelate version 1.18 to identify clusters without relying on population genetic models (Jombart et al. 2010).
An individual-based analysis grounded on detecting deviations from the isolation-by-distance (IBD) models (Keis et al. 2013; Tang et al. 2018) was used to characterise barriers of and corridors for dispersal of each lineage using R package ResDisMapper (Tang et al. 2019). First, distributions of genetic distance (Nei’s standard genetic distance) and geographic distance (both measured in GenAlEx v 6.5) were checked using two modelling methods—linear and nonlinear—before a best-fit method based on R2 value was chosen for IBD residual calculation for each pair of individuals. Resistance values, together with their corresponding statistical significance over the landscape, were then calculated and visualised using default settings.
Identification of distinct lineages
In order to assess the relationship of our samples within the recently uncovered P. speciosa species complex (Bongaerts et al. 2021), we employed the cleaved amplified polymorphic sequence (CAPS) assay. This assay was designed to rapidly assign individuals to one of the three cryptic lineages that occur sympatrically in East Australia. Genomic DNA for each sample was amplified with three separate primer pairs (Pspe-GRN, Pspe-BLU and Pspe-RED), and amplicons were digested with restriction enzymes (HhaI, HaeIII and Taqα1, respectively) following reaction conditions described in Bongaerts et al. (2021). The lengths of restriction fragments were then determined via agarose gel electrophoresis and used to assign samples to one of the three clusters (Bongaerts et al. 2021) (see Fig. S1).
We also compared representative samples from our main clusters through joint variant calling and a maximum likelihood tree. For this comparison, we included three samples from each of our two main genetic clusters and six samples from each of the six main clusters (“green”, “blue”, “red”, “dark green”, “dark blue” and “purple”) in Bongaerts et al. (2021). RAxML version 8.2.12 (Stamatakis 2014) was used to infer a maximum likelihood phylogeny based on the SNP data with invariant sites removed. We applied an ascertainment bias correction for the omission of invariant sites (Lewis 2001; Leaché et al. 2015) to perform tree searches using 50 random starting trees and bootstrap resampling with 1000 replicates.
Results
Sequencing of the nextRAD libraries resulted in an average of 3.01 million reads per sample that mapped to the reference (n = 144) and genotype calling initially yielded 19,716 biallelic SNPs. After quality control to remove SNPs that were linked, deviated from Hardy–Weinberg equilibrium or had more than 1% missing data per SNP, removing colonies with more than 15% missing data per sample, and following minimal representation filtering, we retained 8590 biallelic SNPs (n = 140). clonecorrect under default parameters did not identify any clones. BayeScan identified 32 outlier SNPs. PCA and STRUCTURE analyses with or without SNPs identified as putatively under selection did not alter assignments of individuals to populations, and because of this, we opted to use the larger dataset for subsequent analyses.
PCA and STRUCTURE clearly showed that samples were clustered into two distinct lineages that were not site specific (Fig. 2), possibly representing cryptic species. The CAPS assay yielded clearly distinct restriction fragment length patterns for the two lineages (Fig. S1; Table S1). Specifically, one of the lineages had the diagnostic mutation for the blue cluster described by Bongaerts et al. (2021), as indicated by the restriction digest of the Pspe-BLU amplicon by the HaeIII enzyme into two 100–150 bp bands, whereas no digestion was observed for the other lineage. None of the samples had the diagnostic mutation of the “red” or “green” lineage, as indicated by consistent digestion observed for all samples with Pspe-Red-Taqα1 (not “red”) and no digestion observed for Pspe-GRN HhaI (not “green”) (Fig. S1; Table S1). It must be noted the CAPS assay was designed to target specifically GBR lineages, and was not diagnostic for the “dark green” and “dark blue” lineages in Okinawa (Bongaerts et al. 2021). We therefore conducted joint variant calling and maximum likelihood analysis with representative samples from Bongaerts et al. (2021), which indicated our lineages to be most closely related to the “dark green” and “dark blue” clusters (which appear to be geographic subclusters of the “green” and “blue” lineages) (Fig. 3; Table S1). We therefore analysed the two lineages (green and blue) separately, in order to detect genetic clustering within the two lineages with identical filtering steps as the overall dataset.

Analysis of 8590 SNPs found across samples of Pachyseris speciosa from Singapore. a STRUCTURE plot of ancestry proportions with K = 2 and K = 4 depicting two main clusters (green and blue) that were not site specific. Within the green cluster two subclusters could be observed (dark and light green). b Principal component analyses (PCAs) of the blue and the green clusters showed separation on PC1 (18.9% variations)

Maximum likelihood tree of representative samples of the green and blue lineages from Singapore showing clustering with samples from the “dark green” and “dark blue” clusters in Okinawa from Bongaerts et al. (2021)
Based on 6979 and 6562 biallelic SNPs (including 45 and 38 outlier SNPs detected by BayeScan) for the green (n = 70) and blue (n = 47) lineages, respectively, global mean FST was higher in the green lineage (FST = 0.0142) compared to blue lineage (FST = 0.0037). Evanno’s method and PCA showed that the most likely “K” is two for the green lineage (Fig. 4a, b; Fig. S2a). For this number of populations, STRUCTURE plots showed that St. John comprises exclusively of a dark green subcluster (not to be confused with the “dark green” cluster in Bongaerts et al. 2021) and that Semakau and TPT comprise mainly the light green subcluster (Fig. 4a). PCA and STRUCTURE showed no geographic structuring of colonies within the blue lineage (Fig. 4c, d; Fig. S2b). ResDisMapper identified a dispersal corridor at the southern periphery of the islands along the Singapore Strait for both lineages (Fig. 5a, b).

Analysis of the green and blue clusters with 8590 SNPs. a STRUCTURE plot of ancestry proportions from K = 2 to K = 4 with the best model depicting two main subclusters (dark and light green) that were not site specific. St. John has a 100% proportion of the dark green subcluster. Semakau has the highest proportion of the light green subcluster (54%). b Independent principal components (PCs) are separated primarily into two subclusters on PC1 (11.4% variations) with a few mosaics from Hantu, Sisters and Semakau. c STRUCTURE plot of ancestry proportions from K = 2 to K = 4 with the best model depicting one population across all sites within Singapore’s blue cluster. d Principal components (PCs) showing one cluster with a few mosaics from Kusu and St. John

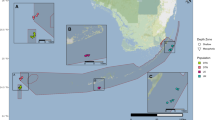
Resistance map produced by ResDisMapper for a green lineage and b blue lineage of Pachyseris speciosa. Areas with resistance values that are higher/lower than those from a null distribution with high probability and lie within the red/green contours represent a significant barrier/corridor. Areas within the blue contours have resistance values with high probability of being positive or negative (high “certainty”). Yellow circles indicate sampling points
Discussion
The aim of this study was to characterise the Pachyseris speciosa species complex and its population genetic structure in Singapore using a genotyping-by-sequencing approach (nextRAD; Russello et al. 2015). Based on sequencing of 144 colonies that were identified as P. speciosa based on conventional taxonomy (Dana 1846; Veron and Pichon 1980; Veron 2000; Terraneo et al. 2014) from seven sites 2–15 km apart, we found two clearly distinct lineages (i.e. green and blue) that were sympatric at each of the seven sampling sites (Fig. 2). This separation into two lineages was supported through a CAPS assay and clustering analyses based on shared variants, with the latter indicating relatedness to the “dark green” and “dark blue” clusters from Okinawa identified in Bongaerts et al. (2021) (Fig. 3). Our results greatly extend the geographic range of these recently uncovered P. speciosa species, further confirming their status as distinct and widespread species.
The population structuring within each of the two lineages reveals an interesting pattern: the blue lineage seems to be one panmictic population, with only a few outliers in St. John and Kusu Island, while the green lineage has unexplained substructuring (Fig. 4). The low genetic structure of the blue lineage across all geographic sites is similar to other broadcasting spawners (e.g. Porto-Hannes et al. 2015; Bongaerts et al. 2017; Eckert et al. 2019) and more specifically in Singapore (Tay et al. 2015). Gene flow mechanisms of coral species are dependent mostly upon their life history traits, as broadcast spawners’ populations are expected to be more connected due to the long pelagic larval duration of broadcasted larvae compared to brooders (Serrano et al. 2016; Underwood et al. 2018). Reproductive patterns of P. speciosa in Singapore have never been documented. Australian P. speciosa species were reported to be gonochoric spawners, and the different lineages showed different timing of gamete release. It is possible that the different genetic patterns of the Singapore lineages could be linked to variations in the timing and mode of gamete release. Furthermore, changes in current regimes associated with tidal cycles, storms and seasonal monsoon along the Singapore Strait (Sin et al. 2016) may influence gene flow and drive the observed differences in genetic structures of the two P. speciosa lineages. The two lineages of P. speciosa—showing such distinct population genetic and gene flow patterns between them—support the species-level divergence inferred from the SNP data in this work and in Bongaerts et al. (2021), which found four distinct lineages including the two found here.
ResDisMapper analyses showing barriers vs. corridors to gene flow between sites indicate that connectivity between the western sites and between eastern and western sites is limited and could be observed only between the eastern sites and Raffles and Hantu (Fig. 5). The population distinction between the western and eastern populations and among the western populations is concerning and could be due to the massive reclamation and development of the area in the last 60 years. Fringing and patch reefs, mangroves, sandy and rocky shores were major coastal features in this area prior to development in the 1960’s (Chou et al. 2019). Yet, the distribution of coral reefs has been greatly reduced by landfill construction (Chou and Tan 2007), the expanding port facilities and terminals (Sin et al. 2016) and the reclamation and development of offshore islands (Tay et al. 2018). These extensive coastal changes have resulted in rapid declines in area and quality of reef habitats (Hilton and Manning 1995), especially those adjacent to the western cluster of islands due to their proximity to the intensive urban activities. The increase in land area could have reduced water flow at the western islands, or increased introduction of sediments, nutrients, heavy metals and organic chemicals (Goh and Chou 1997; Sin et al. 2016) and led to filtering of more tolerant genotypes.
Considering most islands are isolated (i.e. showing barriers between them and the others), there may be a need for these results to inform future developments to consider enhancing physical and biological connectedness among the western island cluster. Such actions will promote biological connectivity and resilience, especially since coral recruitment is at low levels in the Singapore Strait (Bauman et al. 2015). Relatedly, recent genotyping of the endosymbionts of P. speciosa suggests that the two lineages do not have divergent dominant Symbiodiniaceae types (Jain et al. 2020; Smith et al. 2020). Singapore’s reefs provide habitat for a diverse coral assemblage (Guest et al. 2016), yet a strong selective pressure caused by the turbid water along the Singapore Strait possibly limits the diversity of the associated endosymbiont community. Interestingly, detailed characterisation of the bleaching susceptibility of the two lineages also showed no clear difference as both lineages have similar responses to bleaching (Jain et al. 2020).
The subclustering within the green lineage (i.e. Figure 4a, b), possibly promoted by the barriers to gene flow (i.e. Figure 5a), may help explain the cryptic lineages of P. speciosa in other geographic sites. If these processes do scale up to the global level, i.e. at larger geographic scale and over longer time, these minute population differentiations could provide more opportunities for isolation of lineages, especially over the 10-million-year history of the three cryptic lineages (Bongaerts et al. 2021). We note that there are three other nominal species currently considered to be junior synonyms of P. speciosa (Veron and Pichon 1980; Scheer and Pillai 1983). Agaricia levicollis Dana (1846) has a non-specific type locality referring to Southeast Asia (‘East Indies’, Dana 1846, p. 338), while Pachyseris haimei Quelch, 1886, and Pachyseris clementei Nemenzo, 1955, were described from Tahiti (French Polynesia) and Puerto Galera (Philippines), respectively. Localities of Agaricia levicollis and P. clementei are within the range of the green and blue clusters discovered between Bongaerts et al. (2021) and the present study. Detailed taxonomic work integrating these genomic results, morphological differences between these nominal species, and more geographically resolved collections in the Central Indo-Pacific are needed to ascertain potential relationships with these junior synonyms.
Population genetics and connectivity among reefs play a vital role in shaping regional patterns of reef biodiversity and recovery following disturbance (Bongaerts et al. 2010, 2017; Sheets et al. 2018). This research highlights how a small reef system less than 15 km across all sites sampled can have highly sympatric distributions, furthermore showing distinct subclustering of a cryptic lineage.
References
Afiq-Rosli L, Wainwright BJ, Gajanur AR, Lee AC, Ooi SK, Chou LM, Huang D (2021) Barriers and corridors of gene flow in an urbanised tropical reef system. Evol Appl. https://doi.org/10.1111/eva.13276
Andrews KR, Norton EL, Fernandez-Silva I, Portner E, Goetze E (2014) Multilocus evidence for globally distributed cryptic species and distinct populations across ocean gyres in a mesopelagic copepod. Mol Ecol 23:5462–5479
Appeltans W, Ahyong ST, Anderson G, Angel MV, Artois T, Bailly N, Bamber R, Barber A, Bartsch I, Berta A, Błazewicz-Paszkowycz M, Bock P, Boxshall G, Boyko CB, Brandão SN, Bray RA, Bruce NL, Cairns SD, Chan TY, Cheng L, Collins AG, Cribb T, Curini-Galletti M, Dahdouh-Guebas F, Davie PJF, Dawson MN, De Clerck O, Decock W, De Grave S, De Voogd NJ, Domning DP, Emig CC, Erséus C, Eschmeyer W, Fauchald K, Fautin DG, Feist SW, Fransen CHJM, Furuya H, Garcia-Alvarez O, Gerken S, Gibson D, Gittenberger A, Gofas S, Gómez-Daglio L, Gordon DP, Guiry MD, Hernandez F, Hoeksema BW, Hopcroft RR, Jaume D, Kirk P, Koedam N, Koenemann S, Kolb JB, Kristensen RM, Kroh A, Lambert G, Lazarus DB, Lemaitre R, Longshaw M, Lowry J, MacPherson E, Madin LP, Mah C, Mapstone G, McLaughlin PA, Mees J, Meland K, Messing CG, Mills CE, Molodtsova TN, Mooi R, Neuhaus B, Ng PKL, Nielsen C, Norenburg J, Opresko DM, Osawa M, Paulay G, Perrin W, Pilger JF, Poore GCB, Pugh P, Read GB, Reimer JD, Rius M, Rocha RM, Saiz-Salinas JI, Scarabino V, Schierwater B, Schmidt-Rhaesa A, Schnabel KE, Schotte M, Schuchert P, Schwabe E, Segers H, Self-Sullivan C, Shenkar N, Siegel V, Sterrer W, Stöhr S, Swalla B, Tasker ML, Thuesen EV, Timm T, Todaro MA, Turon X, Tyler S, Uetz P, Van Der Land J, Vanhoorne B, Van Ofwegen LP, Van Soest RWM, Vanaverbeke J, Walker-Smith G, Walter TC, Warren A, Williams GC, Wilson SP, Costello MJ (2012) The magnitude of global marine species diversity. Curr Biol 22:2189–2202
Arrigoni R, Terraneo TI, Galli P, Benzoni F (2014) Lobophylliidae (Cnidaria, Scleractinia) reshuffled: Pervasive non-monophyly at genus level. Mol Phylogenet Evol 73:60–64
Arrigoni R, Berumen ML, Stolarski J, Terraneo TI, Benzoni F (2019) Uncovering hidden coral diversity: a new cryptic lobophylliid scleractinian from the Indian Ocean. Cladistics 35:301–328
Arrigoni R, Berumen ML, Terraneo TI, Caragnano A, Bouwmeester J, Benzoni F (2015) Forgotten in the taxonomic literature: Resurrection of the scleractinian coral genus Sclerophyllia (Scleractinia, Lobophylliidae) from the Arabian Peninsula and its phylogenetic relationships. Syst Biodivers 13:140–163
Arrigoni R, Berumen ML, Mariappan KG, Beck PSA, Hulver AM, Montano S, Pichon M, Strona G, Terraneo TI, Benzoni F (2020) Towards a rigorous species delimitation framework for scleractinian corals based on RAD sequencing: the case study of Leptastrea from the Indo-Pacific. Coral Reefs 39:1001–1025
Baird HP, Miller KJ, Stark JS (2011) Evidence of hidden biodiversity, ongoing speciation and diverse patterns of genetic structure in giant Antarctic amphipods. Mol Ecol 20:3439–3454
Bauman AG, Guest JR, Dunshea G, Low J, Todd PA, Steinberg PD (2015) Coral settlement on a highly disturbed equatorial reef system. PLoS One 10:e0127874
Benzoni F, Arrigoni R, Stefani F, Pichon M (2011) Phylogeny of the coral genus Plesiastrea (Cnidaria, Scleractinia). Contrib Zool 80(4):231–249
Benzoni F, Stefani F, Stolarski J, Pichon M, Mitta G, Galli P (2007) Debating phylogenetic relationships of the scleractinian Psammocora: Molecular and morphological evidences. Contrib Zool 76:35–54
Benzoni F, Arrigoni R, Zoologica S, Dohrn A, Waheed Z, Stefani F (2014) Phylogenetic relationships and revision of the genus Blastomussa (Cnidaria: Anthozoa: Scleractinia) with description of a new species. Raffles Bull Zool 62:358–378
Bickford D, Lohman DJ, Sodhi NS, Ng PKL, Meier R, Winker K, Ingram KK, Das I (2007) Cryptic species as a window on diversity and conservation. Trends Ecol Evol 22:148–155
Bongaerts P, Riginos C, Brunner R, Englebert N, Smith SR, Hoegh-Guldberg O (2017) Deep reefs are not universal refuges: Reseeding potential varies among coral species. Sci Adv 3:e1602373
Bongaerts P, Riginos C, Ridgway T, Sampayo EM, van Oppen MJH, Englebert N, Vermeulen F, Hoegh-Guldberg O (2010). Genetic divergence across habitats in the widespread coral Seriatopora hystrix and its associated Symbiodinium. PLoS One 5:e10871
Bongaerts P, Cooke I, Ying H, Wels D, Den SH, Hernandez-Agreda A, Brunner CA, Dove S, Englebert N, Eyal G, Forêt S, Grinblat M, Hay KB, Harii S, Hayward DC, Lin Y, Mihaljević M, Moya A, Muir P, Sinniger F, Smallhorn-West P, Torda G, Ragan MA, Van-Oppen MJH, Hoegh-Guldberg O (2021) Cryptic diversity masks ecologically distinct coral species on tropical reefs. Curr Biol 31:2286–2298
Browne NK, Tay J, Todd PA (2015) Recreating pulsed turbidity events to determine coral-sediment thresholds for active management. J Exp Mar Bio Ecol 466:98–109
Browne NK, Precht E, Last KS, Todd PA (2014) Photo-physiological costs associated with acute sediment stress events in three near-shore turbid water corals. Mar Ecol Prog Ser 502:129–143
Chadè I, Mcdonald-Madden E, Mccarthy MA, Wintle B, Linkie M, Possingham HP (2008) When to stop managing or surveying cryptic threatened species. PNAS 105:13936–13940
Chevalier JP (1975) Les scléractiniaires de la Mélanésie française (Nouvelle Calédonie, Iles Chesterfield, Iles Loyauté, Nouvelles Hébrides). Deuxième partie. Expédition Française sur les Récifs Coralliens de la Nouvelle Calédonie 7:1-407
Chou LM, Tan KPP (2007) Conserving reefs beside a marine landfill in Singapore. Coral Reefs 26:719
Chou LM, Huang D, Tan KS, Toh TC, Goh BPL, Tun K (2019) Singapore. In: Sheppard CRC (ed) World seas: an environmental evaluation, vol II. The Indian Ocean to the Pacific. Academic Press, London, pp 539–558
Chow GSE, Chan YKS, Jain SS, Huang D (2019) Light limitation selects for depth generalists in urbanised reef coral communities. Mar Environ Res 147:101–112
Costello MJ, Wilson S, Houlding B (2012) Predicting total global species richness using rates of species description and estimates of taxonomic effort. Syst Biol 61:871–883
Dana JD (1846) U.S. Exploring Exped (1838–1842). Vol. VII. Zoophytes. C. Sherman. 740 pp. Available from: http://www.sil.si.edu/digitalcollections/usexex/navigation/ScientificText/USExEx19_08select.cfm.
Dikou A, van Woesik R (2006) Survival under chronic stress from sediment load: Spatial patterns of hard coral communities in the southern islands of Singapore. Mar Pollut Bull 52:1340–1354
Eckert RJ, Studivan MS, Voss JD (2019) Populations of the coral species Montastraea cavernosa on the Belize Barrier Reef lack vertical connectivity. Sci Rep 9:1–11
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620
Fišer C, Robinson CT, Malard F (2018) Cryptic species as a window into the paradigm shift of the species concept. Mol Ecol 27:613–635
Foll M, Gaggiotti O (2008) A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics 180:977–993
Frederick Grassle J, Maciolek NJ (1992) Deep-sea species richness: regional and local diversity estimates from quantitative bottom. Am Nat 139:313–341
Fukami H, Budd AF, Paulay G, Sole-Cava A, Allen CC, Iwao K, Knowlton N (2004) Conventional taxonomy obscures deep divergence between Pacific and Atlantic corals. Nature 427:829–832
Fukami H, Chen CA, Budd AF, Collins A, Wallace C, Chuang YY, Chen C, Dai CF, Iwao F, Sheppard C, Knowlton N (2008) Mitochondrial and nuclear genes suggest that stony corals are monophyletic but most families of stony corals are not (Order Scleractinia, Class Anthozoa, Phylum Cnidaria). PLoS One 3:e3222
Gélin P, Postaire B, Fauvelot C, Magalon H (2017) Reevaluating species number, distribution and endemism of the coral genus Pocillopora Lamarck, 1816 using species delimitation methods and microsatellites. Mol Phylogenet Evol 109:430–446
Gilbert KJ, Andrew RL, Bock DG, Franklin MT, Moore B, Kane NC, Rennison DJ, Veen T, Vines TH (2012) Recommendations for utilizing and reporting population genetic analyses: the reproducibility of genetic clustering using the program STRUCTURE. Mol Ecol 604:822–2416
Goh BPL, Chou LM (1997) Heavy metal levels in marine sediments of Singapore. Environ Monit Assess 97:131–141
Grundt HH, Kjølner S, Borgen L, Rieseberg LH, Brochmann C (2006) High biological species diversity in the Arctic flora. PNAS 106:972–975
Guest JR, Tun K, Low J, Vergés A, Marzinelli EM, Campbell AH, Bauman AG, Feary DA, Chou LM, Steinberg PD (2016) 27 years of benthic and coral community dynamics on turbid, highly urbanised reefs off Singapore. Sci Rep 6:36260
Hilton MJ, Manning SS (1995) Conversion of coastal habitats in Singapore: Indications of unsustainable development. Environ Conserv 22:307–322
Hoeksema, BW, Cairns S (2020) World List of Scleractinia. Pachyseris Milne Edwards & Haime, 1849. World Register of Marine Species [http://www.marinespecies.org/aphia.php?p= taxdetails&id=1363]
Huang D, Meier R, Todd PA, Chou LM (2009) More evidence for pervasive paraphyly in scleractinian corals: Systematic study of Southeast Asian Faviidae (Cnidaria; Scleractinia) based on molecular and morphological data. Mol Phylogenet Evol 50:102–116
Huang D, Benzoni F, Arrigoni R, Baird AH, Berumen ML, Bouwmeester J, Chou LM, Fukami H, Licuanan WY, Lovell ER, Meier R, Todd PA, Budd AF (2014) Towards a phylogenetic classification of reef corals: the Indo-Pacific genera Merulina, Goniastrea and Scapophyllia (Scleractinia, Merulinidae). Zool Scr 43:531–548
Hughes TP, Connolly SR, Keith SA (2013) Geographic ranges of reef corals (Cnidaria: Anthozoa: Scleractinia) in the Indo-Pacific. Ecology 94:1659
Jain SS, Afiq-Rosli L, Feldman B, Levy O, Phua JW, Wainwright BJ, Huang D (2020) Homogenization of endosymbiont communities hosted by equatorial corals during the 2016 mass bleaching event. Microorganisms 8:1370
Janes JK, Miller JM, Dupuis JR, Malenfant RM, Gorrell JC, Cullingham CI, Andrew RL (2017) The K=2 conundrum. Mol Ecol 26:3594–3602
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:94
Kamvar ZN, Tabima JF, Grünwald NJ (2014) Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2:e281
Kamvar ZN, Brooks JC, Grünwald NJ (2015) Novel R tools for analysis of genome-wide population genetic data with emphasis on clonality. Front Genet 6:208
Keis M, Remm J, Ho SYW, Davison J, Tammeleht E, Tumanov IL, Saveljev AP, Männil P, Kojola I, Abramov AV, Margus T, Saarma U (2013) Complete mitochondrial genomes and a novel spatial genetic method reveal cryptic phylogeographical structure and migration patterns among brown bears in north-western Eurasia. J Biogeogr 40:915–927
Kerr AM, Baird AH, Hughes TP (2011) Correlated evolution of sex and reproductive mode in corals (Anthozoa: Scleractinia). Proc R Soc B 278:75–81
Kitahara MV, Cairns SD, Stolarski J, Miller DJ (2013) Deltocyathiidae, an early-diverging family of robust corals (Anthozoa, Scleractinia). Zool Scr 42:201–212
Kitahara MV, Cairns SD, Stolarski J, Blair D, Miller DJ (2010) A comprehensive phylogenetic analysis of the Scleractinia (Cnidaria, Anthozoa) based on mitochondrial CO1 sequence data. PLoS One 5:e11490
Kitahara MV, Stolarski J, Cairns SD, Benzoni F, Stake JL, Miller DJ (2012) The first modern solitary Agariciidae (Anthozoa, Scleractinia) revealed by molecular and microstructural analysis. Invertebr Syst 26:303–315
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15:1179–1191
Ladner JT, Palumbi SR (2012) Extensive sympatry, cryptic diversity and introgression throughout the geographic distribution of two coral species complexes. Mol Ecol 21:2224–2238
Lamarck JBP (1815) Les polypes. Hist Nat des animaux sans vertèbres, Paris
Lang JC (1984) Whatever works: the variable importance of skeletal and non-skeletal characters in scleractinian taxonomy. Palaeontogr Am 54:18–44
Leaché AD, Banbury BL, Felsenstein J, de Oca ANM, Stamatakis A (2015) Short tree, long tree, right tree, wrong tree: new acquisition bias corrections for inferring SNP phylogenies. Syst Biol 64:1032–1047
Lewis PO (2001) A likelihood approach to estimating phylogeny from discrete morphological character data. Syst Biol 50:913–925
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079
Lischer HEL, Excoffier L (2012) PGDSpider: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 28:298–299
Mora C, Tittensor DP, Adl S, Simpson AGB, Worm B (2011) How many species are there on earth and in the ocean? PLoS Biol 9:e1001127
Nygren A (2014) Cryptic polychaete diversity: A review. Zool Scr 43:172–183
Paz-García DA, Hellberg ME, García-de-León FJ, Balart EF (2015) Switch between morphospecies of Pocillopora corals. Am Nat 186:434–440
Pfenninger M, Schwenk K (2007) Cryptic animal species are homogeneously distributed among taxa and biogeographical regions. BMC Evol Biol 7:121
Porras-Hurtado L, Ruiz Y, Santos C, Phillips C, Carracedo Á, Lareu MV (2013) An overview of structure: applications, parameter settings, and supporting software. Front Genet 4:1–13
Porto-Hannes I, Zubillaga AL, Shearer TL, Bastidas C, Salazar C, Coffroth MA, Szmant AM (2015) Population structure of the corals Orbicella faveolata and Acropora palmata in the Mesoamerican Barrier Reef System with comparisons over Caribbean basin-wide spatial scale. Mar Biol 162:81–98
Pritchard JK, Wen W (2003) Documentation for STRUCTURE Software:Version 2. Chicago: University of Chicago Press. Retrived from: http://web.stanford.edu/group/pritchardlab/software/structure2_1.html
Richards ZT, Berry O, van Oppen MJH (2016) Cryptic genetic divergence within threatened species of Acropora coral from the Indian and Pacific Oceans. Conserv Genet 17:577–591
Russello MA, Waterhouse MD, Etter PD, Johnson EA (2015) From promise to practice: pairing non-invasive sampling with genomics in conservation. PeerJ 3:e1106
Scheer G, Pillai CSG (1983) Report on the stony corals from the Red Sea. Zoologica (Stuttgart) 133:1–198
Scheffers BR, Joppa LN, Pimm SL, Laurance WF (2012) What we know and don’t know about Earth’s missing biodiversity. Trends Ecol Evol 27:501–510
Schmidt-Roach S, Lundgren P, Miller KJ, Gerlach G, Noreen AME, Andreakis N (2013) Assessing hidden species diversity in the coral Pocillopora damicornis from Eastern Australia. Coral Reefs 32:161–172
Serrano XM, Baums IB, Smith TB, Jones RJ, Shearer TL, Baker AC (2016) Long distance dispersal and vertical gene flow in the Caribbean brooding coral Porites astreoides. Sci Rep 6:21619
Sheets EA, Warner PA, Palumbi SR (2018) Accurate population genetic measurements require cryptic species identification in corals. Coral Reefs 37:549–563
Sheppard CRC, Sheppard ALS (1991) Corals and coral communities of Saudi Arabia. Fauna Arab 12:1–170
Sites JW, Marshall JC (2003) Delimiting species: A Renaissance issue in systematic biology. Trends Ecol Evol 18:462–470
Sin TM, Ang HP, Buurman J, Lee AC, Leong YL, Ooi SK, Steinberg P, Teo SLM (2016) The urban marine environment of Singapore. Reg Stud Mar Sci 8:331–339
Smith EG, Gurskaya A, Hume BCC, Voolstra CR, Todd PA, Bauman AG, Burt JA (2020) Low Symbiodiniaceae diversity in a turbid marginal reef environment. Coral Reefs 39:545–553
Souter P (2010) Hidden genetic diversity in a key model species of coral. Mar Biol 157:875–885
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313
Stefani F, Benzoni F, Yang SY, Pichon M, Galli P, Chen CA (2011) Comparison of morphological and genetic analyses reveals cryptic divergence and morphological plasticity in Stylophora (Cnidaria, Scleractinia). Coral Reefs 30:1033–1049
Tang Q, Fung T, Rheindt FE (2019) ResDisMapper: An R package for fine-scale mapping of resistance to dispersal. Mol Ecol Resour 20:819–831
Tang Q, Low GW, Lim JY, Gwee CY, Rheindt FE (2018) Human activities and landscape features interact to closely define the distribution and dispersal of an urban commensal. Evol Appl 11:1598–1608
Tay JY, Wong SK, Chou LM, Todd PA (2018) Land reclamation and the consequent loss of marine habitats around the Ayer Islands, Singapore. Nature in Singapore 11:1–5
Tay YC, Noreen AME, Suharsono CLM, Todd PA (2015) Genetic connectivity of the broadcast spawning reef coral Platygyra sinensis on impacted reefs, and the description of new microsatellite markers. Coral Reefs 34:301–311
Terraneo TI, Berumen ML, Arrigoni R, Waheed Z, Bouwmeester J, Caragnano A, Stefani F, Benzoni F (2014) Pachyseris inattesa sp. n. (Cnidaria, Anthozoa, Scleractinia): a new reef coral species from the Red Sea and its phylogenetic relationships. ZooKeys 433:1–30
Todd PA (2008) Morphological plasticity in scleractinian corals. Biol Rev 8:315–337
Todd PA, Sidle RC, Lewin-Koh NJI (2004) An aquarium experiment for identifying the physical factors inducing morphological change in two massive scleractinian corals. J Exp Mar Bio Ecol 299:97–113
Toonen RJ, Puritz JB, Forsman ZH, Whitney JL, Fernandez-Silva I, Andrews KR, Bird CE (2013) ezRAD: a simplified method for genomic genotyping in non-model organisms. PeerJ 1:e203.
Underwood JN, Richards ZT, Miller KJ, Puotinen ML, Gilmour JP (2018) Genetic signatures through space, time and multiple disturbances in a ubiquitous brooding coral. Mol Ecol 27:1586–1602
Veron JEN (2000) Corals of the World. Aust Inst Mar Sci. Townsville
Veron JEN (2002) New species described in Corals of the World (Vol. 11). Aust Inst Mar Sci. Townsville
Veron JEN, Pichon M (1980) Scleractinia of Eastern Australia, III: families Agaraciidae, Siderastreidae, Fungiidae, Oculinidae, Merulinidae, Mussidae, Pectiniidae, Caryophylliidae, Dendrophylliidae. Aust Inst Mar Sci Monogr Ser 4:1–422
Warner PA, Van Oppen MJH, Willis BL (2015) Unexpected cryptic species diversity in the widespread coral Seriatopora hystrix masks spatial-genetic patterns of connectivity. Mol Ecol 24:2993–3008
Wells JW (1956) Scleractinia. In: Moore RC (ed) Treatise on invertebrate paleontology, Part F, Coelenterata: Boulder, Colo. Geol Soc Am and Univ Kansas Press, pp F328–F444
Willig MR, Kaufman DM, Stevens RD (2003) Latitudinal gradients of biodiversity: pattern, process, scale, and synthesis. Annu Rev Ecol Evol Syst 34:273–309
Wong JSY, Chan YKS, Ng CSL, Tun KPP, Darling ES, Huang D (2018) Comparing patterns of taxonomic, functional and phylogenetic diversity in reef coral communities. Coral Reefs 37:737–750
Acknowledgements
We thank Sudhanshi S. Jain, Adi Zweifler, Eviatar Weizman and Inbal Ayalon for assistance in field and laboratory work. This research was supported by the National Research Foundation, Prime Minister’s Office, Singapore and the Israel Science Foundation under their 2nd Joint Grant Call (NRF2017NRF-ISF002-2658; Grant Number 2658/17).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Topic Editor: Francesca Benzoni
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Feldman, B., Afiq-Rosli, L., Simon-Blecher, N. et al. Distinct lineages and population genomic structure of the coral Pachyseris speciosa in the small equatorial reef system of Singapore. Coral Reefs 41, 575–585 (2022). https://doi.org/10.1007/s00338-021-02160-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00338-021-02160-4