
Abstract
Objectives
To evaluate the longitudinal changes of chest CT findings in patients with chronic hypersensitivity pneumonitis (HP) and identify risk factors for fibrotic progression and acute exacerbation (AE).
Methods
This retrospective study included patients with chronic HP with follow-up CT. Baseline and serial follow-up CT were evaluated semi-quantitatively. Fibrosis score was defined as the sum of the area with reticulation and honeycombing. The modified CT pattern of Fleischner Society idiopathic pulmonary fibrosis diagnostic guidelines was evaluated. Cox proportional hazards regression was performed to determine significant variables associated with fibrotic progression and AEs.
Results
Of 91 patients, mean age was 59.1 years and 61.5% were women. The median follow-up period was 4.9 years. Seventy-nine patients (86.8%) showed fibrotic progression with persistent areas of mosaic attenuation, finally replaced by fibrosis, and 20 (22.0%) developed AE. Baseline fibrosis score and CT pattern of usual interstitial pneumonia (UIP)/probable UIP were independent risk factors for predicting fibrotic progression (hazard ratio [HR] = 1.05, 95% confidence interval [CI] = 1.02–1.09, p < 0.001, for fibrosis score; HR = 2.50, CI = 1.50–4.16, p < 0.001, for CT pattern) and AEs (HR = 1.07, CI = 1.01–1.13, p = 0.019, for fibrosis score; HR = 5.47, CI = 1.23–24.45, p = 0.026, for CT pattern) after adjusting clinical covariables.
Conclusion
Fibrotic progression and AE were identified in 86.8% and 22.0% of patients with chronic HP. Fibrosis score and CT pattern of UIP/probable UIP on baseline chest CT may predict fibrotic progression and AE.
Key Points
• Most patients (87%) showed fibrotic progression on long-term follow-up with persistent areas of mosaic attenuation that were finally replaced by fibrosis at a later stage.
• One-fifth of patients (22%) experienced acute exacerbation associated with worse prognosis.
• Fibrosis score (sum of reticulation and honeycombing) and CT pattern of UIP/probable UIP on baseline CT were independent predictors for predicting fibrotic progression and acute exacerbation.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Hypersensitivity pneumonitis (HP) is a diffuse interstitial lung disease that results from an immunologic response to various inhaled antigens in susceptible individuals [1]. HP can be classified into two different categories: acute/inflammatory HP and chronic/fibrotic HP [2]. The presence of fibrotic change on high-resolution computed tomography (CT) images or lung tissue indicates chronic fibrotic HP. The clinical course and outcomes of chronic HP are highly variable. Some patients can show stable disease and may also experience partial recovery; however, some patients can show disease progression and manifest acute respiratory decline during the clinical course, similar to acute exacerbation (AE) in patients with idiopathic pulmonary fibrosis (IPF). However, predicting the course of the disease and the risk of fibrotic progression in each individual remains a challenge for clinicians, and it is difficult to decide the initiation of drug treatment.
Previous studies have shown that clinical, histopathologic, and radiologic findings can predict prognosis in chronic HP [1, 3,4,5,6,7] and patients with radiologic and histopathologic evidence of fibrosis or usual interstitial pneumonia (UIP)-like pattern of fibrosis experience worse survival than those without. However, few studies have evaluated the serial CT changes over a period of time in patients with chronic HP and no analysis was presented regarding the fibrotic progression based on imaging as an endpoint, which can be an early prognostic indicator. Acute exacerbation (AE) in IPF is increasingly recognized as a relatively common and highly morbid clinical event. AE is also reported in non-IPF patients and seems to be associated with worse outcomes [8, 9]. However, data in patients with chronic HP are limited, and the risk factors associated with the AE are poorly defined [4, 10].
Therefore, the primary aim of our study was to evaluate longitudinal changes in chest CT features in patients with chronic HP and to identify risk factors for fibrotic progression and AE.
Materials and method
Study population
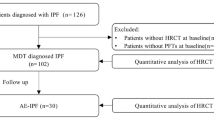
Retrospective analysis of the interstitial lung disease databases of Asan Medical Center (a tertiary referral hospital in Seoul, Republic of Korea) identified 101 consecutive patients with chronic HP between January 2002 and December 2017. All patients underwent surgical or transbronchial lung biopsy and were diagnosed with chronic HP through a multidisciplinary discussion. Among the 101 patients, 10 were excluded since serial follow-up CTs with an interval of more than 1 year were not available. Finally, 91 patients with definite HP and follow-up CT with an interval longer than 1 year were included in this study. This study was approved by the Institutional Review Board (2019–1365) of Asan Medical Center, and the requirement for informed consent was waived due to the study’s retrospective nature.
Clinical data
Clinical and survival data of all patients were obtained from medical records, telephone interviews, and/or the records of the National Health Insurance Service of Korea. Bronchoalveolar lavage (BAL) was performed in accordance with the guidelines [11]. AE of chronic HP was defined as acute, clinically significant respiratory deterioration of unknown cause, typically within one month, and characterized radiologically by new bilateral opacities according to the criteria suggested for IPF [12].
Analysis of CT findings
CT scans were performed at the time of the initial diagnosis and at sequential follow-up. CT scans were considered of diagnostic quality if they included thin-section images with reconstruction interval less than 2 mm, were through the whole thorax, and were free of significant motion artifacts. To evaluate serial changes in chest CT features and to determine risk factors for fibrotic progression and AE, baseline and serial follow-up CTs were evaluated qualitatively and semi-quantitatively. Two chest radiologists (E.J.C. and J.C.), who were blinded to all clinical data except the diagnosis of chronic HP, reviewed and scored the baseline CTs, with discrepancies resolved by consensus. Fibrotic progression was defined as an increase in extent of interstitial fibrosis including reticulation, honeycombing, and/or traction bronchiectasis on CT which was reviewed by consensus between the two observers.
For the semi-quantitative analysis of chest CT, the average extent of reticulation, honeycombing, ground-glass opacity (GGO), mosaic attenuation (on inspiration), consolidation, and nodules were scored to the nearest 10th percentile in 6 lung zones as previously described [13]. The mean extent of reticulation and honeycombing were summed and defined as the total fibrosis score. The severity of bronchiectasis (grade 0–3) was assessed by modifying the previously described CT score in each of the six lung zones [14]. The final bronchiectasis grade was then calculated by summing the grades of each of the lung zones (range 0–18). All serial follow-up CTs acquired during the follow-up period were evaluated semi-quantitatively (J.C.).
Furthermore, to evaluate whether the UIP pattern on CT was associated with fibrotic progression, baseline CT scans were classified using a modification of the Fleischner Society IPF diagnostic guidelines [15]. To apply these diagnostic criteria to the HP population, the previous diagnostic criteria were modified by excluding mosaic attenuation and distribution from the list of features of alternative diagnosis prohibiting a UIP diagnosis, as previously described for patients with rheumatoid arthritis-associated interstitial lung disease [16].
Statistical analysis
Data and statistical analyses were performed using SPSS (SPSS software, version 23.0; IBM) and R software (version 3.6.3, [www.R-project.org]). Univariable and multivariable Cox proportional hazards regression analyses were performed to determine significant variables associated with fibrotic progression and AE occurrence. The definition of fibrotic progression-free survival was time from diagnosis to the fibrotic progression on CT or death. From the multivariable analysis, adjusted hazard ratio (HR) was evaluated for the significant imaging parameters as pre-specified covariables. Additional covariables were selected based on the clinical importance and from significant predictors of univariable analysis, including collinearity and numbers of events. Three models were tested with the addition of age, sex, and pulmonary function (forced vital capacity [FVC] or diffusion capacity for carbon monoxide [DLco], respectively, for fibrotic progression and AE) as covariables to those pre-specified. The overall performance of the imaging parameter predicting fibrotic progression and AE was evaluated using Harrell’s C-index, and the optimal cut-off values of continuous variables predicting fibrotic progression and AE were determined using the algorithm that splits the cohort into two groups by estimation of the optimal cut-off point with the highest log-rank statistics [17]. Time-dependent receiver operating characteristic (ROC) curve was calculated for risk scores and area under the ROC curve (AUC), sensitivity, and specificity using the R package timeROC [18]. To investigate the longitudinal changes of parenchymal abnormalities among patients, we performed linear mixed model for analyzing the repeated measures considering the repeated CT scans per patient, using PROC MIXED procedure. Fibrotic progression-free survival and overall survival were evaluated using Kaplan–Meier curves and the log-rank test. In all analyses, p < 0.05 was considered statistically significant.
Results
Study population
The characteristics of 91 patients included in the study are summarized in Table 1. The mean age was 59.1 years and 61.5% were women. The median follow-up period was 4.9 years (interquartile range, 3.2–8.1 years). Death occurred in 39 (42.9%) patients, and the median survival time was 9.5 years.
Serial changes in CT features
Seventy-nine (86.8%) showed fibrotic progression during the follow-up period. The median imaging follow-up time between the initial and last CT was 3.9 years (range, 1.0–10.9 years), and the median follow-up interval between each CT was 12.7 months (range, 11.0–71.1 months) in all patients. The median imaging follow-up time between the initial and last CT was 2.21 years (range, 1.0–6.3 years) in patients without fibrotic progression and 4.1 years (range 1.0–10.9 years) in those with fibrotic progression. The 1- and 2-year cumulative incidence of fibrotic progression was 15.4% and 41.7%, respectively. In patients with fibrotic progression, the median fibrotic progression-free survival was 2.1 years (95% confidence interval [CI], 1.6–10.9).
When changes in CT features between initial and last follow-up were assessed, reticulation, honeycombing, and traction bronchiectasis showed significant increase (change in extent of reticulation, 10.6 ± 8.6%; honeycombing, 3.9 ± 5.8%; and traction bronchiectasis grade, 3.6 ± 3.4) in patients with fibrotic progression. Areas of mosaic attenuation were mostly persistent in patients with fibrotic progression. Among the 79 patients with fibrotic progression, 24 (24/79, 30.3%) showed a decrease in the extent of areas of mosaic attenuation, which were finally replaced by fibrosis in the course of fibrotic progression (Fig. 1). The other 55 patients showed persistent and unchanged areas of mosaic attenuation. No patient showed significant increase in the extent of mosaic attenuation since the baseline CT. There was a tendency of improvement of GGO and nodules (change in extent of GGO, − 7.5 ± 16.6% and nodule, − 4.7 ± 12.7%) in patients who did not show fibrotic progression during the follow-up period.

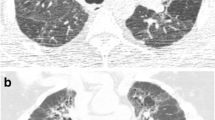
65-year-old female with chronic HP. a Baseline axial thin-section CT scan at the level of the lung base shows bilateral peribronchovascular ground-glass opacities (GGOs), traction bronchiectasis, and reticular opacities admixed with areas of mosaic attenuation (arrows). b One-year follow-up CT shows mild progression of fibrosis with slight increase in extent of reticular opacities and mild aggravation of traction bronchiectasis in both lung bases. However, the areas of mosaic attenuation are spared and persistent (arrows). c Three-year follow-up CT shows marked increase in extent of reticular opacities and honeycombing in both lung bases, also replacing the previous areas of mosaic attenuation in the left lower lobe (arrowhead). However, the areas of mosaic attenuation in the right lower lobe are relatively spared from the increase of fibrosis (arrow)
Risk factors of fibrotic progression
In a Cox proportional hazards model, the univariable analysis identified age, baseline FVC, UIP CT pattern, extent of reticulation, extent of GGO, total fibrosis score, and bronchiectasis grade on baseline CT as significant factors associated with fibrotic progression (Table 2). Neither the presence nor the extent of mosaic attenuation was associated with fibrotic progression. In the multivariable Cox proportional hazards analysis, baseline fibrosis score and UIP or probable UIP CT pattern were independent predictors of fibrotic progression when adjusted by sex, age, and baseline FVC (Table 2).
The C-index for predicting fibrotic progression was 0.66. The AUC for predicting fibrotic progression was 0.69 at 3 years (e-Figure 1). The best cut-off level for fibrosis score was 10%. For 10% cut-off value of baseline fibrosis score, sensitivity and specificity were 69.9% and 57.9% for 3-year fibrotic progression. Figure 2 shows the changes in the overall extent of parenchymal abnormalities during the follow-up period subdivided based on the baseline fibrosis score. There were a significant increase in the extent of honeycombing and fibrosis score (Wilcoxon test, p < 0.001) and a slight decrease in the extent of mosaic attenuation (p = 0.002) on 2-year follow-up CT compared to the baseline CT in patients with baseline fibrosis score ≥ 10% but not in those with baseline fibrosis score < 10% (Table 3).

Serial CT changes in overall extent of parenchymal abnormalities of patients subdivided based on baseline fibrosis score. Serial changes in overall extent of (a) total fibrosis score, (b) reticulation, (c) honeycombing, (d) mosaic attenuation, (e) ground-glass opacity, (f) nodule, and (g) grade of bronchiectasis on CT. Linear mixed model was used for analyzing the repeated measures considering the longitudinal follow-up CT per patient. Patients were subdivided based on baseline fibrosis score (fibrosis score of high-risk group ≥ 10% and fibrosis score of low-risk group < 10%). The large dots indicate the mean of data at each time point, and error bars indicate the standard error of the mean.
Patients could be stratified into high- or low-risk groups of fibrotic progression based on the 10% cut-off value (median fibrotic progression-free survival: 2.0 years [95% CI, 1.6–2.2] for the high-risk group and 3.1 years [95% CI, 2.3–10.9] for the low-risk group; log-rank test, p = 0.001) (Fig. 3). Overall survival was significantly different between the high- and low-risk groups when stratified according to the baseline fibrosis score (log-rank test, p < 0.001; e-Figure 2). When the Fleischner system of IPF was examined for fibrotic progression, there was a significant difference in outcomes among the four groups (log-rank test, p = 0.001) and between patients with UIP or probable UIP CT pattern and the others (log-rank test, p < 0.001; Fig. 3).

Kaplan–Meier curve of fibrotic progression subdivided according to the baseline fibrosis score (cut-off value, 10%) and the usual interstitial pneumonia (UIP) CT pattern based on the Fleischner Society idiopathic pulmonary fibrosis (IPF) diagnostic guidelines. a Risk stratification based on fibrosis score on baseline CT (fibrosis score of high-risk group ≥ 10% and fibrosis score of low-risk group < 10%). Risk stratification based on UIP CT pattern based on the Fleischner Society IPF diagnostic guidelines: b four groups—UIP, probable UIP, indeterminate for UIP, and alternative diagnosis, and (c) two groups—combined UIP and probable UIP CT pattern or the others
Risk factors of acute exacerbation
During the follow-up period, 20 (22.0 %) patients showed AE, and the mortality rate was 36.8% (7/20). The extent of honeycombing and fibrosis score on baseline CT were significantly higher in patients with AE than in those without AE (p = 0.019 for honeycombing and p = 0.037 for fibrosis score; e-Table 2). The extent of GGO on baseline CT was significantly higher in patients without AE than in those with AE (p = 0.003). Among 12 patients with available follow-up chest CT after AE, 66.7% (8/12) showed fibrotic progression on follow-up CT after AE, with marked aggravation of bronchiectasis (change in bronchiectasis grade, median 4 [range 3–8]).
Univariable analysis revealed that older age at diagnosis, presence of honeycombing, UIP CT pattern, higher extent of reticulation and honeycombing, lower extent of GGO, and higher grade of bronchiectasis and fibrosis score on baseline CT were associated with AE events (Table 4). Baseline DLCO showed marginal significance (p = 0.051) as a clinical predictor of AE. Higher baseline fibrosis score and UIP or probable UIP CT pattern were significant independent predictors of AE when adjusted by clinical covariables (Table 4).
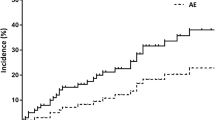
The C-index of fibrosis score for predicting AE was 0.71. The AUC for predicting AE was 0.75 for 3 years (e-Figure 1). The best cut-off for fibrosis score was 13%. For the 13% cut-off value of baseline fibrosis score, sensitivity and specificity were 83.4% and 55.4% for 3-year AE. Patients could be stratified into high- and low-risk groups of AE based on the cut-off value of the baseline fibrosis score (log-rank test, p = 0.036 for 10% cut-off and p = 0.001 for 13% cut-off; Fig. 4). Patients could also be stratified based on the presence of the UIP CT pattern showing different outcomes for AE occurrence (Fig. 4).

Cumulative incidence of acute exacerbation subdivided according to the baseline fibrosis score and the usual interstitial pneumonia (UIP) CT pattern based on the Fleischner Society idiopathic pulmonary fibrosis (IPF) diagnostic guidelines. Risk stratification based on fibrosis score on baseline CT: a 10% cut-off of fibrosis score (high-risk group ≥ 10% and fibrosis score of low-risk group < 10%) and (b) 13% cut-off of fibrosis score (high-risk group ≥ 13% and fibrosis score of low-risk group < 13%). Risk stratification based on UIP CT pattern based on the Fleischner Society IPF diagnostic guidelines: c four groups—UIP, probable UIP, indeterminate for UIP, and alternative diagnosis, and (d) two groups—combined UIP and probable UIP or the others
Discussion
Our study assessed the disease trajectory of patients with chronic HP on serial follow-up CT. Although chronic HP has been known to have a variable clinical course, in our study, most of the patients (86.6%) showed fibrotic progression on CT. Serial follow-up CTs showed increased extent of fibrosis (reticulation, honeycombing, and traction bronchiectasis) with relatively spared areas of mosaic attenuation that were finally replaced by fibrosis in the later stage of progression. Moreover, a substantial proportion of patients (22.0%) experienced AE during the follow-up period, which resulted in worse prognosis and progression of fibrosis. Fibrosis score and UIP or probable UIP CT pattern on baseline CT were significant risk factors for predicting both fibrotic progression and AE events.
Previous studies have reported that a proportion of patients with chronic HP exhibit a rapid decline in lung function and progression to death, as in the case of patients with IPF [5, 19]. Numerous analyses highlight the prognostic value of identification and/or quantification of radiologic fibrosis in patients with chronic HP in relation to survival [3, 5, 20, 21]. In previous studies, honeycombing and fibrosis were associated with a worse prognosis, while the presence of mosaic attenuation or air trapping was associated with a better prognosis. Our findings are consistent with previous studies demonstrating that the presence of pulmonary fibrosis on CT is independently associated with worse survival. In this study, we paid particular attention to the assessment of fibrotic progression as an endpoint to identify a progressive fibrotic phenotype that might need early initiation of treatment and more active follow-up. We found that reticulation, bronchiectasis, and fibrosis score, which is a sum of the extent of reticulation and honeycombing, were significant risk factors for early fibrotic progression. In multivariable analysis, the total extent of fibrosis on baseline CT, whether it was honeycombing or not (i.e., fibrosis score) were powerful independent risk factors predicting fibrosis progression, along with FVC.
Chung et al [3] evaluated the associations between mosaic attenuation and air trapping with a reduced hazard of death. In our study, the extent of mosaic attenuation was not a significant prognostic factor predicting fibrotic progression. The discrepancy in results can be due to a different time point of observation, and it is uncertain whether dichotomized presence or extent of mosaic attenuation is an independent prognostic factor or not [3, 5, 22]. However, it is evident that patients who show predominant acute HP-like CT features, such as centrilobular nodules and GGOs which might reflect the active/inflammatory cellular phase of HP with only minimal or absence of fibrosis on initial CT, are less likely to show early fibrotic progression and less likely to experience AE, which were also supported by our results.
On serial follow-up, CTs showed an increase in the extent of reticulation and honeycombing, with the development of honeycombing in many patients who had no honeycombing on baseline CT, and aggravation of traction bronchiectasis, consistent with a previous study [23]. The extent of centrilobular nodules and GGOs decreased on serial CT, with the difference being more profound in patients without fibrotic progression or with low baseline fibrosis score (< 10%), which might reflect an acute inflammatory component for HP underlying chronic fibrosis. Intriguingly, mosaic attenuation in chronic HP tended to remain constant over time and seemed to be preserved from pulmonary fibrosis that fibrosis preferentially involving and extending to the other areas in the early phase. During the course of fibrotic progression, fibrosis finally replaced areas of mosaic attenuation (areas of decreased attenuation). Further study is needed to clarify if and how the presence of mosaic attenuation, airway changes, and vascular changes can independently modify the process of development and progression of interstitial fibrosis in chronic HP.
The fibrosis score and UIP/probable UIP CT patterns were significant independent risk factors for AE. Miyazaki et al reported low baseline DLCO as a risk factor for AE in chronic HP [10]. In addition, they also reported low lymphocyte levels in BAL fluid and a UIP-like pattern on histology at the time of diagnosis as risk factors for AE. In our study, both baseline DLCO and percentage of lymphocytes showed borderline significance as predictors of AE (p = 0.051 and p = 0.086, respectively). Regarding UIP pattern, we evaluated the CT pattern by modifying the Fleischner system of IPF diagnosis; the risk of AE can be stratified according to CT pattern regardless of the presence of mosaic attenuation and whether the distribution was IPF-like or not. We also found that the incidence of idiopathic AE increased in association with a UIP CT pattern and presence of honeycombing, consistent with results from the previous study based on pathology.
There are a number of limitations to the current study. First, because of its retrospective nature, a variety of treatment options were used in patients who had progressive disease, and so we were unable to confidently make inferences about the effects of therapy. However, the long-term benefits and the effects of immunosuppressive drugs remain limited and treatment with steroids and/or immunosuppressive drugs were not significant risk factors for both fibrotic progression and AE on univariable analysis in our study. Another limitation of this study is the potential for the CT scorers to have unconscious biases in the scoring (i.e., different sensitive thresholds for mosaic attenuation on visual analysis).
In conclusion, fibrotic progression and acute exacerbation were identified in 86.8% and 22.0% of patients with chronic HP. Our data suggest that fibrosis score and CT pattern of UIP or probable UIP on baseline chest CT may predict fibrotic progression and AE.
Abbreviations
- AE:
-
Acute exacerbation
- AUC:
-
Area under the receiver operating characteristic curve
- BAL:
-
Bronchoalveolar lavage
- CI:
-
Confidence interval
- CT:
-
Computed tomography
- DLCO :
-
Diffusing capacity for carbon monoxide
- FEV1 :
-
Forced expiratory volume in 1 s
- FVC:
-
Forced vital capacity
- GGO:
-
Ground-glass opacity
- HP:
-
Hypersensitivity pneumonitis
- IPF:
-
Idiopathic pulmonary fibrosis
- ROC:
-
Receiver operating characteristic
- UIP:
-
Usual interstitial pneumonia
References
Selman M, Pardo A, King TE Jr (2012) Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med 186:314–324
Vasakova M, Morell F, Walsh S, Leslie K, Raghu G (2017) Hypersensitivity pneumonitis: perspectives in diagnosis and management. Am J Respir Crit Care Med 196:680–689
Chung JH, Zhan X, Cao M et al (2017) Presence of air trapping and mosaic attenuation on chest computed tomography predicts survival in chronic hypersensitivity pneumonitis. Ann Am Thorac Soc 14:1533–1538
Chiba S, Tsuchiya K, Akashi T et al (2016) Chronic hypersensitivity pneumonitis with a usual interstitial pneumonia-like pattern: correlation between histopathologic and clinical findings. Chest 149:1473–1481
Salisbury ML, Gu T, Murray S et al (2019) Hypersensitivity pneumonitis: radiologic phenotypes are associated with distinct survival time and pulmonary function trajectory. Chest 155:699–711
Walsh SL, Sverzellati N, Devaraj A, Wells AU, Hansell DM (2012) Chronic hypersensitivity pneumonitis: high resolution computed tomography patterns and pulmonary function indices as prognostic determinants. Eur Radiol 22:1672–1679
Wang P, Jones KD, Urisman A et al (2017) Pathologic findings and prognosis in a large prospective cohort of chronic hypersensitivity pneumonitis. Chest 152:502–509
Kolb M, Bondue B, Pesci A et al (2018) Acute exacerbations of progressive-fibrosing interstitial lung diseases. Eur Respir Rev 27:180071
Moua T, Westerly BD, Dulohery MM, Daniels CE, Ryu JH, Lim KG (2016) Patients with fibrotic interstitial lung disease hospitalized for acute respiratory worsening: a large cohort analysis. Chest 149:1205–1214
Miyazaki Y, Tateishi T, Akashi T, Ohtani Y, Inase N, Yoshizawa Y (2008) Clinical predictors and histologic appearance of acute exacerbations in chronic hypersensitivity pneumonitis. Chest 134:1265–1270
Meyer KC, Raghu G, Baughman RP et al (2012) An official American Thoracic Society clinical practice guideline: the clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am J Respir Crit Care Med 185:1004–1014
Collard HR, Moore BB, Flaherty KR et al (2007) Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 176:636–643
Best AC, Meng J, Lynch AM et al (2008) Idiopathic pulmonary fibrosis: physiologic tests, quantitative CT indexes, and CT visual scores as predictors of mortality. Radiology 246:935–940
Bhalla M, Turcios N, Aponte V et al (1991) Cystic fibrosis: scoring system with thin-section CT. Radiology 179:783–788
Lynch DA, Sverzellati N, Travis WD et al (2018) Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society white paper. Lancet Respir Med 6:138–153
Jacob J, Hirani N, van Moorsel CHM et al (2019) Predicting outcomes in rheumatoid arthritis related interstitial lung disease. Eur Respir J 53:1800869
Ogluszka M, Orzechowska M, Jedroszka D, Witas P, Bednarek AK (2019) Evaluate cutpoints: adaptable continuous data distribution system for determining survival in Kaplan-Meier estimator. Comput Methods Programs Biomed 177:133–139
Heagerty PJ, Lumley T, Pepe MS (2000) Time-dependent roc curves for censored survival data and a diagnostic marker. Biometrics 56:337–344
Churg A, Sin DD, Everett D, Brown K, Cool C (2009) Pathologic patterns and survival in chronic hypersensitivity pneumonitis. Am J Surg Pathol 33:1765–1770
Hanak V, Golbin JM, Hartman TE, Ryu JH (2008) High-resolution ct findings of parenchymal fibrosis correlate with prognosis in hypersensitivity pneumonitis. Chest 134:133–138
Chung JH, Montner SM, Adegunsoye A et al (2017) CT findings associated with survival in chronic hypersensitivity pneumonitis. Eur Radiol 27:5127–5135
Jacob J, Bartholmai BJ, Egashira R et al (2017) Chronic hypersensitivity pneumonitis: identification of key prognostic determinants using automated CT analysis. BMC Pulm Med 17:81
Tateishi T, Ohtani Y, Takemura T et al (2011) Serial high-resolution computed tomography findings of acute and chronic hypersensitivity pneumonitis induced by avian antigen. J Comput Assist Tomogr 35:272–279
Acknowledgments
We express our gratitude to Seonok Kim and Hwa Jung Kim of the Department of Clinical Epidemiology and Biostatistics, Asan Medical Center, for valuable advice regarding the statistical analysis.
Funding
The authors state that this work has not received any funding.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Guarantor
The scientific guarantor of this publication is Eun Jin Chae.
Conflict of interest
The authors of this manuscript declare no relationships with any companies whose products or services may be related to the subject matter of the article.
Statistics and biometry
Seonok Kim of the Department of Clinical Epidemiology and Biostatistics, Asan Medical Center, kindly provided statistical advice for this manuscript.
Informed consent
Written informed consent was waived by the Institutional Review Board.
Ethical approval
Institutional Review Board approval was obtained.
Methodology
• retrospective
• diagnostic or prognostic study
• performed at one institution
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(DOCX 588 kb)
Rights and permissions
About this article

Cite this article
Choe, J., Chae, E.J., Kim, Y.J. et al. Serial changes of CT findings in patients with chronic hypersensitivity pneumonitis: imaging trajectories and predictors of fibrotic progression and acute exacerbation. Eur Radiol 31, 3993–4003 (2021). https://doi.org/10.1007/s00330-020-07469-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00330-020-07469-2