
Abstract
Relatively small volumes of water may contain sufficient environmental DNA (eDNA) to detect target aquatic organisms via genetic sequencing. We therefore assessed the utility of eDNA to document the diversity of coral reef fishes in the central Red Sea. DNA from seawater samples was extracted, amplified using fish-specific 16S mitochondrial DNA primers, and sequenced using a metabarcoding workflow. DNA sequences were assigned to taxa using available genetic repositories or custom genetic databases generated from reference fishes. Our approach revealed a diversity of conspicuous, cryptobenthic, and commercially relevant reef fish at the genus level, with select genera in the family Labridae over-represented. Our approach, however, failed to capture a significant fraction of the fish fauna known to inhabit the Red Sea, which we attribute to limited spatial sampling, amplification stochasticity, and an apparent lack of sequencing depth. Given an increase in fish species descriptions, completeness of taxonomic checklists, and improvement in species-level assignment with custom genetic databases as shown here, we suggest that the Red Sea region may be ideal for further testing of the eDNA approach.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Coral reefs represent less than 0.02% of the surface area of our oceans and yet host more than 25% of the recognized marine species (Spalding et al. 2001). In the Red Sea, coral reefs are characterized by their high and unique biodiversity (DiBattista et al. 2016b), historical isolation (DiBattista et al. 2016a), and extreme seasonal and spatial fluctuations in abiotic and biotic factors (Raitsos et al. 2013). Yet compared to other major global reef systems (e.g., Great Barrier Reef), the associated communities of the Red Sea are considerably understudied (Berumen et al. 2013).
Community assemblages of reef fishes have historically been documented through diver-operated underwater visual census methods (UVC; Roberts et al. 1992) or baited remote underwater video surveys (BRUVs; Harvey et al. 2012). Even though these standardized survey methods are ideal for identifying large, mobile, and conspicuous species, the smaller, more cryptic species are often missed (see Ackerman and Bellwood 2000). In recent years, genomic approaches have shown considerable potential for identifying diversity in marine environments, although many of these studies are based on fixed, benthic sampling methods (e.g., Leray and Knowlton 2015). Environmental DNA (eDNA, i.e., genetic material sourced from microbes or that which has been shed from multi-cellular organisms), on the other hand, has proven useful in monitoring the presence/absence of invasive species and assessing biodiversity in aquatic environments (e.g., Thomsen et al. 2012).
The majority of macrobial eDNA investigations to date have been species-specific, but multi-species PCR in combination with high-throughput sequencing (metabarcoding) can extend these methods to reveal a greater diversity of species in our oceans (e.g., Miya et al. 2015). Even though the sensitivity of these high-throughput assays is likely influenced by the frequency of cell shedding, water flow (currents or tides), and rates of DNA degradation, in most cases this approach has advantages when combined with existing monitoring efforts, including operating at a reduced cost. Given that small volumes of water may contain sufficient eDNA to sequence and detect target aquatic organisms (Thomsen et al. 2012; Miya et al. 2015), our study is the first to apply this noninvasive survey method and assess its utility to document the diversity of fishes inhabiting Red Sea coral reefs.
Materials and methods
Study site
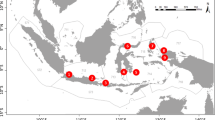
Sampling was conducted in the central Red Sea over a 3-d period in November 2015 near Thuwal, Saudi Arabia (Fig. 1). Seven sampling sites were located on inshore, midshore, and offshore reefs, with six replicate seawater samples (500 mL per sample) taken from both the exposed and sheltered sides of the reef (a total of 12 replicates from each reef). Four replicate samples were also taken from the King Abdullah University of Science and Technology (KAUST) boat harbor, where there are relatively fewer reef fish species. Multiple precautions were taken to minimize contamination with exogenous eDNA, including rinsing all water bottles and the filtering apparatus in a 10% bleach solution prior to use and between each sampling. Moreover, seawater was collected only at the surface (top 20 cm), on the windward side of the research vessel, and within 5 m of the reef, changing latex gloves in between samples. To minimize eDNA degradation, the samples were filtered immediately on the research vessel using a peristaltic pump (Sentino microbiology pump, Pall Life Sciences, MI) and 47 mm (0.45 µm) micronylon sterilized membranes (Pall Life Sciences), which were then placed in individual plastic bags and stored on ice prior to being frozen at –20 °C until eDNA extraction. All extractions were performed within 5 d of collection to limit the decay of template material on the membranes.

Seawater sampling sites at inshore (orange), midshore (green), and offshore (purple) coral reefs, and a boat harbor (red) along the central Red Sea coastline near Thuwal, Saudi Arabia
DNA extraction
Genomic DNA was extracted in a sterile environment at KAUST using a DNeasy blood and tissue kit (Qiagen Inc., CA) following the manufacturer’s protocol with a few modifications: (1) 360 µL of ATL tissue lysis buffer and 40 µL proteinase K were added to 1/3 of the filter membrane that had been cut up in a UV hood; (2) the filter membrane was digested for 3 h; (3) the supernatant following digestion was removed and transferred to a sterile 2-mL tube where 400 µL of absolute ethanol and 400 µL of AL lysis buffer was added; (4) two separate microcentrifuge spins with the digested solution were used to pass all of the DNA through the filter column; (5) all samples were eluted in one step using 100 µL of AE buffer; and (6) extraction controls, for which all steps remained the same except for the addition of the filter membrane, were included for each set of 12 samples.
Fusion-tag qPCR
Quantitative PCR (qPCR) experiments were conducted in a separate, dedicated laboratory at Curtin University in Australia where all benches and utensils were routinely cleaned with bleach and UV sterilized. This approach allowed us to: (1) recover fish 16S mitochondrial DNA barcode sequences of ~200 bp from mixed samples with the primers 16SF/D (5′-GACCCTATGGAGCTTTAGAC-3′) and 16S2R-degenerate (5′-CGCTGTTATCCCTADRGTAACT-3′) (modified from Deagle et al. 2007); (2) add multiple forward (N = 8) and reverse (N = 25) fusion tags to the qPCR products that each contained a unique 10-bp Illumina adaptor sequence; and (3) estimate final concentrations of the DNA amplicons using SYBR Green as a reporter of fluorescence. All qPCRs were run in triplicate and included “no template” controls to check for sample cross-contamination. qPCR reactions (25 mL) consisted of the following: 10 × PCR Buffer (Bioline, London, UK), 0.25 mM MgCl2, 10 mM dNTPs, 0.4 mg mL−1 bovine serum albumin, 1.25 U Ampli-Taq Gold (Applied Biosystems, CA), 20 mM primers, and 2 μL undiluted DNA extract. Cycling conditions were as follows: 95 °C for 5 min, followed by 45 cycles of 95 °C for 30 s, 54 °C for 30 s, and 72 °C for 45 s, with a final step of 72 °C for 10 min. Failed amplifications were not repeated, resulting in fewer than 12 replicates per sampling station. DNA amplicons from each sample were pooled in equimolar concentration based on qPCR endpoints and quantification on a LabChip GX Touch (PerkinElmer Health Sciences, MA), size-selected using a Pippen Prep (Sage Science, MA), and then purified using a QIAquick PCR Purification Kit (Qiagen Inc., CA) following the manufacturer’s protocol. The final library was quantified on a LabChip GX Touch followed by sequencing in the TrEnD Laboratory at Curtin University on an Illumina MiSeq platform using 300 cycle MiSeq V2 reagent kits and nanoflow cells. All unfiltered sequence data are accessible from DRYAD (doi:10.5061/dryad.1pm20).
Bioinformatic filtering
Sequences were assigned to samples, quality filtered, and trimmed in Geneious Pro v 4.8.4 (Drummond et al. 2009) using the following workflow: (1) reads containing imperfect Illumina adaptor sequences were discarded; (2) reads containing imperfect forward or reverse fish 16S primer sequences were discarded; (3) reads were de-multiplexed based on the combined forward and reverse adaptor sequences; (4) reads with imperfect adaptor barcode sequences were discarded; (5) reads smaller than 160 bp were discarded; (6) singletons were discarded; and (7) chimeric sequences were flagged and discarded using USEARCH v 8.0.1623 (Edgar 2010).
Taxonomic assignment
We interrogated the NCBI BLASTn database (March 2016) on the Magnus Cray XC40 system located at the Pawsey Supercomputing Centre at Technology Park in Western Australia, with our 16S sequences in FASTA format. BLASTn results were imported into MEtaGenome ANalyzer (MEGAN) v 5.11.3 (Huson and Weber 2013), and taxonomic identities were assigned to genus or species (where possible) based on the lowest common ancestor (LCA) algorithm using the following settings: minimum number of reads = 2; minimum bit score = 300; top percent to be considered = 10%; and minimum complexity = not considered. A similarity of 95% was accepted for a genus-level match and 98% for a species-level match (Meyer and Paulay 2005). Taxonomic assignments to species with LCA were further evaluated against expert knowledge of species distributions and the most up-to-date checklist of Red Sea fishes (DiBattista et al. 2016b and references therein). To be conservative, matches at the species level were not accepted if that taxon was not known from the Red Sea or if 16S sequences from all species within that genus were not available on NCBI or in our custom genetic database.
Given multiple hits to poorly archived fish 16S sequences on NCBI, an issue raised by Pleijel et al. (2008), in addition to hits to fish species not found in the Red Sea (16 of the 25 species assigned), a modified BLASTn search was performed against a curated custom genetic database of Red Sea fishes, within which we selected a single representative 16S sequence from all reef-fish species found in the Red Sea that were available on NCBI. Where possible, sequences were extracted from datasets published by trusted sources and those that had vouchered their whole specimens at museums (Electronic supplementary material, ESM4. fasta). We additionally generated 16S sequences for 38 fish species sampled in the Red Sea to add to the custom database, which provided a total of 474 of the 1071 known reef-fish species from the region (DiBattista et al. 2016b).
Results and discussion
Our 16S eDNA metabarcoding approach at seven reef sites and a boat harbor in the central Red Sea revealed between 26 and 46 genera of fish (out of 511 known genera; DiBattista et al. 2016b) based on 250,145 total DNA sequences. These genera represent a diversity of conspicuous (e.g., Chaetodon), cryptobenthic (e.g., Eviota), numerically abundant (e.g., Pseudanthias), and commercially relevant shorefish (e.g., Cephalopholis, Epinephelus, Lethrinus, and Lutjanus) known to inhabit the Red Sea (DiBattista et al. 2016b). Our eDNA assignment approach using the NCBI database and our custom genetic database identified 12 or 6 out of the 21 most abundant reef fish genera identified by a recent UVC study (Roberts et al. 2016), respectively, and between 17 and 42% of these genera were only detected at a single site. This difference in detection capability may be due to the limited spatial scale of our sampling (<1% of the eastern Red Sea coastline) or our lack of ability to resolve fish taxa based on short but amplifiable 16S fragments of DNA.
DNA sequencing was successful for 53 of the 88 replicate seawater samples collected at reefs in the central Red Sea (Fig. 1). Of these replicates, there was a modest bias toward amplification success on the sheltered side versus the exposed side of the reefs (31 vs. 22), which may indicate increased suspension of organic material or water residency time in the former; sampling effort should be focused here in future studies. However, given the lack of data for the exposed side of the reefs, we combined data from both sides of the reef for all downstream analyses. DNA degradation, primer biases, or a lack of template DNA may have all played a role in the samples that failed to amplify. Moreover, for eDNA surveys such as this, it is difficult to know a priori what the DNA template concentration will be in the water column. In some cases we recovered limited amounts of template, a finding that contrasts with other studies (Miya et al. 2015), but that is important to consider when designing eDNA surveys in an environment for the first time.
Our noninvasive survey method revealed a trend in sampling effort at some (but not all) reefs based on the number of cumulative fish genera assigned per replicate (Fig. 2a). For example, no new genera were assigned after as few as one replicate out of four total replicate seawater samples taken from KAUST harbor, whereas new genera continued to be assigned for two reefs that had at least eight or nine replicates included in the analysis. Indeed, taxonomic assignment to genus had not yet reached saturation at four of the eight reefs based on the upward trajectory of the accumulation curves. This suggests that 12 or more replicates, based on the volume of seawater that we filtered in this study (500 mL), may be required. In contrast, there was no clear bias in this “saturation trend” among inshore, midshore, or offshore reefs. No relationship was observed between the number of 16S sequences generated per reef and the number of genera assigned (r 2 = 0.037, p = 0.17; Fig. 2b), although when we removed two outlier data points that appeared to drive the regression (>2 SD, both from Al Fahal South), this relationship became significant (r 2 = 0.45, p < 0.0001), indicating a clear benefit of more replicates and increased sequencing coverage per replicate, also highlighted by rarefaction analysis of randomly selected samples (ESM Fig. S1).

Number of cumulative (a) or absolute (b) genera assigned per replicate as a function of amplifiable 16S mitochondrial DNA sequences detected at seven inshore (orange), midshore (green), and offshore (purple) coral reefs, and a boat harbor (red) along the central Red Sea coastline near Thuwal, Saudi Arabia. SN Shi’b Nazar; RR Rose Reef; UAB Um Al Balam; AFS Al Fahal South; IF Inner Fsar; TN Tahla North; AS Abu Shusha; KH KAUST Harbor
When the number of unique sequences per genus was averaged across each reef we identified over-represented genera (e.g., Chlorurus, Halichoeres, Scarus) at some, but not all, reefs based on assignments made using the NCBI database (Fig. 3a) and our custom genetic database (Fig. 3b). This may indicate biases in the workflow that preferentially target these genera or increased DNA concentrations of these genera present in seawater samples. Moreover, some of the common genera in the Red Sea were notably absent from our data (e.g., Apogon, Coris, Gobiodon, Pseudocheilinus, Pygoplites; Roberts et al. 2016). Low values for the average number of unique sequences in most cases (Fig. 3a: mean = 8.08 ± 3.65 SE, median = 0.67; Fig. 3b: mean = 5.02 ± 2.65, median = 0.65) further suggest that there may be a clear benefit of increased coverage per reef by either increasing the number of replicates (>12 replicates), increasing the volume of water filtered (>500 mL), decreasing the pore size of the filter membrane to capture smaller particles (<0.45 µm), or generating higher numbers of metabarcoding reads. The benefit of generating higher numbers of metabarcoding reads is supported by our rarefaction analysis of randomly selected samples from a single reef, where there was considerable variability in the level of sequencing at which saturation was reached for assignment of genera (ESM Fig. S1). Based on heatmaps (Fig. 3), there was also an indication that different reefs had different compositions of fish. Although previous aquatic studies have attempted to infer the abundance of taxa from the number of sequence reads (amphibians: Pilliod et al. 2013; fish: Mahon et al. 2013; Lacoursière-Roussel et al. 2016), we chose to avoid this inference due to confounding factors potentially skewing the proportions of reads, including fluctuating environmental conditions, variable rates of DNA degradation (Dejean et al. 2011), and low template numbers (Murray et al. 2015).


Heatmap showing the mean number of unique 16S mitochondrial DNA sequences for each genus (a, b) detected at seven inshore (orange), midshore (green), and offshore (purple) coral reefs, and a boat harbor (red) along the central Red Sea coastline near Thuwal, Saudi Arabia. Analyses were independently conducted using the National Center for Biotechnology Information (NCBI) database (a) or our custom genetic database (b). Numbers in parentheses are the number of seawater replicates that amplified per sample site. See Fig. 1 for summary of sample sites; abbreviations as in Fig. 2
Even with our custom genetic database, less than half of all Red Sea species were represented with a 16S barcode sequence (474 of the 1071 species). Indeed, some speciose genera were under-represented in our custom database, with ≤10% of the species in their respective genera with a 16S barcode sequence (ESM File S1; ESM Fig. S2). Nonetheless, almost twice as many taxa were confidently assigned to species using our custom database (N = 16) versus the NCBI database (N = 9), despite the same conservative criteria being used in both cases (Table 1).
Caveats to consider when barcoding seawater samples based on 16S fish DNA include misidentification in the NCBI reference database, incomplete lineage sorting or hybridization between species, or (in our case) a dearth of vouchered sequences from reef fish sampled in the Red Sea. Also, because 16S is a much shorter read for eDNA applications, the taxonomic resolution at the species level is weak, particularly in the Red Sea. Indeed, we have previously shown that intraspecific divergence of reef fish from adjacent Indian Ocean populations can be comparable to interspecific comparisons (DiBattista et al. 2013) and that cryptic lineages are not uncommon (DiBattista et al. 2017). These caveats are not unique to our study or restricted to reef fish, but can be mitigated with increased regional barcoding initiatives, and may be less of an issue for better-characterized coral reefs (e.g., Great Barrier Reef). The benefit of this approach despite its equivocal outcome is that it may prove to be more cost-effective than investing in expensive field time and also that eDNA studies are not limited by poor environmental conditions (e.g., visibility, hazardous conditions) or a lack of taxonomic expertise. These initial eDNA trials from the Red Sea show that the method has several potential applications but that surveys will need to be carefully designed given the number of potentially confounding factors.
References
Ackerman JL, Bellwood DR (2000) Reef fish assemblages: a re-evaluation using enclosed rotenone stations. Mar Ecol Prog Ser 206:227–237
Berumen ML, Hoey AS, Bass WH, Bouwmeester J, Catania D, Cochran JEM, Khalil MT, Miyake S, Mughal MR, Spaet JLY, Saenz-Agudelo P (2013) The status of coral reef ecology research in the Red Sea. Coral Reefs 32:737–748
Deagle BE, Gales NJ, Evans K, Jarman SN, Robinson S, Trebilco R, Hindell MA (2007) Studying seabird diet through genetic analysis of faeces: a case study on macaroni penguins (Eudyptes chrysolophus). PLoS One 2:e831
Dejean T, Valentini A, Duparc A, Pellier-Cuit S, Pompanon F, Taberlet P, Miaud C (2011) Persistence of environmental DNA in freshwater ecosystems. PLoS One 6:e23398
DiBattista JD, Berumen ML, Gaither MR, Rocha LA, Eble JA, Choat JH, Craig MT, Skillings DJ, Bowen BW (2013) After continents divide: comparative phylogeography of reef fishes from the Red Sea and Indian Ocean. J Biogeogr 40:1170–1181
DiBattista JD, Choat JH, Gaither MR, Hobbs JP, Lozano-Cortés DF, Myers RF, Paulay G, Rocha LA, Toonen RJ, Westneat M, Berumen ML (2016a) On the origin of endemic species in the Red Sea. J Biogeogr 43:13–30
DiBattista JD, Roberts M, Bouwmeester J, Bowen BW, Coker DF, Lozano-Cortés DF, Choat JH, Gaither MR, Hobbs JP, Khalil M, Kochzius M, Myers R, Paulay G, Robitzch V, Saenz-Agudelo P, Salas E, Sinclair-Taylor TH, Toonen RJ, Westneat M, Williams S, Berumen ML (2016b) A review of contemporary patterns of endemism for shallow water reef fauna in the Red Sea. J Biogeogr 43:423–439
DiBattista JD, Bowen BW, Gaither MR, Hobbs J-PA, Saenz-Agudelo P, Piatek M, Rocha LA, Choat JH, McIlwain J, Priest MA, Sinclair-Taylor TH, Berumen ML (2017) Comparative phylogeography of reef fishes from the Gulf of Aden to the Arabian Sea reveals two cryptic lineages. Coral Reefs 36:625–638
Drummond AJ, Ashton B, Cheung M, Heled J, Kearse M, Moir R, Stones-Havas S, Thierer T, Wilson A (2009) Geneious v 4.8. www.geneious.com/
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461
Harvey ES, Newman SJ, McLean DL, Cappo M, Meeuwig JJ, Skepper CL (2012) Comparison of the relative efficiencies of stereo-BRUVs and traps for sampling tropical continental shelf demersal fishes. Fish Res 125:108–120
Huson DH, Weber N (2013) Microbial community analysis using MEGAN. Methods Enzymol 531:465–485
Lacoursière-Roussel A, Côté G, Leclerc V, Bernatchez L (2016) Quantifying relative fish abundance with eDNA: a promising tool for fisheries management. J Appl Ecol 53:1148–1157
Leray M, Knowlton N (2015) DNA barcoding and metabarcoding of standardized samples reveal patterns of marine benthic diversity. Proc Natl Acad Sci U S A 112:2076–2081
Mahon AR, Jerde CL, Galaska M, Bergner JL, Chadderton WL, Lodge DM, Hunter ME, Nico LG (2013) Validation of eDNA surveillance sensitivity for detection of Asian carps in controlled and field experiments. PLoS One 8:e58316
Meyer CP, Paulay G (2005) DNA barcoding: error rates based on comprehensive sampling. PLoS Biol 3:e422
Miya M, Sato Y, Fukunaga T, Sado T, Poulsen JY, Sato K, Minamoto T, Yamamoto S, Yamanaka H, Araki H, Kondoh M (2015) MiFish, a set of universal PCR primers for metabarcoding environmental DNA from fishes: detection of more than 230 subtropical marine species. R Soc Open Sci 2:150088
Murray DC, Coghlan ML, Bunce M (2015) From benchtop to desktop: important considerations when designing amplicon sequencing workflows. PLoS One 10:e0124671
Pilliod DS, Goldberg CS, Arkle RS, Waits LP (2013) Estimating occupancy and abundance of stream amphibians using environmental DNA from filtered water samples. Can J Fish Aquat Sci 70:1123–1130
Pleijel F, Jondelius U, Norlinder E, Nygren A, Oxelman B, Schander C, Sundberg P, Thollesson M (2008) Phylogenies without roots? A plea for the use of vouchers in molecular phylogenetic studies. Mol Phylogenet Evol 48:369–371
Raitsos DE, Pradhan Y, Brewin RJ, Stenchikov G, Hoteit I (2013) Remote sensing the phytoplankton seasonal succession of the Red Sea. PLoS One 8:e64909
Roberts CM, Shepherd ARD, Ormond RF (1992) Large-scale variation in assemblage structure of Red Sea butterflyfishes and angelfishes. J Biogeogr 1992:239–250
Roberts MB, Jones GP, McCormick MI, Munday PL, Neale S, Thorrold S, Robitzch VS, Berumen ML (2016) Homogeneity of coral reef communities across 8 degrees of latitude in the Saudi Arabian Red Sea. Mar Pollut Bull 105:558–565
Spalding MD, Ravilious C, Green EP (2001) World atlas of coral reefs. University of California Press, Berkeley
Thomsen PF, Kielgest J, Iversen LL, Møller PR, Rasmussen M, Willerslev E (2012) Detection of a diverse marine fish fauna using environmental DNA from seawater samples. PLoS One 7:e41732
Acknowledgements
This study was funded by KAUST Award No. CRG-1-2012-BER-002 and baseline research funds to M.L.B., as well as a Curtin University Early Career Research Fellowship to J.D.D., M.S., and M.B. acknowledge the support of ARC Linkage Project (LP160100839) to explore marine metabarcoding applications. The authors would also like to acknowledge KAUST’s Coastal and Marine Resources Core Lab for logistical support, as well as Matthew Power, Megan Coghlan, and Brendan Chapman for DNA sequencing assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Editor-in-Chief Prof. Morgan S. Pratchett
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
DiBattista, J.D., Coker, D.J., Sinclair-Taylor, T.H. et al. Assessing the utility of eDNA as a tool to survey reef-fish communities in the Red Sea. Coral Reefs 36, 1245–1252 (2017). https://doi.org/10.1007/s00338-017-1618-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00338-017-1618-1