
Abstract
Kawasaki disease (KD) is a medium vessel vasculitis that predominantly affects children below 5. Diagnosis of KD is based on the presence of characteristic clinical manifestations as there are no definite diagnostic laboratory investigations for the diagnosis of this disease. Presence of atypical clinical features such as myositis often pose diagnostic challenge for the treating physicians. Presence of myositis and severe muscular weakness in KD is distinctly unusual and may lead to delays in diagnosis and administration of definite therapy. We report a 10-year-old boy who presented with fever, rash and proximal muscle and pharyngeal weakness. A clinical possibility of toxic shock syndrome or juvenile dermatomyositis was initially considered. However, he continued to have fever and developed periungual peeling of skin in fingers. Hence, a possibility of KD with myositis was considered. He showed prompt response to intravenous immunoglobulin and methylprednisolone. We also provide a review of similarly reported cases of KD myositis. It is important for clinicians to be aware of this atypical clinical presentation to avoid delays in diagnosis and treatment of KD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Kawasaki disease (KD) is a medium vessel vasculitis that predominantly affects children below 5. Diagnosis of KD is based on presence of characteristic clinical manifestations as there are no definite diagnostic laboratory investigations for the diagnosis of this disease [1, 2]. Presence of atypical clinical manifestations of KD often pose diagnostic challenge for the treating physicians. “Atypical Kawasaki disease” is the term used for patients who have an unusual clinical manifestation of disease such as arthralgia or arthritis, aseptic meningitis, hepatic dysfunction, gallbladder hydrops, gastrointestinal involvement, uveitis, pneumonitis, peripheral gangrene, renal involvement and neuromuscular involvement.
Myositis presenting as severe muscular weakness is an unusual clinical presentation of KD that may lead to delays in diagnosis and administration of definite therapy. There are no guidelines for management of myositis in patients with KD. Objective of the study is to report a case of severe muscle weakness in a child with KD and review all previously reported cases of myositis and muscular weakness in patients with KD.
Case description
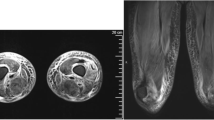
A 10-year-old boy presented with left scrotal swelling and fever for 7 days. On day 3 of illness, he developed erythematous rash over limbs and abdomen and redness of eyes. He also had diffuse painful swelling in his hands and feet. He developed progressive difficulty in getting up from squatting and supine position and was bed ridden by day 6 of illness. A clinical possibility of toxic shock syndrome (TSS) was initially considered. Laboratory investigations showed anemia, neutrophilic leucocytosis, elevated creatinine kinase (CK-NAC) and lactate dehydrogenase (LDH) and high c-reactive protein (CRP) (Table 1). He was treated with intravenous antimicrobials (ceftriaxone and cloxacillin) and intravenous immunoglobulin (IVIg) [1 gm/kg]. His rash disappeared, he became afebrile, and scrotal swelling reduced. On day 18 of illness, the child was brought to us with complaints of persistent muscle weakness and inability to move or roll in bed. On examination, he had pallor, periungual peeling of skin in the fingers (Fig. 1), swollen and tender calf muscles, neck flexor muscle weakness, symmetric proximal muscle weakness of upper and lower limbs (power 2/5), absent gag reflex and preserved deep tendon reflexes. Small amount of pus discharge from the left sided scrotal swelling was also noted. Rest of the examination was unremarkable. Clinical possibility of KD with myositis, juvenile dermatomyositis (JDM) and infections associated myositis was considered. Laboratory investigations showed anaemia, neutrophilic leucocytosis, thrombocytosis, elevated LDH and pro-brain natriuretic peptide (Pro-BNP) (Table 1). Transthoracic 2D echocardiography showed normal coronary artery Z scores. Incision and drainage of scrotal abscess was done. Pus culture and blood cultures were sterile. In view of persistent fever, periungual peeling of skin, neutrophilic leucocytosis, thrombocytosis and elevated pro-BNP, a clinical possibility of ‘incomplete’ and ‘atypical’ KD with myositis was considered. He was initiated on IVIg (2 g/kg). In view of severe muscle weakness, he was also given pulse intravenous methyl prednisolone (30 mg/kg/day) for 3 days. He showed rapid clinical recovery with improvement in muscle weakness by day 4. He was discharged on aspirin (4 mg/kg/day) and tapering doses of oral prednisolone (initial dose 2 mg/kg/day). A repeat 2D echocardiography at 6 weeks of follow-up revealed normal coronary artery Z scores. Aspirin and oral prednisolone were discontinued at this time. He remains clinically well on follow-up.

Periungual peeling of skin in both hands
Search strategy
We searched PubMed, MEDLINE, Embase, Scopus databases for published literature using following search term: Kawasaki disease and myositis; Kawasaki disease and muscle weakness; Kawasaki disease and Juvenile dermatomyositis; Kawasaki disease and JDM as on December 02, 2020. We found nine papers using this search strategy. One case was excluded as it showed neuromuscular involvement in KD predominantly in the form of neuropathy. Of the remaining eight papers, data were not available for one. Hence, seven papers were included in the final literature review of myositis in Kawasaki disease (Table 2).
Discussion
Although a clinical possibility of JDM was initially considered in view of symmetric proximal muscle weakness including neck flexor muscles with poor gag reflex, he showed brisk improvement following IVIg and had no pathognomonic cutaneous manifestations of JDM. Presence of fever, rash, edema over hands and feet, red eyes along with periungual desquamation of skin and rapid improvement following administration of IVIg suggests a diagnosis of KD with myositis.
KD is a medium vessel systemic vasculitis with predilection to involve coronary arteries. Diagnosis of KD is clinical and is based on the American Heart Association (AHA) diagnostic guidelines and European consensus-based recommendations for the diagnosis and treatment of Kawasaki disease—the SHARE initiative [1, 2]. The AHA guidelines [3] suggest the term “atypical Kawasaki disease” for patients who have an unusual clinical manifestation of the disease that may include arthralgia or arthritis, aseptic meningitis, hepatic dysfunction, gallbladder hydrops, gastrointestinal involvement, uveitis, pneumonitis, peripheral gangrene, renal involvement and neuromuscular involvement [4]. Myositis and muscle weakness have rarely been reported in patients with KD [4]. Presence of myositis and muscular weakness often creates diagnostic conundrum for the treating physician thereby leading to delays in diagnosis and initiation of therapy.
The first two cases of myositis in children with KD were reported by Koutras et al. in 1980 and 1982, respectively [5, 6]. Subsequently, few more cases of KD have been reported wherein muscular involvement was a predominant clinical presentation [7,8,9,10,11,12].
Herein we have reviewed all cases of KD myositis and muscular weakness reported till date in the literature (Table 2). We could retrieve the details of seven such cases (three boys and three girls while gender was not mentioned for one). Median age at onset of symptoms was 6 years (range 8 months–10 years). Five cases had complete KD. Diffuse muscle weakness involving the limbs (proximal > distal) was noted in four. Focal myositis involving calf [11] and iliopsoas muscle [12] was reported in one each. Orbital myositis was reported in one patient (that developed 35 days after giving IVIg) [9]. One patient was also noted to have dysphagia and dysphonia [6], while severe muscular weakness leading to respiratory failure and requiring ventilation was reported in one [8]. Median duration from onset of first symptom to the onset of muscle weakness and myositis was 9 days (range 36 h–35 days). In all previously reported cases, the diagnosis of KD preceded the onset of muscle weakness except one case wherein myositis was the presenting manifestation of KD [12]. The present case developed proximal muscle weakness on day 6 of illness. He also had neck muscles involvement.
Laboratory investigations in previously reported patients showed elevated muscle enzymes in three children who had diffuse muscle weakness, while muscle enzymes were found to be normal in two and were not reported in remaining two. Higher levels of muscle enzymes may correlate with disease severity as was seen in a patient who had respiratory failure [8]. It appears that patients with KD myositis who have relatively higher CK levels may be at risk of developing respiratory failure and hence need to be monitored closely. In the first three reported patients, electromyography (EMG) and/or muscle biopsy was performed to document evidence of myositis. Muscle biopsy revealed type 2 fibre atrophy, inflammatory infiltrates in perivascular and peri-endomysium spaces and features of vasculopathy. Histopathological evidence of myositis and pan-arteritis was reported in one patient who had orbital myositis [9]. In all recently diagnosed patients, MRI was used to assess the myositis. Subcutaneous edema with thickening and increased T2 weighted signals in the pre-muscular and intermuscular fascia in the medial and posterior aspects of the proximal leg were reported in these patients. Present case was found to have elevated muscle enzyme. However, MRI of muscles, EMG or muscle biopsy could not be performed as he showed prompt clinical recovery with treatment and these investigations were not considered clinically useful at that time.
The underlying pathophysiological basis for KD and myositis has not been studied in detail because of paucity of the literature on this subject. However, it has been suggested that immune-complex deposits in affected muscles may lead to muscle weakness.
In previously reported patients with KD myositis, coronary artery aneurysms (CAA) were reported in 4/7 patients [7,8,9, 12]. Coronary arteries were normal in two, while it was not assessed in one. Thus, children with KD myositis appear to be at relatively higher risk of development of CAA (~ 57%). However, this interpretation is based on a review of seven cases only. Delay in diagnosis and initiation of therapy may be the reason for higher risk of CAA seen in these patients. However, it must also be noted that two patients were not given IVIg and were treated with aspirin only [6, 7]. One of these two patients developed CAAs [7]. Corticosteroids along with IVIg were used in three children [9, 10, 12]. Indications for use of corticosteroids were IVIg resistance and presence of muscle weakness.
Both AHA and European consensus-based recommendations for the diagnosis and treatment of Kawasaki disease (the SHARE initiative) have stressed on early treatment in both complete and incomplete forms of KD using intravenous immunoglobulin and aspirin [1, 2]. Corticosteroids have been recommended for high-risk cases. We used IVIg, aspirin and intravenous methylprednisolone followed by oral prednisolone in the present case.
Prognosis of muscle involvement in patients with KD has been reported to be good and full recovery was noted in all. While muscle weakness in six previously reported patients recovered at a median duration of 2.25 months (range: 18 days to 3 months), it recovered after 12 months in one patient with orbital myositis [9]. The present case showed complete recovery of muscle weakness within 1 week of initiation of therapy.
To conclude, we report a child with KD who presented with severe muscle weakness mimicking JDM. Myositis and muscle weakness are rare manifestations of KD and may sometime be the predominant presenting clinical manifestation. KD should be considered in children presenting with muscle weakness with prolonged fever, rash and other suggestive clinical manifestations. These patients may be at high risk of development of CAAs, likely because of delays in diagnosis and initiation of therapy. There are no guidelines for management of myositis in patients with KD. These patients apparently respond well to IVIg and steroid therapy. Recovery of muscle weakness is often complete. However, at times, this may take a few weeks to recover. However, these observations are based on a review of only seven cases reported till date and need more data in future to draw a strong conclusion.
References
McCrindle BW, Rowley AH, Newburger JW, Burns JC, Bolger AF, Gewitz M et al (2017) Diagnosis, treatment, and long-term management of kawasaki disease: a scientific statement for health professionals from the American Heart Association. Circulation 135(17):e927–e999. https://doi.org/10.1161/CIR.0000000000000484
de Graeff N, Groot N, Ozen S, Eleftheriou D, Avcin T, Bader-Meunier B et al (2019) European consensus-based recommendations for the diagnosis and treatment of Kawasaki disease—the SHARE initiative. Rheumatology (Oxford) 58(4):672–682. https://doi.org/10.1093/rheumatology/key344
Newburger JW, Takahashi M, Gerber MA, Gewitz MH, Tani LY, Burns JC et al (2004) Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Circulation 110(17):2747–2771. https://doi.org/10.1161/01.CIR.0000145143.19711.78
Jindal AK, Pilania RK, Prithvi A, Guleria S, Singh S (2019) Kawasaki disease: characteristics, diagnosis, and unusual presentations. Expert Rev ClinImmunol 15(10):1089–1104. https://doi.org/10.1080/1744666X.2019.1659726
Koutras AK (1980) Myositis with mucocutaneous lymph-node syndrome. N Y State J Med 80(7 Pt 1):1138–1139
Koutras A (1982) Myositis with Kawasaki’s disease. Am J Dis Child 136(1):78–79. https://doi.org/10.1001/archpedi.1982.03970370080025
Sugie H, Sugie Y, Ichimura M, Mizuno Y, Nishida M, Igarashi Y (1985) A case of polymyositis associated with Kawasaki disease. Brain Dev 7(5):513–515. https://doi.org/10.1016/s0387-7604(85)80120-0
Gama C, Breeden K, Miller R (1990) Myositis in Kawasaki disease. PediatrNeurol 6(2):135–136. https://doi.org/10.1016/0887-8994(90)90048-6
Lin H, Burton EM, Felz MW (1999) Orbital myositis due to Kawasaki’s disease. PediatrRadiol 29(8):634–636. https://doi.org/10.1007/s002470050665
Agarwal S, Gupta A, Suri D, Rawat A, Singh S (2015) Proximal muscle weakness in a child with Kawasaki Disease. Indian J Pediatr 82(9):866. https://doi.org/10.1007/s12098-015-1709-3
Lee EY, Oh JY, Chong CY, Choo JTL, Mahadev A, Tan NWH (2015) A case of atypical Kawasaki disease with myositis. Glob Pediatr Health. https://doi.org/10.1177/2333794X15599649
Vigil-Vázquez S, Butragueño-Laiseca L, López-González J, García-San Prudencio M, Rincón-López E (2019) A case of Kawasaki disease presenting as severe myositis. Indian J Pediatr. 86(11):1066–1067. https://doi.org/10.1007/s12098-019-03009-z
Funding
None.
Author information
Authors and Affiliations
Contributions
GA: patient management, concept and draft of manuscript, critical review of literature; NJ, AN, RS: patient management, critical review of literature; AKJ: patient management, concept of the manuscript, critical review of manuscript for important intellectual content and final approval of the version to be published; SS: patient management, critical review of manuscript for important intellectual content. All co-authors take full responsibility for the integrity of the study and the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they do not have any conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Anjani, G., Johnson, N., Navid, A. et al. Kawasaki disease malingering as juvenile dermatomyositis: case-based review . Rheumatol Int 42, 913–919 (2022). https://doi.org/10.1007/s00296-021-04826-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00296-021-04826-2