
Abstract
Juvenile dermatomyositis (JDM) is a rare chronic inflammatory disease of unknown etiology and primarily involves muscle and skin. It is the most common idiopathic inflammatory myopathy of childhood. This study aimed to evaluate demographic and clinical features, laboratory data, treatment modalities, and outcome of patients with JDM at a referral pediatric rheumatology center in Turkey. We retrospectively reviewed medical records of patients diagnosed with JDM between the years 2003–2016 at the Pediatric Rheumatology Department Cerrahpasa Medical Faculty. A total of 50 patients (35 females), median age at the onset 6.1 ± 4.1 years, were identified. Mean follow-up period was 74.5 ± 49.7 months. Presenting clinical symptoms included heliotrope rash (100%), Gottron papule (96%), muscle weakness (90%), erythroderma (88%), and calcinosis (38%). All patients had elevated muscle enzymes at the disease onset. Sixty-eight percent of the patients had anti-nuclear antibody positivity. Electromyography on 27 patients and muscle biopsy on 14 patients were performed, and all of them showed signs of juvenile dermatomyositis. Early aggressive treatment with corticosteroids mostly in combination with methotrexate was used. Cyclosporine was added to 48% of the patients’ treatment regimen in case of severe or refractory disease. All patients except two cases, who were referred to our clinic after long disease duration with widespread calcinosis, achieved remission. Early diagnosis and early initiation of intensive therapy are important in reducing JDM complications. International collaboration is needed in order to better understanding and management of the disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Juvenile dermatomyositis (JDM) is the most common among the idiopathic inflammatory myopathies of childhood involving primarily striated muscle and skin [1–3]. It is characterized by various severities of vasculitis early in disease course and the development of calcinosis in later stages [1]. Estimated annual incidence of JDM is 0.19–4.1 cases per million children [4, 5]. Although the pathogenesis remains still unclear, environmental factors and infectious agents in genetically susceptible individuals are considered to be related to the disease pathogenesis [6].
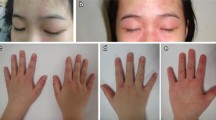
The pathognomonic rashes and proximal muscle weakness are characteristic features of the disease. The disease may further affect other organs, including gastrointestinal tract, heart, lungs, kidneys, and eyes [7, 8]. Secondary malignancies are rarely associated with JDM differentiated from adult myositis [9].
The Bohan and Peter criteria are used for the diagnosis of juvenile dermatomyositis which include typical cutaneous rashes and two or more of the following symptoms and signs: (1) symmetric weakness of the proximal musculature, (2) elevated serum muscle enzyme levels, (3) electromyography changes of myositis, and (4) biopsy-proven myositis. A diagnosis of JDM requires exclusion of all other inflammatory myopathies [10, 11]. Muscle weakness and typical skin rashes are present in most cases at the disease onset. Elevations of serum muscle enzyme levels, electromyographic demonstration of myopathy, and muscle biopsy contribute additional support for the diagnosis [12]. After widespread use of imaging methods magnetic resonance imaging (MRI) gained important role in demonstrating muscle inflammation. Incorporating new imaging techniques such as MRI findings were revealed to be added to diagnostic criteria [12, 13]. Magnetic resonance imaging is currently preferred to electromyography (EMG) and muscle biopsy as a noninvasive technique [14].
The mainstay of treatment for many years is corticosteroid and methotrexate. The introduction of biological therapies accommodates the treatment and outcome [15–17]. However calcinosis, lipodystrophy, and contractures still challenge physicians. Mortality rate in the recent years was reported low [18].
This study was established a retrospective chart review of patients diagnosed with JDM between the years 2003 and 2016. All patients initially diagnosed in our center or referred from other hospitals were included in the study. Our purpose was to evaluate the demographic and laboratory features, treatment methods, and outcome of the patients with JDM.
Materials and methods
We reviewed the medical charts of patients diagnosed with JDM and followed up at the Pediatric Rheumatology Department of Istanbul University Cerrahpasa Medical Faculty between the years 2003–2016. All patients initially diagnosed or referred were included in the study. Patients not admitting clinical visits after the diagnosis or patients getting different diagnosis after the disease reevaluation were excluded from the study. In conclusion, totally 50 patients diagnosed with JDM meeting Bohan and Peter criteria [10, 11] were enrolled in the study.
All medical charts were retrospectively investigated for demographic, initial and follow-up clinical and laboratory findings, electromyography and muscle biopsy results, treatment modalities, treatment responses, complications, and calcinosis course after treatment initiation. All patients were following-up by the same pediatric rheumatologists. The muscle strength was determined with standard manual muscle testing. Electromyography and muscle biopsy were performed along with facilities when needed. Clinical remission was defined as the absence of active myositis and skin rashes. The absence of active myositis was determined with normal muscle strength and serum muscle enzyme levels. Active and inactive diseases were described according to the PRINTO criteria for clinically inactive disease in juvenile dermatomyositis [19]. Magnetic resonance imaging was not included in the study because it was not performed routinely.
Statistical analysis
Statistical analysis was performed by using the IBM SPSS Statistics 21.0 software (IBM Corp. Released 2012. IBM SPSS Statistics for Windows, Version 21.0. Armonk, NY: IBM Corp.). Distributional properties of continuous variables were expressed in terms of mean ± standard deviation and median (min–max:minimum–maximum). Categorical variables were presented with frequency and percentage (%). The Shapiro–Wilk test was used to examine the differences in the distributions of continuous variables.
The Mann–Whitney U and the Chi-Square tests were used to determine differences between continuous and categorical variables of two groups. Statistical significance level was obtained p ≤ 0.05.
Results
Demographic data
Totally 50 patients with juvenile dermatomyositis were reviewed. The ratio of female (n: 35) to male (n: 15) was 2.3:1. The mean age at the onset of symptoms was 6.1 ± 4.1 years, and the mean age at the diagnosis was 6.6 ± 4.1 years. The mean follow-up period was 74.5 ± 49.7 months (6–166 months). Demographics and clinical features are summarized in Table 1.
Clinical findings
The most common initial presentations were typical skin rashes (100%). Muscle weakness was present in 90% of patients at the onset of the disease. The mean duration to achieve complete muscle strength was 6.7 ± 9.3 months. However, two of the patients are excluded while their muscle strength could not be examined appropriately because of diffuse joint contractures. Erythematous rash (88%) and calcinosis (38%) were other common clinical features.
Calcinosis was developed in 19 patients (38%). The mean onset of calcinosis was 25.5 ± 26.2 months of the disease. It commonly involves all body or the extremities. The mean age at which calcinosis was distinguished was 6.6 ± 3.9 years. The mean duration between the onset of symptoms and diagnosis of the patients with calcinosis was 7.1 ± 2.3 months while the patients without calcinosis were diagnosed meanly at the age of 6.6 ± 4.1 years, and the mean duration of symptoms before diagnosis was 5.2 ± 2.5 months. No statistical difference was found between the patients with or without calcinosis in terms of the mean age at the disease onset or the symptomatic duration before diagnosis (p > 0.05). Although no statistical difference was found between the patients with or without calcinosis in terms of the mean age at the disease onset or the symptomatic duration before diagnosis (p > 0.05), the mean duration to gain complete muscle strength for the patients developing calcinosis was 10.71 ± 11.41 months, and it was statistically longer than the patients without calcinosis (p = 0.046).
Laboratory evaluation
The initial hemoglobin level was 11.5 ± 1.4 g/dL, leukocyte count 9.616 ± 3.393/mm3, thrombocyte count 332.250 ± 121.179/mm3, and erythrocyte sedimentation rate 35 ± 22.1 mm/h. C-reactive protein positivity was present in 19 patients (38%). Laboratory findings at the disease onset were demonstrated in Table 2.
Elevation of serum levels of muscle enzymes at the disease onset was noted in all patients (100%). The mean creatine kinase level was 223,326 ± 329,201 IU/L as the median level was 538.00 IU/L. The mean duration to return normal levels was 3.8 ± 5.8 months while the median was 2 months. There was no statistical difference between the mean durations of gaining complete muscle strength and normal muscle enzyme levels (p > 0.05). In addition, no statistical difference was found between the patients with or without calcinosis in terms of the laboratory data (p > 0.05).
Initial antinuclear antibody titers were elevated in 34 patients (68%). Antibodies against extractable nuclear antigens and histidyl-tRNA synthetase (anti-jo 1) were not identified in any patients.
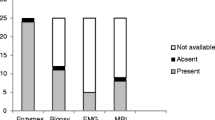
Needle electromyography on extremities was performed in 27 patients (54%); all of them demonstrated signs of myopathy as showing polyphasic contractions with decreased amplitude and short duration with signs of denervation. Muscle biopsy was performed in 14 patients (28%); all histopathology reports were consistent with JDM as demonstrating perivascular muscular atrophy, perivascular fibrosis, capillary damage, and perivascular chronic inflammatory cell infiltration.
Treatment and response
All patients received corticosteroid treatment (oral steroids or intravenous pulse for 3 consecutive days followed by oral steroids) for the mean duration of 33.9 ± 40.6 months. In almost all patients (96%), glucocorticoids were combined with methotrexate at the mean dosage of 13.6 ± 5.8 mg/week (5–60 mg/week) for the mean duration of 46.6 ± 37.8 months. Patients nonresponsive to steroid and methotrexate or patients steroid dependent (48%) were treated with additional cyclosporine therapy. Other immunosuppressive treatments were as follows in decreasing frequencies: intravenous immunoglobulin (18%), cyclophosphamide (10%), and infliximab (4%). Physical therapy was initiated at the time of diagnosis to improve muscle strength and reduce the risk of development of contractures. More aggressive immunosuppressive therapies were applied to the patients with developed calcinosis for considerable duration of time. The mean duration of steroid and methotrexate therapies were significantly longer (p = 0.001). Seventy-four percent of them received cyclophosphamide contrary to 29% of the patients without calcinosis. In addition, 17 patients with calcinosis (89.4% of the patients with calcinosis) received alendronate to alter calcium metabolism and bone mineralization. Most of these patients showed decreased severity of calcinosis. Except two patients, who were referred after long disease duration and had diffused calcinosis, all patients achieved complete remission.
Complications
The most common complication was calcinosis (38%); two patients with calcinosis developed severe joint contractures (4%). Three patients developed skin ulcerations (6%). Lipodystrophy was found in six patients (12%). Dysphagia and dysphonia were present in only one patient; otherwise cardiac, pulmonary or gastrointestinal involvements were not described in any other patients.
Outcomes
The mean duration for follow-up of the study group was 74.5 ± 49.7 months (6–166 months). Considering 5 and 10 year follow-up periods, clinical and laboratory remissions were similar excluding calcinosis. Calcinosis was more commonly seen in patients with longer follow-up periods; the mean follow-up duration was 90 ± 51 months for patients developed calcinosis in opposite to 64 ± 46 months without calcinosis. Neither malignancy nor death was observed in our study group.
Discussion
Juvenile dermatomyositis is the most common idiopathic inflammatory myopathy of childhood despite its very low incidence and primarily affects skin and striated muscle [1, 2]. Initial clinical findings are typical skin rashes and muscle weakness. Myocardium, lung, and gastrointestinal involvements are rarely seen [7, 8]. Although the diagnostic criteria proposed by Bohan and Peter in 1975 still remain standard for the diagnosis they are needed to be revised to adjust new developing autoantibodies, biomarkers, imaging modalities, and outcome tools. Some of the new suggested criteria are the presence of calcinosis, nailbed capillary changes, and signs of myositis on MRI [13, 14].
Current study retrospectively described a single-center JDM experience in 2003–2016. Juvenile dermatomyositis affects girls 2–5 times more frequently than boys. The female-to-male ratio was reported 1.1:1 in an 85 patient-included study from Iran [20], 1.4:1 in a European multi-centered study [21], and 2.3:1 in a survey from USA [5]. The ratio in our cohort was 2.3:1, similar to the studies in the literature. Disease onset is especially common for the ages 5–14 years. The mean age was defined 7.5 years (range 1.5–15) in the study from Iran [20], 7.1 years (range 2.2–15.3) in the study of Gowdie et al. [15], and 6.1 years (range 1.5–16) in our study. Although the disease presents more frequently after the age 5, younger children were reported in the previous studies and in our cohort.
Proximal muscle weakness and typical skin rashes are pathognomonic clinical findings of JDM [1]. However, clinical manifestations may vary widely in terms of frequency. In the study by Gowdie et al., Gottron papule (91%), heliotrope rash (73%), and muscle weakness (95%) were the most common presentations [15]. Malek et al. reported Gottron papule (59%), heliotrope rash (71%), and muscle weakness (96%) as preceding manifestations [20]. In our study, heliotrope rash (100%) and Gottron papule (96%) were described in almost every patient, and muscle weakness was defined in 90% of patients. Because the diagnosis of JDM requires cutaneous changes, patients with muscle weakness should be evaluated for the presence of typical skin rashes.
Calcinosis occurs in approximately 18–27.7% of children with JDM and is one of the important complications affecting morbidity. It is less frequently seen at the disease onset and usually develops later [15, 19–22]. In our study, calcinosis was developed in 38% of patients. Our higher percentage might be due to longer follow-up period which average was 6.2 years (6–166 months). Other studies defined above had 2–5 years follow-up period. Contrary, joint contractures were seen rarer in our cohort considering only two patients who were referred to us with delay and already had diffuse calcinosis and joint contractures [23–27]. Early diagnosis, early initiation of immunosuppressive medications with physical therapy, and combination of drugs commonly including prednisolone, intravenous methylprednisolone and methotrexate, followed by cyclosporine provided lower frequencies of contractures in our cohort. Prasad et al. related early onset methotrexate therapy to better clinical results compared with their past studies [25]. Early initiation of combined immunosuppressive medications and physical therapy minimize the risk for development of calcinosis and contractures [3, 28]. Similarly, combined immunosuppressive regimen was administered early in our cohort. Patients with calcinosis received methotrexate and steroid treatments for longer duration. Cyclophosphamide was more frequently applied to the patients with calcinosis. Additionally, cyclosporine and alendronate therapies were used in severe and refractory cases. All patients achieved remission except two patients referred after long-term disease duration. Only one patient had complaints of dysphagia and dysphonia. Otherwise, none of our patients had myocardium or lung involvement.
Measurement of serum levels of muscle enzymes is one of the most useful laboratory findings for JDM diagnosis. Malek et al. reported elevated creatine kinase levels in 87% of the study population [20]. All of our patients had elevated CK levels at the disease onset. No statistical difference was found between mean durations of achieving complete muscle strength and normal muscle enzyme levels. Thus, duration of elevated creatine kinase levels did not correlate with the improvement of muscle strength in our study. Serum levels of muscle enzymes should be investigated in addition to clinical findings for the diagnosis of JDM and should be followed to measure disease activity.
Antinuclear antibody positivity was evaluated 68% in our cohort that was similar to other studies [13, 24, 25]. Malek et al. suggested that ANA positivity was an important risk factor for disease complications such as lipodystrophy and growth retardation [20]. Six patients (12%) developed lipodystrophy and none of our patients had growth retardation. No statistical significance was found between ANA positivity and gender distribution, lipodystrophy, and calcinosis.
The positivity for antibodies against extractable nuclear antigens and histidyl-tRNA synthetase (anti-jo 1) was not detected in any patients in our study. Anti-jo 1 positivity was reported 15% in the study of Malek et al. [20] and 11% in the study of Prasad et al. [25]. In our study, failure to determine these antibodies was related to their uncommon presence during JDM and the laboratory methods used.
Electromyography and muscle biopsy were not performed for all patients in case of the lack of need or facilities. All applied patients showed signs of juvenile dermatomyositis. Malek et al. performed EMG to all study group and muscle biopsy to most of the patients [20]. They found signs of inflammatory myositis in 96% of cases on EMG and in 93.7% of cases on muscle biopsy. Since the majority of the patients meet clinical and laboratory criteria for diagnosis, invasive tests can be considered in patients in whom the diagnosis of JDM is not clear. The myopathological features of JDM determine the severity of disease in addition to providing a definitive diagnosis [29, 30]. Prominent vasculopathy is a sign of severe JDM and requires more intensive therapy [31].
Magnetic resonance imaging was not included into this study because it was not routinely performed. MRI demonstrates the extent and the features of muscle disease [32]. It provides a reliable baseline demonstration of disease, can detect most severely affected muscles for biopsy, and can be used for follow-up for disease progression and treatment response [33]. It is clear that the application of improving imaging techniques decreases the need for invasive procedures.
A recently published consensus on the management of JDM recommends the combination of corticosteroids (intravenous methylprednisolone 15–30 mg/kg/dose for 3 consecutive days followed by oral prednisolone 1–2 mg/kg/day) with methotrexate 15–20 mg/m2/week. After improvement is seen which means normal muscle strength, absence of skin rashes and major organ involvements, considering the weaning of steroids and ongoing therapy with methotrexate for a minimum 1 year remission and off-steroids are recommended. For severe disease in newly diagnosed patients or refractory patients, intensified treatment with cyclophosphamide 500–1000 mg/m2 for every 3–6 months is suggested. When improvement is not seen despite all these treatments, rituximab or infliximab with high dose methotrexate, cyclosporine, or intravenous immunoglobulin are recommended [34]. Similar to the consensus, we initiated therapy with corticosteroids which were almost always combined with methotrexate, and we added cyclosporine in the evidence of severe or refractory disease. Cyclophosphamide, infliximab, and IVIG were used for refractory cases. The treatment algorithm of our clinic is shown in supplementary file 1. While the new consensus recommends treatment with cyclosporine in case of resistance to cyclophosphamide, cyclosporine combined with steroid and methotrexate treatments was used in 48% of patients in our cohort. Recently published study of Aggarwal et al. [35] showed improvement of refractory skin rashes after the addition of rituximab to the standard regimen. Rituximab was not applied in our cohort. Our patients achieved remission except two cases that had delay in diagnosis and initiation of treatments, and referred to us after long term disease period with severe flexion contractures and calcinosis.
The present study had some limitations due to its retrospective design. Measurement of other serum muscle enzymes in addition to creatine kinase and detection of autoantibodies could not be done. Treatment responses as complete, partial response, or nonresponse and disease courses as monocyclic, polycyclic, or chronic continuous course could not be achieved. Because of the possible limitations of a retrospective study, future prospective multi-centered studies are needed regarding the disease rarity.
In conclusion, juvenile dermatomyositis, as a rare and serious inflammatory myopathy should be diagnosed and treated early with intensive immunosuppressive medications and physical therapy. Close monitoring of the disease status and treatment response is important for clinical evaluation and early clarification of disease complications. International collaboration is essential in order to better understanding of the prognosis and more efficient evaluation of the disease and treatment response.
Abbrevations: ANA, anti-nuclear antibody; CK, creatine kinase; EMG, electromyography; ENA, extractable nuclear antibody; IVIG, intravenous immunoglobulin; JDM, juvenile dermatomyositis; MRI magnetic resonance imaging; MTX, methotrexate
References
Huber A, Feldman BM (2013) An update on inflammatory myositis in children. Curr Opin Rheumatol 25:630–635
Rider LG, Lindsley CB, Miller FW (2016) Juvenile Dermatomyositis. In: Petty RE, Laxer RM, Lindsley CB, Wedderburn L (eds) Textbook of pediatric rheumatology, 7th edn. Elsevier, Philadelphia, pp 351–383
Rider LG, Katz JD, Jones OY (2013) Developments in the classification and treatment of the juvenile idiopathic inflammatory myopathies. Rheum Dis Clin N Am 39:877–904
Symmons DP, Sills JA, Davis SM (1995) The incidence of juvenile dermatomyositis: results from a nation-wide study. Br J Rheumatol 34:732–736
Mendez EP, Lipton R, Ramsey-Goldman R, Roettcher P, Bowyer S, Dyer A, Pachman LM (2003) US incidence of juvenile dermatomyositis, 1995–1998: results from the National Institute of Arthritis and Musculoskeletal and Skin Diseases registry. Arthritis Rheum 49:300–305
Batthish M, Feldman BM (2011) Juvenile Dermatomyositis. Curr Rheumatol Rep 13:216–224
Shah M, Mamyrova G, Targoff IN, Huber AM, Malley JD, Rice MM, Miller FW, Rider LG (2013) The clinical phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine (Baltimore) 92:25–41
Ramanan AV, Feldman BM (2002) Clinical features and outcomes of juvenile dermatomyositis and other childhood onset myositis syndromes. Rheum Dis Clin N Am 28:833–857
Morris P, Dare J (2010) Juvenile dermatomyositis as a paraneoplastic phenomenon: an update. J Pediatr Hematol Oncol 32:189–191
Bohan A, Peter JB (1975) Polymyositis and dermatomyositis (first of two parts). N Engl J Med 292:344–347
Bohan A, Peter JB (1975) Polymyositis and dermatomyositis (second of two parts). N Engl J Med 292:403–407
Robinson AB, Hoeltzel MF, Wahezi DM, Becker ML, Kessler EA, Schmeling H et al (2013) Clinical characteristics of children with juvenile dermatomyositis: the childhood arthritis and rheumatology research alliance (CARRA) registry. Arthritis Care Res 66:404–410
Brown VE, Pilkington CA, Feldman BM, Davidson JE (2006) An international consensus survey of the diagnostic criteria for juvenile dermatomyositis (JDM). Rheumatology (Oxford) 45:990–993
Gowdie PJ, Allen RC, Kornberg AJ, Akikusa JD (2013) Clinical features and disease course of patients with juvenile dermatomyositis. Int J Rheum Dis 16:561–567
Martin N, Li CK, Wedderburn LR (2012) Juvenile dermatomyositis: new insights and new treatment strategies. The Adv Musculoskelet Dis 4:41–50
Ernste FC, Reed AM (2013) Idiopathic inflammatory myopathies: current trends in pathogenesis, clinical features, and up-to-date treatment recommendations. Mayo Clin Proc 88:83–105
Carstens PO, Schmidt J (2014) Diagnosis, pathogenesis and treatment of myositis: recent advances. Clin Exp Immunol 175:349–358
Hashkes PJ, Wright BM, Lauer MS, Worley SE, Tang AS, Roettcher PA, Bowyer SL (2010) Mortality outcomes in pediatric rheumatology in the US. Arthritis Rheum 62:599–608
Lazarevic D, Pistorio A, Palmisani E, Miettunen P, Ravelli A, Pilkington C et al (2013) The PRINTO criteria for clinically inactive disease in juvenile dermatomyositis. Ann Rheum Dis 72:686–693
Malek A, Raeeskarami SR, Ziaee V, Aghighi Y, Moradinejad MH (2014) Clinical course and outcomes of Iranian children with juvenile dermatomyositis and polymyositis. Clin Rheumatol 33:1113–1118
Ruperto N, Pistorio A, Oliveira S, Zulian F, Cuttica R, Ravelli A et al (2016) Prednisone versus prednisone plus cyclosporine versus prednisone plus methotrexate in new-onset juvenile dermatomyositis: a randomised trial. Lancet 387:671–678
Faller G, Mistry BJ, Tikly M (2014) Juvenile dermatomyositis in South African child is characterized by frequent dystrophic calcification: a cross sectional study. Pediatr Rheumatol Online J 12:2
McCann LJ, Juggins AD, Maillard SM, Wedderburn LR, Davidson JE, Murray KJ, Pilkington CA (2006) The Juvenile Dermatomyositis National Registry and Repository (UK and Ireland)—clinical characteristics of children recruited within 5 yr. Rheumatology (Oxford) 45:1255–1260
Ravelli A, Trail L, Ferrari C, Ruperto N, Pistorio A, Pilkington C et al (2010) Long term outcome and prognostic factors of juvenile dermatomyositis: a multinational, multicenter study of 490 patients. Arthritis Care Res (Hoboken) 62:63–72
Prasad S, Misra R, Agarwal V (2013) Juvenile dermatomyositis at a tertiary care hospital: is there any change in the last decade? Int J Rheum Dis 16:556–560
Saini I, Kalaivani M, Kabra SK (2016) Calcinosis in juvenile dermatomyositis: frequency, risk factors and outcome. Rheumatol Int 36:961–965
Kishi T, Miyamae T, Hara R, Nakajima S, Imagawa T, Mori M, Yokota S (2013) Clinical analyses of 50 children with juvenile dermatomyositis. Mod Rheumatol 23:311–317
Kim S, El-Hallak M, Dedeoglu F, Zurakowski D, Fuhlbrigge RC, Sundel RP (2009) Complete and sustained remission of juvenile dermatomyositis resulting from aggressive treatment. Arthritis Rheum 60:1825–1830
Estruch R, Grau JM, Fernandez-Sola J, Casademont J, Monforte R, Urbano-Marquez A (1992) Microvascular changes in skeletal muscle in idiopathic inflammatory myopathy. Hum Pathol 23:888–895
Dorph C, Englund P, Nennesmo I, Lundberg IE (2006) Signs of inflammation in both symptomatic and asymptomatic muscles from patients with polymyositis and dermatomyositis. Ann Rheum Dis 65:1565–1571
Gitiaux C, De Antonio M, Aouizerate J, Gherardi RK, Guilbert T, Barnerias C et al (2016) Vaculopathy-related clinical and pathological features are associated with severe juvenile dermatomyositis. Rheumatology (Oxford) 55:470–479
Kolasinski SL, Chi AS, Lopez-Garib AJ (2016) Current perspectives on imaging for systemic lupus erythematosus, systemic sclerosis, and dermatomyositis/polymyositis. Rheum Dis Clin N Am 42:711–732
Elessawy SS, Abdelsalam EM, Abdel Razek E, Tharwat S (2016) Whole-body MRI for full assessment and characterization of diffuse inflammatory myopathy. Acta Radiol Open 5:2058460116668216 eCollection 2016
Enders FB, Bader-Meunier B, Baildam E, Constantin T, Dolezalova P, Feldman BM et al (2016) Consensus-based recommendations for the management of juvenile dermatomyositis. Ann Rheum Dis. doi:10.1136/annrheumdis-2016-209247 [Epub ahead of print]
Aggarwal R, Loganathan P, Koontz D, Qi Z, Reed AM, Oddis CV (2016) Cutaneous improvement in refractory adult and juvenile dermatomyositis after treatment with rituximab. Rheumatology (Oxford). Nov 11. pii: kew396. [Epub ahead of print]
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosures
None.
Electronic supplementary material
Supplementary file 1
The treatment algorithm of our clinic for Juvenile dermatomyositis (DOCX 38 kb)
Rights and permissions
About this article

Cite this article
Barut, K., Aydin, P.O.A., Adrovic, A. et al. Juvenile dermatomyositis: a tertiary center experience. Clin Rheumatol 36, 361–366 (2017). https://doi.org/10.1007/s10067-016-3530-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-016-3530-4