
Abstract
Hexactinellida (glass sponges) are abundant and important components of Antarctic benthic communities. However, the relationships and systematics within the common genus Rossella Carter, 1872 (Lyssacinosida: Rossellidae), are unclear and in need of revision. The species content of this genus has changed dramatically over the years depending on the criteria used by the taxonomic authority consulted. Rossella was formerly regarded as a putatively monophyletic group distributed in the Southern Ocean and the North Atlantic. However, molecular phylogenetic analyses have shown that Rossella is restricted to the Southern Ocean, where it shows a circum-Antarctic and subantarctic distribution. Herein, we provide a molecular phylogenetic analysis of the genus Rossella, based on mitochondrial (16S rDNA and COI) and nuclear (28S rDNA) markers. We corroborate the monophyly of Rossella and provide evidence supporting the existence of one species, namely Rossella antarctica Carter, 1872 and a species flock including specimens determined as Rossella racovitzae Topsent, 1901, Rossella nuda Topsent, 1901, Rossella fibulata Schulze and Kirkpatrick 1910, and Rossella levis (Kirkpatrick 1907).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glass sponges (class Hexactinellida) are key components of Antarctic suspension-feeder communities (Arnaud et al. 1998; Gutt 2007). Antarctic hexactinellids, particularly species of the genera Rossella and Anoxycalyx (Scolymastra), can reach remarkable size, biomass, and abundance (Barthel and Tendal 1994; McClintock et al. 2005; Janussen and Tendal 2007). Rossella species are the most abundant and biomass-rich benthic organisms in many habitats of the Antarctic shelf, covering up to 50% of the seafloor and increasing its spatial complexity (Fig. 1) by forming biogenic structures which can be used by other species (Gutt and Starmans 1998; Starmans et al. 1999; Janussen and Reiswig 2009; Dayton et al. 2013; Fillinger et al. 2013). Rossella have been also reported to structure benthic communities (Barthel 1992a, b) and to play a major role in local silicon cycling (Gatti 2002; Gutt et al. 2013). Large Rossella specimens can harbor a diverse community of invertebrates and juvenile stages of many other organisms, and serve as substratum for various taxa of other sessile invertebrates (epibionts and endobionts) (Kunzmann 1996; Barthel 1997; Gutt and Schickan 1998; Kersken et al. 2014).

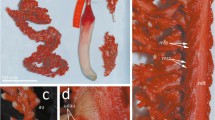
Photo Gutt, J. and Starmans, A. (2004): Sea-bed photographs (benthos) along ROV profile PS56/127-1. doi:10.1594/PANGAEA.198695. Inset calycocome of Rossella antarctica; Photo C. Göcke
Seafloor dominated by Rossella spp. in the Weddell Sea, Antarctica (latitude −70.8717, longitude −10.5233; depth −245 m)
From a morphological perspective, Rossella is characterized by the presence of calycocomes (Fig. 1) and its typical microhexasters (Tabachnick 2002), although similar spicules also occur in a few other genera (see Dohrmann et al. 2012). In contrast to the relatively stable genus-level systematics, intra-generic relationships remain unclear and most Rossella species still require a revised and clear morphological delineation (Barthel and Tendal 1994; Göcke and Janussen 2013; Göcke et al. 2015). As a result, the number of species recognized for the genus has varied in the past, ranging from 2 to 21 species depending on the taxonomic authority (e.g., Burton 1929; Koltun 1976; Barthel and Tendal 1994; see also van Soest et al. 2017). The great variation of the number of recognized species is, to some extent, not surprising. With the sole exception of Rossella antarctica, all remaining species lack clear morphological apomorphies (cf. Barthel and Tendal 1994), or their diagnostic characters are weak (e.g., external morphology, shape, and size of dermal megascleres), although different morphs are clearly present. In addition, the majority of characters used for species delimitation in the genus are continuous, making differences between species mainly gradual and subject to diverse interpretations. Calycocome sizes, for instance, tend to overlap between species, as does the size of other taxonomically important spicules (Barthel and Tendal 1994; Göcke and Janussen 2013). External body shape is variable even within species (Tabachnick 2002). Finally (and paradoxically), the lack of appropriate sampling has, to some extent, hampered the systematic evaluation of the variability of the main characters used for distinguishing different species. Many species were originally sampled from the Ross Sea, whereas most recent studies (e.g., Barthel and Tendal 1994; Göcke and Janussen 2013) are based on material from the Weddell Sea. Recently Göcke et al. (2015) have re-established Rossella podagrosa based on new material from its type locality, which is in almost full accordance with its type. In material from the Weddell Sea only fuzzy matches with the type are visible, if any, so that the species was considered a synonym for several decades (Barthel and Tendal 1994). Unfortunately, the new specimen of R. podagrosa was not suitable for molecular work and not included in this study. Fresh material from the Ross Sea in general is barely accessible today.
Clarifying the systematic relationships within the genus Rossella is an important task of potential benefit to other areas of Antarctic research. Rossella species are ecosystem engineers in the Antarctic benthos (see above), and their distribution, as that of many other Antarctic sponges, is thought to be circum-antarctic (Sara et al. 1992; Janussen and Reiswig 2009), although it is not clear yet, whether some species or “morphs” occur only locally. The role that different Rossella species play in structuring Antarctic communities, as well as whether some or all species are, indeed, a circumpolar cohesive unit, strongly depends upon the clear delineation of those species. Here, we provide a first phylogenetic analysis of the genus Rossella based on mitochondrial (16S rDNA, COI) and nuclear (28S rDNA) markers and including five of the eight species recognized as valid by Barthel and Tendal (1994), a system we chose as a working basis, at it seems to us the most justified of all Rossella concepts, although it is certainly not perfect (e.g., lack of R. podagrosa, see above). We aim to test different morphology-based taxonomic arrangements that have been proposed for the genus throughout its taxonomic history to cope with its morphological diversity and attempt to reconcile the current morphology-based classification of Rossella spp. from the Antarctic Weddell Sea with the molecular results obtained here. We aim to provide an evolutionary framework for the study of this important Southern Ocean (SO) taxon and hope that our contribution helps to develop future studies on the systematics of this genus of Antarctic sponges.
Materials and methods
Specimens and laboratory procedures
Specimens (Table 1) were collected by trawling during the German ANT XXIII/8 (2006/2007) and ANT XXIV/2-SYSTCO expedition (2007/2008) to the Weddell Sea (Atlantic sector of West Antarctica), photographed and fixed in 96% ethanol. Sponges were determined to species level using standard procedures (e.g., Janussen et al. 2004) and pertinent literature. DNA was extracted from small pieces of tissue with the NucleoSpin DNA tissue extraction kit (Macherey–Nagel) following the manufacturer’s protocol.
Three different molecular markers—partial 28S rDNA (ca. 1.2 kb), partial 16S rDNA (ca. 0.5 kb), and the standard barcoding fragment (Folmer et al. 1994) of COI (ca. 0.6 kb)—were amplified using 12.5 µl reaction volumes of GoTaq (Promega) supplemented with BSA. Three-step PCR protocols, including an initial denaturation step of 94 °C 3 min, 35–40 cycles of 94 °C 30 s, 50 °C/40 °C 30 s, 72 °C 60 s, and a final extension of 5 min at 72 °C, were used for all markers (see Online Resource 1 for details on the annealing temperature for each primer). For 28S rDNA and COI, we designed Rossella-specific primers (Online Resource 1) to avoid co-amplification of non-target organisms; 16S rDNA primers were as in Dohrmann et al. (2008). PCR products were cleaned by standard ammonium acetate–ethanol precipitation or ExoSAP-IT (Affymetrix) enzymatic PCR clean-up and sequenced in both directions using the same primers used for PCR and the BigDye Terminator 3.1 chemistry (Applied Biosystems). Sequencing reactions were precipitated with sodium acetate–ethanol and subsequently analyzed on an ABI 3700 genetic analyzer at the sequencing service of the Department of Biology, LMU München. Trace files were assembled in CodonCode Aligner (CodonCode Corporation); hexactinellid origin of all obtained sequences was verified using NCBI BLAST (Johnson et al. 2008). Sequences are deposited at EMBL under accession numbers HE80191–HE80223.
Outgroup choice and sequence alignment
New sequences were manually aligned in SeaView 4 (Gouy et al. 2010) to published alignments (Dohrmann et al. 2012b). However, we restricted the taxon set to representatives of the families Leucopsacidae and Rossellidae, as well as Clathrochone clathroclada (Lyssacinosida incertae sedis). Leucopsacidae and C. clathroclada have been shown to be successive sister groups to Rossellidae (Dohrmann et al. 2011), and were therefore used as outgroups. Alignments were concatenated into a supermatrix and ambiguously alignable regions removed. The final alignment is ~1.2 kb long and is available at Open Data LMU (doi…).
Phylogenetic analysis
Using the concatenated alignment, we inferred both Maximum likelihood (ML) and Bayesian phylogenies with RAxML 7.2.8 (Stamatakis 2006) and PHASE 2.0 (http://www.bioinf.manchester.ac.uk/resources/phase/), respectively. The GTR model of nucleotide substitution (Tavaré 1986) was used for 16S rRNA, COI as well as for 28S rRNA single-stranded regions (loops). Among-site rate variation was modeled using a discrete approximation of a gamma distribution with 4 categories (+G; Yang 1994, 1996). For the stem regions (paired sites) of the 28S rRNA, we used the S16 and S7A models of sequence evolution (Savill et al. 2001) for the ML analysis. We searched for the ML tree using 20 independent tree-search replicates and assessed branch support with 1000 bootstrap pseudoreplicates (Felsenstein 1985), using the “rapid bootstrap” algorithm described by Stamatakis et al. (2008). In the Bayesian analysis, two independent Markov Chain Monte Carlo (MCMC) chains were run for 10,000,000 generations after a burn-in of 250,000 generations, sampling every 100 generations. Model specifications for the Bayesian analysis were the same as for the ML analysis (i.e., GTR+G for 16SrDNA and COI); however, we only used the S7A model for the 28S rRNA stem regions because it was difficult to achieve chain convergence using the S16 model and because the S7A model provides a good compromise between biological plausibility and computational complexity.
Partition addition bootstrap and alternative lineage attachment analysis
To assess the influence of the individual partitions or combinations thereof on the ML topology inferred from the concatenated data matrix (i.e., the concatenated molecular data ML tree), we performed ML bootstrap analyses (1000 pseudoreplicates) for each individual marker and for all combinations of two markers using RAxML 7.2.8. For all these analyses, we used the same model settings as in the concatenated molecular data analysis for the corresponding partition (e.g., GTR+G for 16S rDNA and S16 for 28S rDNA stems). After each analysis, we determined the partition-specific bootstrap support (sensu Struck et al. 2006) for the branches present in the concatenated molecular data tree using consensus from the phyutility package (Smith and Dunn 2008).
We also assessed alternative branching positions of different Rossella species using linmove from the phyutility package. In brief, linmove screens a set of phylogenetic trees and reports the frequency with which alternative placements of a branch occur in that set. The analysis facilitates the visualization of alternative branching positions of a lineage showing low bootstrap support values, which allows to determine whether poorly supported branches have only a few attachment points occurring with high frequency or branch off at several multiple positions with low frequency.
Testing hypotheses of relationships within Rossella
Different taxonomic arrangements proposed for Rossella can be translated into specific phylogenetic hypotheses and evaluated with available statistical tests (e.g., Huelsenbeck 1997; Huelsenbeck and Crandall 1997; Goldman et al. 2000; Whelan et al. 2001). We used the AU test implemented in CONSEL (Shimodaira and Hasegawa 2001; Shimodaira 2002) with site-wise log-likelihood values obtained from RAxML 8.2.4 to test the monophyly of R. nuda, and R. racovitzae, the two species for which more than one specimen was available. Briefly, ML analyses constrained to enforce the monophyly of only R. nuda, only R. racovitzae, or these two species simultaneously were performed in RAxML, and the ML values of the best (constrained) trees were compared against the best unconstrained ML phylogeny in CONSEL.
Results
Phylogenetic analysis of the concatenated molecular dataset
We recovered a phylogenetic tree congruent with published analyses of the class Hexactinellida (Dohrmann et al. 2008, 2009, 2012a, b). Bayesian and ML analyses recovered generally similar trees, but the Bayesian phylogeny did not include a clade of R. fibulata + R. racovitzae, which was present in the ML phylogeny with moderate support. Both independent MCMC runs of the Bayesian analysis converged to the same consensus topology. The ML topology was not sensitive to model choice for the 28S rDNA stem sites, as analyses using S7A and S16 resulted in the same phylogeny.
Our phylogenetic analyses (Fig. 2) recovered a well-supported clade comprising all Rossella spp. which nested deeply within the family Rossellidae. Rossella antarctica specimens formed a highly supported clade in both Bayesian and ML analyses. Specimens belonging to other morphologically defined Rossella species formed a large clade hereafter named the R. racovitzae clade. Within this clade, other morphologically defined Rossella species were not recovered as monophyletic, but were polyphyletic in both the ML and Bayesian tree. Support values within the R. racovitzae clade were generally low (<50%), with only some branches showing moderate (50–80%) bootstrap support in the ML analysis. In contrast, the Bayesian analysis assigned high posterior probabilities (PP > 0.95) to most branches within this clade.

Phylogenetic relationships (cladogram) of Rossella. The tree corresponds to the concatenated molecular data maximum likelihood topology. The vertices of the stars above the branches show the bootstrap value obtained for a given branch when using a single partition or a combination of partitions (see inset). The centers of the stars show, on the left, the bootstrap value of the maximum likelihood concatenated molecular data analysis, and on the right, the posterior probability obtained for the branch in the Bayesian analysis. Dark gray bars on the right annotate the family Rossellidae (R) and Leucopsacidae (L); Clathrochone is currently incertae sedis in Lyssacinosida. Within Rossellidae, Rossella is indicated with a black bar and the R. racovitzae species flock is highlighted in light gray. Information about other specimens included in the analysis can be found in Dohrmann et al. (2008, 2009). T.A. = R. racovitzae specimen from Terre Adélie
Partition addition bootstrap analysis and lineage movement
Bootstrap values assigned to the branches of the concatenated molecular data ML tree varied between different partitions or combinations thereof (Fig. 2). In general, bootstrap support increased when more data were added to the analysis. However, there was conflict between partitions in some specific cases. For instance, the R. antarctica clade was not supported by 16S rDNA sequences alone but received high bootstrap support from the COI partition. When the two markers were combined, bootstrap support was only moderate (50–70%) in contrast to the high support (>70%) assigned to this clade in the concatenated molecular data analysis. Within the R. racovitzae clade, support was low when single partitions or combinations of two partitions were used for the analysis, and was only moderate in the concatenated molecular data phylogeny. Lineage movement analysis revealed that morphospecies included in the R. racovitzae clade were not monophyletic in any of the bootstrap pseudoreplicates, invariably forming clades with specimens belonging to different morphospecies.
Hypothesis testing
All constrained phylogenetic hypotheses explored in this study were found to be significantly worse (p < 0.001) than the unconstrained ML tree (Table 2). The decay in the likelihood values of the constrained ML phylogenies was highly related to the number of constraints. Trees constrained to make single species monophyletic (e.g., R. nuda or R. racovitzae) showed higher log-likelihood values than trees constrained to make all species monophyletic (e.g., R. nuda and R. racovitzae monophyletic). These results were insensitive to model selection, leading to identical conclusions when either the S16 or the S7A model was applied to 28S rDNA stems.
Discussion
The last 50 years of taxonomic history have seen genus Rossella expanding from two species, R. antarctica and R. racovitzae (Rossella-concept of Koltun 1976), to eight species (Rossella-concept of Barthel and Tendal 1994) to 20 species currently accepted as valid in the World Porifera Database (van Soest et al. 2017). All six species resurrected by Barthel and Tendal (1994) were included in the broad and ‘highly polymorphic’ R. racovitzae by Koltun (1976). Here, we have sequenced two mitochondrial markers and one nuclear marker in an attempt to clarify the systematics of Rossella using an independent set of characters not used by previous authors. Our results reveal that genus Rossella divides into two main clades corresponding to Koltun’s Rossella species: a well-supported R. antarctica clade was recovered as sister to a moderately supported group of specimens assigned to various nominal species and here referred to as the R. racovitzae clade. Both clades have clear diagnostic molecular characters in their COI sequences, i.e., molecular synapomorphies (Fig. 3). The morphology-based taxonomy of the species included within the R. racovitzae clade is not straightforward. Most of its species lack clear apomorphic characters, and many of the characters used for species delimitation inside this clade overlap or are prone to authoritative (subjective) interpretation (Table 3). In addition, broad morphological variation in both external and spicule morphology is found in the R. racovitzae clade (Barthel and Tendal 1994; Göcke and Janussen 2013). Yet, the R. racovitzae clade can be roughly subdivided into several groups that display morphological cohesiveness and resemble the described species included in it (Göcke and Janussen 2013). The existence of these morphological groups and the large morphological variability within the R. racovitzae clade preclude lumping its species into a Rossella species (like Koltun did) without making its diagnosis essentially meaningless. In contrast, R. antarctica can be readily identified and can be clearly distinguished from all other Rossella species based on morphology as well as their clear diagnostic molecular characters; note that no diagnostic molecular characters were found for all the other species within the R. racovitzae clade in the standard barcoding partition.

Maximum likelihood phylogram of Rossella based on the concatenated molecular data matrix. For support ranges of the nodes see Fig. 1. Highlighted are the two main Rossella clades obtained with their corresponding COI diagnostic characters: consensus sequence on top of the alignment, positions identical to the consensus represented with dots. Scale bar, expected number of substitutions per site. T.A. = R. racovitzae specimen from Terre Adélie
The analysis of the alternative branching positions of specimens included within the R. racovitzae clade revealed that specimens morphologically assigned to the same nominal species were not monophyletic in any bootstrap tree of the concatenated molecular data ML analysis or in the Bayesian tree; the AU test of monophyly applied to some phylogenetic hypotheses constrained to group different Rossella species together also rejected the monophyly of the species tested. Species are expected to be poly- or paraphyletic after or during speciation. Therefore, the non-monophyly within the Rossella racovitzae clade in the molecular phylogeny presented here could reflect a recent or ongoing speciation process in the genus. Consequently, we propose that the R. racovitzae clade, in contrast to the well-defined R. antarctica, is a species flock (Lecointre et al. 2013). Species flocks are monophyletic, diverse (morphologically, ecologically, and taxonomically) assemblages of closely related species which evolved rapidly within an area where they are endemic and ecologically dominant (Lecointre et al. 2013). The R. racovitzae species flock includes four out of five species here sampled, these species are endemic to Antarctica, are morphologically diverse and appear to have evolved rapidly as judged by their poly- or paraphyletic status (observed here) and their biogeographic history. Molecular divergence time estimations resulted in a mean age of ~40 ± 20 Ma for crown-group Rossella (Dohrmann et al. 2013). This age accords well with the opening of the Drake Passage (~30 Ma), a geological event that resulted in the final isolation of Antarctica (Lawver and Gahagan 2003) and could have caused the rapid diversification of Rossella in this region. We consider this “Rossella-concept,” including one clear species and a species flock, to currently best reconcile all available evidence (i.e., morphological and molecular) and to provide an evolutionary framework to interpret the high levels of variation within the R. racovitzae clade without requiring the synonymization of most Rossella species in a “highly polymorphic,” and for practical purposes, undefined Rossella species (i.e., R. racovitzae s.l.). We think that synonymizing the species within the R. racovitzae clade would be simplistic and would ignore their idiosyncratic ecologies and life histories. The concept of a species flock, on the contrary, allows for their specific recognition (general differences are definitely present, although so far hard to define), while making the difficulties associated with their systematic status transparent and explicit.
From a biogeographic perspective, the circum-Antarctic cohesiveness of both R. antarctica and members of the R. racovitzae clade remains to be tested. In this study, the only specimen of R. racovitzae from east Antarctica (collected in Terre Adélie) included in the analysis was sister to specimens from the Weddell Sea. However, any conclusion about the biogeography of Rossella species in the SO derived from our current dataset seems premature given the restricted geographic coverage of our sample.
Finally, we would like to highlight the difficulties in getting access to fresh material of all valid species of Rossella for molecular phylogenetic analyses. Rossella is well known for its abundance in Antarctica; however, most specimens collected belong to R. racovitzae, while specimens belonging to other species are collected less frequently. Rosella levis was only collected 3–5 times in four expeditions to the Antarctica by DJ and something similar occurs to R. fibulata (cf. Göcke and Janussen 2013). Rossella racovitzae and R. nuda are more often collected and more material is generally available from these species. Two other species, R. vanhoeffeni and R. villosa, have not been collected after years of field work in the Weddell Sea. Rossella podagrosa is so far only known from the Ross Sea, where it seems to be the most common sponge, although easily overlooked because if its hidden lifestyle (Göcke et al. 2015). Samples of R. podagrosa are currently not available for molecular analysis. These difficulties, somewhat normal in the deep-sea, but paradoxical given the reported abundance of Rossella spp. in Antarctic waters, hamper the thorough testing of the monophyly of most species in the genus. We provide here the first molecular study of the phylogenetic relationships within this important sponge genus in Antarctica and pursue to reconcile the morphological and molecular evidence given the available material and to open new avenues for future work to further clarify the phylogeny of Rossella.
Conclusion
We have obtained a phylogeny of the genus Rossella corroborating its monophyly and showing the existence of two clades corresponding to the well-defined species R. antarctica and a diverse assemblage of species, here considered a species flock and termed R. racovitzae flock. Future sampling and the use of genome-wide molecular markers will certainly contribute to expanding our understanding of these important Antarctic species, in particular about the circumpolar distribution of Rossella, the causes of the high morphological diversity, and the relationships within the R. racovitzae species flock.
References
Arnaud P, Lopez C, Olaso I, Ramil F, Ramos-Espla A, Ramos A (1998) Semi-quantitative study of macrobenthic fauna in the region of the South Shetland Islands and the Antarctic Peninsula. Polar Biol 19:160–166
Barthel D (1992a) Do hexactinellids structure Antarctic sponge associations? Ophelia 36:111–118
Barthel D (1992b) Antarctic hexactinellids: a taxonomically difficult, but ecologically important benthic component. Verhandl Deutschen Zool Ges 58:271–276
Barthel D (1997) Fish eggs and pentacrinoids in Weddell Sea hexactinellids: further examples for the structuring role of sponges in Antarctic benthic ecosystems. Polar Biol 17:91–94
Barthel D, Tendal OS (1994) Antarctic Hexactinellida. In: Wägele JM, Sieg J (eds) Synopses of the Antarctic Benthos, vol 6. Koeltx Scientific Books, Champaign, p 154
Burton M (1929) Porifera. Part II. Antarctic sponges. British Antarctic (‘Terra Nova’) expedition, 1910. Natural History Report, London, British Museum (Natural History). Zoology 6:393–458, pls. I–V
Dayton PK, Kim S, Jarrell SC, Oliver JS, Hammerstrom K, Fisher JL, O’Connor K, Barber JS, Robilliard G, Barry J et al (2013) Recruitment, growth and mortality of an Antarctic hexactinellid sponge, Anoxycalyx joubini. PLoS ONE 8:e56939
Dohrmann M, Janussen D, Reitner J, Collins A, Wörheide G (2008) Phylogeny and evolution of glass sponges (Porifera, Hexactinellida). Syst Biol 57:388–405
Dohrmann M, Collins AG, Wörheide G (2009) New insights into the phylogeny of glass sponges (Porifera, Hexactinellida): monophyly of Lyssacinosida and Euplectellinae, and the phylogenetic position of Euretidae. Mol Phylogenet Evol 52:257–262
Dohrmann M, Göcke C, Reed J, Janussen D (2012a) Integrative taxonomy justifies a new genus, Nodastrella gen. nov., for North Atlantic “Rossella” species (Porifera: Hexactinellida: Rossellidae). Zootaxa 3383:1–13
Dohrmann M, Haen KM, Lavrov DV, Wörheide G (2012b) Molecular phylogeny of glass sponges (Porifera, Hexactinellida): increased taxon sampling and inclusion of the mitochondrial protein-coding gene, cytochrome oxidase subunit I. Hydrobiologia 687:11–20
Dohrmann M, Vargas S, Janussen D, Collins AG, Wörheide G (2013) Molecular paleobiology of early-branching animals: integrating DNA and fossils elucidates the evolutionary history of hexactinellid sponges. Paleobiology 39:95–108
Felsenstein J (1985) Confidence limits on phylogenies—an approach using the bootstrap. Evolution 39:783–791
Fillinger L, Janussen D, Lundälv T, Richter C (2013) Rapid glass sponge expansion after climate-induced Antarctic ice shelf collapse. Curr Biol 23:1–5
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R (1994) DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotech 3:294–299
Gatti S (2002) The role of sponges in high-Antarctic carbon and silicon cycling—a modelling approach. Berl Polar Meeresforsch 434:1–134
Göcke C, Janussen D (2013) Hexactinellida of the genus Rossella, of ANT XXIV/2 (SYSTCO I) expedition—Antarctic Eastern Weddell Sea. Zootaxa 3692:102–122
Göcke C, Janussen D, Reiswig HM, Jarrell SC, Dayton PK (2015) Rossella podagrosa Kirkpatrick, 1907—a valid species after all. Zootaxa 4021(1):169–177
Goldman N, Anderson J, Rodrigo A (2000) Likelihood-based tests of topologies in phylogenetics. Syst Biol 49:652–670
Gouy M, Guindon S, Gascuel O (2010) SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 27:221–224
Gutt J (2007) Antarctic macro-zoobenthic communities: a review and an ecological classification. Antarct Sci 19:165–182
Gutt J, Schickan T (1998) Epibiotic relationships in the Antarctic benthos. Antarct Sci 10:398–405
Gutt J, Starmans A (1998) Structure and biodiversity of megabenthos in the Weddell and Lazarev Seas (Antarctica): ecological role of physical parameters and biological interactions. Polar Biol 20:229–247
Gutt J, Böhmer A, Dimmler W (2013) Antarctic sponge spicule mats shape macrobenthic diversity and act as a silicon trap. Mar Ecol Prog Ser 480:57–71
Huelsenbeck JP (1997) Phylogenetic methods come of age: testing hypotheses in an evolutionary context. Science 276:227–232
Huelsenbeck JP, Crandall K (1997) Phylogeny estimation and hypothesis testing using maximum likelihood. Annu Rev Ecol Syst 28:437–466
Janussen D, Reiswig HM (2009) Hexactinellida (Porifera) from the ANDEEP III expedition to the Weddell Sea, Antarctica. Zootaxa 2136:1–20
Janussen D, Tendal OS (2007) Diversity and distribution of Porifera in the bathyal and abyssal Weddell Sea and adjacent areas. Deep-Sea Res Pt II 54:1864–1875
Janussen D, Tabachnick K, Tendal O (2004) Deep-sea Hexactinellida (Porifera) of the Weddell Sea. Deep-Sea Res Pt II 51:1857–1882
Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, Mcginnis S, Madden TL (2008) NCBI BLAST: a better web interface. Nucleic Acids Res 36:W5–W9
Kersken D, Göcke C, Brandt A, Lejzerowicz F, Schwabe E, Seefeldt MA, Veit-Köhler G, Janussen D (2014) The infauna of three widely distributed sponge species (Hexactinellida and Demospongiae) from the deep Ekström Shelf in the Weddell Sea, Antarctica. Deep-Sea Res Pt II 108:101–112
Koltun VM (1976) Porifera—Part 1: Antarctic sponges. Report B.A.N.Z. Antarctic research expedition 1929–1931 (B, Zoology and Botany) 5: 153–198
Kunzmann K (1996) Associated fauna of selected sponges (Hexactinellida and Demospongiae) from the Weddell Sea, Antarctica. Berl Polarforsch 210:1–93
Lawver L, Gahagan L (2003) Evolution of Cenozoic seaways in the circum-Antarctic region. Palaeogeogr Palaeoclimatol Palaeecol 198:11–37
Lecointre G, Améziane N, Boisselier M-C, Bonillo C, Busson F et al (2013) Is the species flock concept operational? The Antarctic Shelf case. PLoS ONE 8:e68787
McClintock J, Amsler C, Baker B, van Soest R (2005) Ecology of Antarctic marine sponges: an overview. Integr Comp Biol 45:359–368
Sara M, Balduzzi A, Barbieri M, Bavestrello G, Burlando B (1992) Biogeographic traits and checklist of Antarctic demosponges. Polar Biol 12:559–585
Savill N, Hoyle D, Higgs P (2001) RNA sequence evolution with secondary structure constraints: comparison of substitution rate models using maximum-likelihood methods. Genetics 157:399–411
Shimodaira H (2002) An approximately unbiased test of phylogenetic tree selection. Syst Biol 51:492–508
Shimodaira H, Hasegawa M (2001) CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics 17:1246–1247
Smith SA, Dunn CW (2008) Phyutility: a phyloinformatics tool for trees, alignments and molecular data. Bioinformatics 24:715–716
Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690
Stamatakis A, Hoover P, Rougemont J (2008) A rapid bootstrap algorithm for the RAxML web servers. Syst Biol 57:758–771
Starmans A, Gutt J, Arntz W (1999) Mega-epibenthic communities in Arctic and Antarctic shelf areas. Mar Biol 135:269–280
Struck T, Purschke G, Halanych K (2006) Phylogeny of Eunicida (Annelida) and exploring data congruence using a partition addition bootstrap alteration (PABA) approach. Syst Biol 55:1–20
Tabachnick KR (2002) Family Rossellidae Schulze, 1885. In: Hooper JNA, van Soest RWM (eds) Systema porifera: a guide to the classification of sponges. Kluewer Academic/Plenum Publishers, New York, pp 1441–1505
Tavaré S (1986) Some probabilistic and statistical problems in the analysis of DNA sequences. Lect Math Life Sci 17:57–86
van Soest RWM, Boury-Esnault N, Hooper JNA, Rützler K, de Voogd NJ, Alvarez de Glasby B, Hajdu E, Pisera AB, Manconi R, Schönberg C, Janussen D, Tabachnick KR, Klautau M, Picton B, Kelly M, Vacelet J, Dohrmann M, Díaz C, Cárdenas P (2017) World porifera database. http://www.marinespecies.org/porifera
Whelan S, Lio P, Goldman N (2001) Molecular phylogenetics: state-of-the-art methods for looking into the past. Trends Genet 17:262–272
Yang Z (1994) Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J Mol Evol 39:306–314
Yang Z (1996) Among-site rate variation and its impact on phylogenetic analyses. Trends Ecol Evol 11:367–372
Acknowledgements
We thank Annamarie Gabrenya and Astrid Schuster for assistance and support in the laboratory. Constructive comments of past and present members of the Molecular Geo- and Palaeobiology Lab, LMU München, greatly improved the project. This study was possible thanks to funding of the German Science Foundation (DFG), through the SPP 1158 “Antarktisforschung,” grants Wo896/9-1,2 to G. Wörheide, and DJ1063/14-2,2 and DJ1063/17-1 to D. Janussen, respectively. SV is indebted to N. Villalobos Trigueros, M. Vargas Villalobos, S. Vargas Villalobos, and S. Vargas Villalobos for their constant support during the course of the study.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Vargas, S., Dohrmann, M., Göcke, C. et al. Nuclear and mitochondrial phylogeny of Rossella (Hexactinellida: Lyssacinosida, Rossellidae): a species and a species flock in the Southern Ocean. Polar Biol 40, 2435–2444 (2017). https://doi.org/10.1007/s00300-017-2155-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00300-017-2155-7