
Abstract
Objectives
Currently, the BRAF status of pediatric low-grade glioma (pLGG) patients is determined through a biopsy. We established a nomogram to predict BRAF status non-invasively using clinical and radiomic factors. Additionally, we assessed an advanced thresholding method to provide only high-confidence predictions for the molecular subtype. Finally, we tested whether radiomic features provide additional predictive information for this classification task, beyond that which is embedded in the location of the tumor.
Methods
Random forest (RF) models were trained on radiomic and clinical features both separately and together, to evaluate the utility of each feature set. Instead of using the traditional single threshold technique to convert the model outputs to class predictions, we implemented a double threshold mechanism that accounted for uncertainty. Additionally, a linear model was trained and depicted graphically as a nomogram.
Results
The combined RF (AUC: 0.925) outperformed the RFs trained on radiomic (AUC: 0.863) or clinical (AUC: 0.889) features alone. The linear model had a comparable AUC (0.916), despite its lower complexity. Traditional thresholding produced an accuracy of 84.5%, while the double threshold approach yielded 92.2% accuracy on the 80.7% of patients with the highest confidence predictions.
Conclusion
Models that included radiomic features outperformed, underscoring their importance for the prediction of BRAF status. A linear model performed similarly to RF but with the added benefit that it can be visualized as a nomogram, improving the explainability of the model. The double threshold technique was able to identify uncertain predictions, enhancing the clinical utility of the model.
Clinical relevance statement
Radiomic features and tumor location are both predictive of BRAF status in pLGG patients. We show that they contain complementary information and depict the optimal model as a nomogram, which can be used as a non-invasive alternative to biopsy.
Key Points
• Radiomic features provide additional predictive information for the determination of the molecular subtype of pediatric low-grade gliomas patients, beyond what is embedded in the location of the tumor, which has an established relationship with genetic status.
• An advanced thresholding method can help to distinguish cases where machine learning models have a high chance of being (in)correct, improving the utility of these models.
• A simple linear model performs similarly to a more powerful random forest model at classifying the molecular subtype of pediatric low-grade gliomas but has the added benefit that it can be converted into a nomogram, which may facilitate clinical implementation by improving the explainability of the model.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Pediatric low-grade gliomas (pLGG) are the most common brain tumor in children [1].
pLGG are a diverse set of tumors that arise from glial or precursor cells and can occur anywhere in the central nervous system [1, 2]. They include a variety of histopathological diagnoses, including pilocytic astrocytoma, ganglioglioma, dysembryoplastic neuroepithelial tumor, and diffuse glioma, among others [3]. Where total resection is possible, surgical excision can be curative [3]. In partial resection, or if resection is not feasible, the chances of progression or relapse are substantial [3]. Unlike in adult gliomas, the malignant progression of pLGGs is rare [3] and 10-year overall survival (OS) is high, upwards of 90% [4]. However, with 10-year progression-free survival at around 50%, adjuvant therapy is often necessary and morbidity is high [4].
Molecular characterization has identified frequent alterations to the mitogen-activated protein kinase pathway in pLGG, the two most common gene alterations being BRAF fusion and BRAF V600E point mutation (BRAF mutation), which also correspond to prognosis [5,6,7]. This led to the development of targeted therapies, such as BRAF V600E and MEK inhibitors which can supplement or replace the classic cytotoxic adjuvant therapies [4, 5]. The use of these targeted therapies depends on ascertaining the molecular status of pLGG, usually obtained through a biopsy which has its inherent risk, and on occasion yields insufficient material for adequate molecular diagnosis.
Radiomics is a method of extracting information from images in the form of quantitative features [8]. Previously, it was shown that distinguishing between BRAF fusion and mutation in pLGG patients is feasible using machine learning (ML) models trained on radiomics features extracted from T2-weighted fluid-attenuated inversion recovery (FLAIR) MR images [5]. In this study, in order to facilitate translation into the clinical setting, we aimed to establish a nomogram to predict BRAF status (fusion or mutation) based on large internal and external datasets of pLGG, taking clinical factors into account. In addition, we assessed a thresholding method that allows the model to provide predictions for the molecular subtype only when it is confident and abstains otherwise. Finally, compared to the previous work [5], we use a larger dataset (253 vs 115), and a more robust ML pipeline; together these produce more reliable results.
Materials and methods
Patients and data
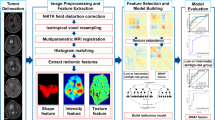
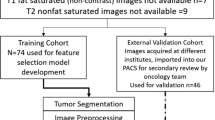
This study was approved by the institutional review board or research ethics board of both participating academic institutions: The Hospital for Sick Children (Toronto, Ontario, Canada) and the Lucile Packard Children’s Hospital (Stanford University, Palo Alto, California). This study was retrospective, and informed consent was waived by the local research ethics boards. An interinstitutional data-transfer agreement was obtained for data-sharing. All patients were identified from the electronic health record database at Toronto and Stanford from 1999 to 2018. Patient inclusion criteria (Fig. 1) were (1) less than 18 years of age, (2) availability of molecular information on BRAF status in histopathologically confirmed pLGG, (3) BRAF fusion or mutation identified, and (4) availability of preoperative brain MR imaging with a non-motion-degraded FLAIR sequence. Spinal cord tumors were excluded from this study. A total of 253 patients were included. In total, 215 were from the Hospital for Sick Children (internal dataset), and 38 were from the Lucile Packard Children’s Hospital (external dataset). Patient information consisted of age at diagnosis, sex, histologic diagnosis, molecular diagnosis regarding the BRAF status, and anatomic location of the tumor (Tables 1, 2, and 3). Figure 2 shows examples of the images in our dataset.

Flowchart of participant selection criteria

Representative examples of patients in our internal dataset. Segmented tumor regions are highlighted in green. On the left is an axial FLAIR MR image of a 6-year-old male with a BRAF-fused pilocytic astrocytoma in the cerebellum. On the right is an axial FLAIR MR image of a 16-year-old female with a BRAF-mutated ganglioglioma in the temporal lobe
Molecular analysis
The molecular characterization of pediatric low-grade gliomas was done based on a tiered approach, with IHC used to detect BRAF mutations followed by FISH or a Nanostring panel to determine BRAF fusions. If sufficient sample quantity and quality allowed, samples that were negative to the aforementioned tests were tested with additional sequencing strategies including panel DNA or RNA sequencing, as described in [7]. For the vast majority of samples, the molecular analysis was done on paraffin-embedded tissue obtained at the time of diagnosis.
MR imaging acquisition, data retrieval, image segmentation, and radiomic feature extraction
All patients from the internal dataset underwent brain MR imaging at 1.5 T (Signa, GE Healthcare) or 3 T (Achieva, Philips Healthcare or MAGNETOM Skyra, Siemens Healthineers). Patients from the external dataset underwent brain MR imaging at 1.5 T or 3 T from a single vendor (Signa or Discovery 750; GE Healthcare). Sequences acquired included a 2D axial and coronal T2 FLAIR, 2D axial and coronal T2-weighted fast spin-echo, 3D axial or sagittal precontrast, and 3D axial gadolinium-based contrast agent–enhanced T1-weighted turbo or fast-field echo.
T2-FLAIR acquisition parameters were TR/TE, 9002/157.5–165 ms; 5-mm section thickness; 2.5-mm gap for the 1.5-T Signa GE MRI scanner, TR/TE, 7000–10,000/140–141 ms; 3- to 5-mm section thickness; 0- to 1-mm gap for the 3-T Achieva MRI scanner, and TR/TE, 9080–9600/83–90 ms; 2.5- to 4-mm section thickness; 1.2- to 2.5-mm gap for the 3-T MAGNETOM Skyra MRI scanner. All MR imaging data were extracted in DICOM format from the respective PACS and were de-identified for further analyses.
Tumor segmentation was performed by a pediatric neuroradiologist (M.W.W.) using 3D Slicer (Version 4.10.2; http://www.slicer.org). Semiautomated tumor segmentation on axial FLAIR images was performed with the level tracing-effect tool. The whole tumor region of interest (ROI) was segmented on axial FLAIR images. The entire tumor was segmented including cystic components. Axial and coronal T2-weighted images were reviewed when tumor borders were difficult to identify. The final placement of the ROI was confirmed by a board-certified neuroradiologist (B.B.E.-W.).
3D Slicer was used to normalize, resample, and bias correct the images; then, the PyRadiomics library [9] was used to calculate the radiomics features through the SlicerRadiomics extension of 3D Slicer using the same pipeline as in [5]. This extension automatically resamples the images to the same voxel size when extracting radiomic features. The default bin width was used (25 voxels), and a symmetric gray-level co-occurrence matrix was enforced. In total, 851 radiomic features were extracted from the ROIs of the FLAIR image for each patient. Radiomic features included histogram, shape, and texture features with and without wavelet-based filters.
Machine learning models
There are known associations between specific BRAF alterations and clinical data such as tumor location [4]. Moreover, random forest models [10] have become popular in radiomics research due to their high transparency and performance, and have proven effective at classifying BRAF status in pLGG using radiomics features [11]. In this study, we trained a random forest on two different sets of features: clinical only and radiomic only. Furthermore, we created an ensemble model, which uses as its prediction the average of the predictions from the models trained on clinical features only, and radiomics features only. We compared the performance of these models to determine whether radiomics features provide additional useful information for this classification task, beyond that which is contained in the clinical data alone. If radiomics features do indeed contain added helpful information, the models relying on radiomics and clinical features together should outperform those relying on either feature set alone.
The radiomics features used included all of the 851 extracted features from the whole tumor ROIs of the FLAIR images from each patient as detailed above. The clinical features used were age, sex, and tumor location. Location was defined using two variables, one less granular (infratentorial vs supratentorial), and one more precise, including categories like cerebellum and temporal lobe. See Table 3 for a full breakdown of tumor locations. The Scikit-learn Python library [12] was used to implement machine-learning models, run experiments and to compute area under the ROC curve (AUC), accuracy, sensitivity, specificity, and Youden’s J statistic.
Cross-validation
We used a nested-cross-validation scheme to train and evaluate our models (Fig. 3). This is computationally expensive; however, it is a robust approach that is not susceptible to biased results based on (un)lucky training/testing splits. An 80/20 split was used for the outer loop which was run 100 times. The inner loop consisted of fivefold cross-validation. Each of the five folds took a turn being held out. The model was trained using a wide array of hyperparameters on the four folds which were not held out. Each of these configurations was evaluated on the held-out fold. After going through this procedure five times, the hyperparameter configuration which performed the best, on average, across the five held-out folds, was determined to be optimal. A random forest model was then trained on the entire inner-loop dataset using these hyperparameters. Finally, we made predictions on the data which was held out in the outer fold to evaluate the model. The model did not encounter this data while being trained in the inner loop.

Visual depiction of nested cross-validation procedure. At the top, in the outer resampling section, orange represents the outer test set, and blue represents the outer training set. In the inner resampling section, light blue represents the inner test set, and dark blue represents the inner training set
Data configuration
While inspecting our data, we observed that there were significant distributional differences between the two datasets. The pathological statistics in Table 2 illustrate that the internal dataset is more heterogeneous than the external dataset. There are a total of 12 separate pathologies in the internal dataset while the external dataset consists of only three. The lack of diversity in the external dataset results in a much easier classification task based on location alone. Indeed, 89.5% of patients in the external dataset have either a supratentorial BRAF-mutated tumor or a BRAF-fused tumor in the infratentorial region. In the internal dataset, just 76.8% of patients have a tumor that follows this typical relationship between molecular subtype and tumor location. The lack of diversity in the external dataset makes it an unreliable test set to evaluate our model with. Rather than discarding the external dataset, which would waste valuable data points, we mixed it with the internal dataset to create a combined dataset. This combined dataset was used in the nested cross-validation procedure.
Thresholding
The output of a binary classifier is usually a value between 0 and 1, where 0 represents one class (negative) and 1 represents the other class (positive). It is up to the user of the model to interpret the class prediction for model outputs between 0 and 1. We used two distinct thresholding approaches for converting the output of our models to class predictions. The first was the classic approach, where a single threshold between 0 and 1 was chosen. Everything above/below the threshold was considered a prediction for the positive/negative class.
This single threshold approach does not account for the model’s confidence in its prediction; a model output just slightly over the threshold and of 1 both result in a positive class prediction. To remedy this, we introduced a thresholding approach that uses two thresholds (upper and lower) to divide the output space into three regions (Fig. 4). An upper region close to 1, above the upper threshold, where the model has high confidence that the true class is positive, a lower region close to 0, below the lower threshold, where the model has high confidence that the true class is negative, and a middle range, where the model does not have high confidence in its prediction. Using this approach, the model says “I don’t know” when the output is in the middle region (between the two thresholds). The model will no longer make a prediction for all patients, but on the subset of patients where it does make a prediction, the expected accuracy is higher than the accuracy across all patients using the single threshold. “High confidence” is a relative term, which depends on the task. Here, we set the double thresholds aiming for 90% accuracy, but this can be adjusted according to performance requirements. If we had chosen a lower target accuracy, the middle region would be smaller and the model would abstain less frequently. Thresholds were tuned on the training data. For the double threshold approach, thresholds that minimized the size of the middle region while satisfying the accuracy condition were selected. Specifically, the lowest (highest) value between 0 and 1, for which 90% of cases above (below) that value was of the positive (negative) class, was selected as the upper (lower) threshold. For the single threshold approach, the threshold that produced maximum accuracy was chosen.

Example depicting the difference between the two thresholding techniques. The placement of the points on the line represents the model output, between 0 and 1, for true negative class points (red) and true positive class points (blue). The single threshold (green) results in a prediction for 100% of patients, with an accuracy of 80%. The double threshold results in a prediction for 80% of patients, with an accuracy of 100%
Nomogram
A nomogram is a visual representation of a mathematical model that generates a probability estimate for an outcome [13]. We present our model in the form of a nomogram, using the Python library Pynomo, to facilitate clinical translation [14]. Due to their high level of complexity, the random forest models we used are not amenable to pictorial representation. For binary classification problems, such as this one, a linear model typically underlies the nomogram. Thus, we trained an additional logistic regression model on both radiomics and clinical features together. L1 regularization, a penalization technique that encourages regression coefficients of 0, was implemented in order to create an interpretable linear regression model with limited non-zero coefficients. We applied z-score normalization to all features prior to training the linear regression model, this was not done for the random forest as it does not impact that model.
Results
Random forest
Table 4 and Fig. 5 list the results for the random forest models trained on the combined internal and external datasets using both radiomics and clinical features alone, along with the ensemble of these two models. The ensemble model performed best according to all but one of these metrics. The clinical and ensemble models had similar sensitivity. By the other four performance metrics, the ensemble model was better than the others; notably, none of the confidence intervals overlapped. For example, the ensemble model had a mean AUC of 0.925 (95% CI: 0.916, 0.932), while the radiomics model had a mean AUC of 0.863 (95% CI: 0.852, 0.874), and the clinical model had a mean AUC of 0.889 (95% CI: 0.881, 0.898).

Distribution over 100 trials of AUC, accuracy, sensitivity, and specificity for the model trained and tested on the combined internal and external datasets. The solid line depicts the median, while the dotted line represents the mean values, which are also illustrated in Table 4
Linear model, thresholding, and nomogram
L1 regularization was used to train the linear model on the combined internal and external datasets, resulting in a compact model with five non-zero coefficients. There were two binary location variables, temporal lobe and cerebellum, and three radiomics features: flatness, surface to volume ratio, and dependence non-uniformity normalized. Both thresholding methods are included in the nomogram (Fig. 6). Table 5 shows that the linear model had a mean AUC of 0.916 (95% CI: 0.908, 0.924). The single threshold resulted in a mean accuracy of 84.5% (95% CI: 83.7%, 85.2%) across all patients while the double threshold approach produced a mean accuracy of 92.2% (95% CI: 91.4%, 93.0%) on the 80.7% (95% CI: 79.3%, 82.2%) of patients for which it made a prediction.

Nomogram representation of the linear model which uses cerebellum, temporal lobe, flatness, surface to volume ratio, and dependence non-uniformity normalized. The corresponding coefficients in the regression formula are − 0.264, 0.679, − 0.083, 0.233, and − 0.073. Both thresholding methods are depicted. Using the single threshold method, the example would result in a prediction of mutation. Under the double threshold method, the model would abstain from making a prediction
Discussion
This study expanded on [5], which showed that an ML model trained on radiomics features could predict the BRAF status of pLGG patients. To confirm the prior results, we repeated the experiments from [5] on a larger dataset (253 vs 115), using a more robust ML pipeline. BRAF mutations tend to be associated with supratentorial lesions, and BRAF fusions tend to be associated with infratentorial lesions [4]. Our results showed that an ensemble RF trained on both radiomics and clinical features (0.925 AUC) performed better than a model trained on either radiomics (0.863 AUC) or clinical features (0.889 AUC) alone. Thus, we concluded that radiomics adds additional predictive power beyond the established relationship between the genetic alteration and the location of the tumor. Additionally, we created a nomogram to facilitate the translation into the clinical setting using a linear model (AUC 0.916), which despite its lower representational capacity performed similarly to the RF. Finally, we introduced a thresholding method that enables the model to provide predictions only when confidence is high. Traditional thresholding produced an accuracy of 84.5% across all patients, while the more sophisticated thresholding method resulted in an accuracy of 92.2% on the 80.7% of patients where a prediction was made. To the best of our knowledge, this work is the first to show that thresholding of the model output can be used to exclude unreliable predictions in medical imaging. Oftentimes, ML models get stuck at the research stage because accuracy across all patients is inadequate for clinical use. The double threshold method can be used to identify a subset of patients for which the model is confident, and accuracy is sufficient, enhancing the utility of these models. Notably, this thresholding method is flexible and can accommodate the end user’s desired level of accuracy. For example, in cases where an incorrect prediction is unacceptable, thresholds can be set such that the model says “I don’t know” often but is rarely incorrect when it makes a class prediction.
Supratentorial pLGGs are most often BRAF-mutated, while infratentorial pLGGs are usually BRAF-fused. In our combined dataset, 74.7% of patients followed this typical relationship between genetic status and tumor location. It should be noted that all of our predictive models produced a higher accuracy than one would expect to achieve from predicting genetic status based on whether the pLGG is located in the supratentorial or infratentorial region. Our models achieved high accuracy despite the substantial variation within the images in our dataset, including different scanners, scan parameters, and resolutions. Strong performance across diverse data means our model is robust; it has great flexibility in terms of the diversity of input images it can process.
There are limitations to this work. First, we observed that the diversity of our external dataset was limited. We confirmed this observation by running a preliminary set of experiments where the model was trained on the internal dataset and tested on the external dataset. We found that the model relying on only clinical features classified the external data nearly perfectly, highlighting the lack of diversity in the external dataset. Thus, for this study, we combined the internal and external datasets. Second, the model differentiates between BRAF fusion and mutation; other potential molecular alterations are not accounted for. However, this molecular differentiation is currently the most important one for prognostication and therapeutic decision-making. Finally, although the double threshold method was successful in identifying uncertain predictions, it is not a replacement for building better ML models. Where model performance is inadequate overall, this approach can help to specify a subset of inputs where the model could still be useful. A more comprehensive approach would be to collect additional data and design superior learning algorithms which would improve performance across all inputs.
Conclusion
In this study, we designed a pipeline to differentiate BRAF status in pediatric low-grade glioma based on a combination of imaging and clinical features and evaluated the performance of this pipeline using a bi-institutional dataset. We developed a nomogram to support the translation of our predictive model into the clinical setting. Additionally, we evaluated an advanced thresholding method that successfully identified uncertain predictions, enhancing the clinical utility of our model. Further studies with larger and more complex external datasets are needed to augment diagnostic accuracy and incorporate additional molecular markers.
Abbreviations
- AUC:
-
Area under the ROC curve
- BRAF mutation:
-
BRAF V600E point mutation
- CI:
-
Confidence interval
- ML:
-
Machine learning
- pLGG:
-
Pediatric low-grade gliomas
- RF:
-
Random forest
- ROI:
-
Region of interest
- FLAIR:
-
T2-weighted fluid-attenuated inversion recovery
References
Ostrom QT, Patil N, Cioffi G et al (2020) CBTRUS Statistical Report: primary brain and other central nervous system tumors diagnosed in the United States in 2013–2017. Neuro Oncol 22 (Supplement_1):iv1–iv96
Sievert AJ, Fisher MJ (2009) Pediatric low-grade gliomas. J Child Neurol 24:1397–1408
Sturm D, Pfister SM, Jones DTW (2017) Pediatric gliomas: current concepts on diagnosis, biology, and clinical management. J Clin Oncol 35:2370–2377
Ryall S, Tabori U, Hawkins C (2020) Pediatric low-grade glioma in the era of molecular diagnostics. Acta Neuropathol Commun 8:30
Wagner MW, Hainc N, Khalvati F et al (2021) Radiomics of pediatric low-grade gliomas: toward a pretherapeutic differentiation of BRAF-mutated and BRAF-fused tumors. AJNR Am J Neuroradiol 42:759–765
Lassaletta A, Zapotocky M, Mistry M et al (2017) Therapeutic and prognostic implications of BRAF V600E in pediatric low-grade gliomas. J Clin Oncol 35:2934–2941
Ryall S, Zapotocky M, Fukuoka K et al (2020) Integrated molecular and clinical analysis of 1,000 pediatric low-grade gliomas. Cancer Cell 37:569-583.e5
Khalvati, F, Zhang Y, Wong A et al (2019) Radiomics. Encyclopedia of Biomedical Engineering:597–603
van Griethuysen JJM, Fedorov A, Parmar C et al (2017) Computational radiomics system to decode the radiographic phenotype. Cancer Res 77:e104–e107
Breiman L (2001) Random Forests. Mach Learn 45:5–32
Wagner MW, Namdar K, Alqabbani A et al (2022) Dataset size sensitivity analysis of machine learning classifiers to differentiate molecular markers of paediatric low-grade gliomas based on MRI. Oncology and Radiotherapy 16:01–06
Pedregosa F, Varoquaux G, Gramfort A et al (2011) Scikit-learn: machine learning in Python. J Mach Learn Res 12:2825–2830
Iasonos A, Schrag D, Raj GV, Panageas KS (2008) How to build and interpret a nomogram for cancer prognosis. J Clin Oncol 26:1364–1370
Hart WE, Laird CD, Watson J-P et al (2017) Pyomo — optimization modeling in Python. Springer, Cham
Acknowledgements
This research has been made possible with the financial support of the Canadian Institutes of Health Research (CIHR) (Funding Reference Number: 184015).
Funding
This research has been made possible with the financial support of the Canadian Institutes of Health Research (CIHR) (Funding Reference Number: 184015).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Guarantor
The scientific guarantor of this publication is Dr. Farzad Khalvati.
Conflict of interest
The authors of this manuscript declare no relationships with any companies, whose products or services may be related to the subject matter of the article.
Statistics and biometry
No complex statistical methods were necessary for this paper.
Informed consent
Written informed consent was waived by the Institutional Review Board.
Ethical approval
Institutional Review Board approval was obtained (The Hospital for Sick Children (Toronto, Ontario, Canada) and the Lucile Packard Children’s Hospital (Stanford University, Palo Alto, California).
Study subjects or cohorts overlap
Some study subjects or cohorts have been previously reported in two previous papers. The first, “Radiomics of Pediatric Low-Grade Gliomas: Toward a Pretherapeutic Differentiation of BRAF-Mutated and BRAF-Fused Tumors,” https://doi.org/10.3174/ajnr.A6998, was an exploratory study that relied on 115 of our 253 patients. The second, “Dataset size sensitivity analysis of machine learning classifiers to differentiate molecular markers of pediatric low-grade gliomas based on MRI,” Oncology and Radiotherapy 16 (S1) 2022: 01–06, relied on 251 of our 253 patients. These previous studies aimed to establish a relationship between radiomics features and BRAF status, and to determine the best machine learning model on this classification task.
Methodology
• Retrospective
• Diagnostic or prognostic study
• Multicenter study
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Birgit B. Ertl-Wagner and Farzad Khalvati are co-senior authors.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kudus, K., Wagner, M.W., Namdar, K. et al. Increased confidence of radiomics facilitating pretherapeutic differentiation of BRAF-altered pediatric low-grade glioma. Eur Radiol 34, 2772–2781 (2024). https://doi.org/10.1007/s00330-023-10267-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00330-023-10267-1