
Abstract
The idiopathic inflammatory myopathies (IIM) are a group of autoimmune diseases resulting from inflammation of muscle and manifesting as weakness, though a range of extra-muscular manifestations are observed. These are often correlated closely with disease subtype and the presence of myositis-specific/myositis-associated antibodies. IIM are notoriously difficult to treat and often refractory to glucocorticoid therapy and synthetic immunosuppressants. Both the innate and adaptive immune systems are implicated in the pathogenesis of IIM. A growing understanding of the key cytokines as well as the cell-mediated and antibody effectors of disease has identified multiple potential targets for biologic therapy. The most widely used of these is B-cell depletion via rituximab though the tumour necrosis factor inhibitors and other biologic therapies used in diseases such as rheumatoid arthritis, systemic lupus erythematosus and multiple sclerosis have also been trialled. This review summarises the literature thus far on biologic therapy in IIM, highlighting both the significant trials that influence current treatment regimens and also the continuing need for further research to inform more effective therapies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The idiopathic inflammatory myopathies (IIM) are a heterogenous group of systemic autoimmune conditions with dominant effects on skeletal muscle, though a range of extra-muscular features may be seen. The main subtypes are defined histologically as polymyositis (PM), dermatomyositis (DM), inclusion body myositis (IBM) and immune mediated necrotising myopathy (IMNM). IIM are associated with significant disability from progressive weakness as well as increased mortality especially from pulmonary and cardiac complications [1].
Historically, IIM have been difficult to treat. Standard therapies involve high-dose glucocorticoids and immunosuppressive agents such as methotrexate, azathioprine, cyclosporine or mycophenolate mofetil [2]. A significant proportion of patients have an incomplete response, needing prolonged glucocorticoid treatment with its inherent side effects and the consequences of incomplete disease control, namely ongoing muscle damage [3]. Literature published on IIM patients treated with these traditional approaches has found that at least a third have some degree of disability despite treatment [4, 5].
The advent of biologic therapies has held great promise for autoimmune diseases, allowing the translation of our understanding of specific processes in disease pathophysiology to therapeutics targeting these autoimmune aberrancies.
In this narrative review, we aim to provide an up to date summary of the current evidence for biologic therapy in IIM.
Diagnosis
The diagnosis of IIM is based on a constellation of clinical, laboratory and imaging findings. In general, muscle weakness, elevated levels of muscle enzymes (creatine kinase—CK, lactate dehydrogenase, aspartate aminotransferase, alanine aminotransferaseand aldolase), a myopathic triad on electromyography (small polyphasic motor unit potentials, fibrillation potentials even at rest and bizarre high-frequency repetitive discharges) and characteristic changes on muscle MRI (such as muscle oedema on T2/STIR sequences [6]) are all suggestive of IIM. There is also a growing range of auto-antibodies identified, including both myositis-specific antibodies (MSA) and myositis-associated antibodies (MAA) which are linked to particular clinical and pathological manifestations [7]. The most definitive diagnostic test remains the muscle biopsy and the histopathology reflects underlying differences in pathogenesis between the IIM subtypes [8].
Clinical features
Muscle weakness is the most prominent finding in patients with IIM. PM and DM typically cause symmetrical proximal weakness of the upper and lower limbs while IBM is uniquely characterised by finger flexor and quadriceps weakness. DM also presents with skin manifestations including a heliotrope rash, Gottron’s papules (erythematous papules/plaques over the bony prominences of joints) and Gottron’s sign (erythema of the extensor surfaces of the finger joints).
Antisynthetase syndrome (ASS) is a unique collection of clinical features in the presence of antibodies directed against aminoacyl transfer RNA (tRNA) synthetases [9]. It is characterised by myositis (PM or DM), interstitial lung disease (ILD), inflammatory arthritis, Raynaud’s phenomenon and thickening/cracking of the skin on the fingertips known as mechanic’s hands [10]. Distinct antisynthetase antibodies have different predilections for ILD and myositis [11].
Pathophysiology
Polymyositis is characterised as the prototypical T-cell driven autoimmune myopathy, predominantly involving the interaction of CD8+ cells and macrophages with MHC1-expressing muscle fibres [12].
Dermatomyositis, on the other hand, involves a cellular infiltrate composed of B-cells, T-cells (CD4+ more than CD8+) and macrophages [8] with a histological appearance of microvascular ischaemia via damage to the muscle capillaries though the mechanisms for this remain unclear [13].
Though IBM shares some features on muscle biopsy with PM such as the CD8+ T-cell infiltrate, it is additionally characterised by a parallel degenerative process, likened to the “Alzheimer’s Disease of the muscle” with the finding of rimmed vacuoles lined with granular material and mitochondrial changes, reflected by an increase in the number of cytochrome c oxidase-negative fibres [14]. Special stains reveal protein aggregates including amyloid, TDP43 and p62 thought to represent altered cellular autophagy of debris. Electron microscopy has characteristic findings of cytomembranous whorls and tubofilamentous inclusions [15].
Unlike the other subtypes, IMNM shows dominant myofibre necrosis and a paucity of inflammatory cell infiltration. Though it is associated with multiple causes (e.g. viruses, statin medications and malignancy), the finding of autoantibodies, namely to signal recognition particle (SRP) and 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) and response to immunosuppression are supportive of an autoimmune contribution to pathogenesis in some patients [16].
Beyond microscopy, a milieu of cytokines is implicated in IIM. A cytokine of significant interest is interferon 1 (IFN 1). Serum interferons as well as interferon-induced genes (referred to as the “IFN signature”) in tissue samples are upregulated in DM [17]. In vitro, IFN inhibits myotube formation [18] and causes myofibre atrophy as well as vascular disruption of dermal microvascular epithelial cells [19] accounting for the muscle and skin manifestations of DM. Furthermore, IFN has been shown to stimulate the translocation of high-motility group box 1 (HMGB1) protein from the nucleus to the cytoplasm in muscle cells inducing the abnormal expression of MHC-1, a key feature of PM and DM [20].
Other pro-inflammatory cytokines implicated include the IL-1 family as well as tumour necrosis factor α (TNFα) both of which are significantly upregulated in IIM [21, 22]. In high concentrations, TNFα has a directly myopathic effect and inhibits myofibre recovery [23].
A greater understanding of the immune mechanisms involved in the pathogenesis of IIM has led to an interest in exploiting these pathways as a therapeutic strategy. The last few years have identified multiple targets for biologic therapy.
Biologics targeting cellular pathways include: rituximab which targets CD20+ B-cells and antibody effects, abatacept which prevents T-cell activation by binding CD80/CD86 on antigen presenting cells, and alemtuzumab which depletes both B- and T-cells populations.
Targets for cytokine effectors include: TNF inhibitors which limit the direct toxic effect of TNFα as well as its amplification of inflammation, basiliximab and tocilizumab which target the IL-2 and IL-6 receptors, respectively, and sifalimumab which targets interferon pathways.
Search strategy
We analysed the evidence for the use of biologic therapeutics in IIM described in articles published up to May 2019. This was performed using the PubMed database and the following key terms: inflammatory myopathies OR myositis OR polymyositis OR dermatomyositis OR anti-synthetase syndrome AND the following agent specific terms: rituximab OR TNF inhibitors (OR infliximab OR adalimumab OR etanercept OR golimumab OR certolizumab) OR abatacept OR alemtuzumab OR basiliximab OR sifalimumab OR tocilizumab.
Of these biologics, rituximab is the agent which has been published about most prolifically. Fifty-one articles were identified on rituximab in IIM including one randomised controlled trial, four open label trials, twenty-three case reports, ten case series, four prospective studies and nine retrospective studies (two of which were registry based) (Fig. 1).Table 1 summarises the key trials for biologic agents in IIM.

Number of original articles published by biologic agent
Rituximab
Molecular mechanisms
Rituximab is a chimaeric murine/human monoclonal antibody targeting CD20 expressing cells. CD20 is found exclusively on pre-B and mature B lymphocytes. While its precise function is still largely unknown, its specificity to these cells has made it a useful candidate for targeted therapies.
Circulating antibodies are found in up to 80% of patients with IIM [24] and the role of B-cells in IIM is a key rationale in the use of rituximab for B-cell depletion. DM is classically viewed as a humorally driven disorder given the finding of B-cells in the inflammatory infiltrate on muscle biopsy [8]. However, whilst PM and IBM are traditionally conceptualised as primarily T-cell mediated diseases, autoantibodies are still detected [25] and immunohistochemical studies confirm the presence of differentiated B-cells/CD138+ plasma cells in muscle tissue [26], providing a rationale for B-cell directed therapies.
The depletion of B-cells that occurs with rituximab is not permanent and repopulation typically occurs between 6 and 12 months though can be as delayed as 42 months [27]. Though clinical relapse (defined as increasing weakness or rising CK) appears to follow B-cell repopulation in many cases [28, 29], there are reports of patients who maintain clinical remission despite immune reconstitution [27, 30]. This may be reflective of the long-term modulation of B-cell subsets caused by rituximab. In the rheumatoid arthritis population treated with rituximab, analyses of B-cell reconstitution have demonstrated lasting reductions of CD27+ memory B-cells compared with pre-treatment levels [31]. In patients with IIM relapse, retreatment with rituximab appears to be an effective strategy [30, 32].
Evidence for use
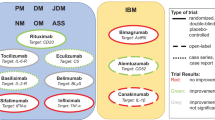
The only randomised controlled trial thus far has been the rituximab in myositis (RIM) trial, published by Oddis et al. [33]. This trial involved 200 patients (195 included in the final analysis) who had treatment-refractory PM, DM or juvenile DM, defined as intolerance or inadequate response to glucocorticoids and at least one other immunosuppressive agent. Patients were randomised to either the “rituximab early” (receiving ritxumab on Day 1 and placebo 8 weeks later) or “rituximab late” (receiving placebo on Day 1 and rituximab 8 weeks later) streams of treatment. Improvement was assessed by a composite measure, definition of improvement (DOI) based on the International Myositis Assessment and Clinical Studies (IMACS) criteria— ≥ 20% improvement in three of any six core set measures (muscle weakness using the Manual Muscle Testing 8 (MMT-8) score, patient global assessment of disease activity, physician global assessment of disease activity, health assessment questionnaire, muscle enzyme levels and global extra-muscular disease activity) with no more than two of these measures worsening by ≥ 25% on two consecutive monthly visits. Though there was no significant difference between the streams in the primary endpoint (time to achieving DOI), it is highly noteworthy that overall, 83% of patients achieved DOI by the conclusion of the trial at 44 weeks. Figure 2 summarises the key findings of RIM.

The RIM trial has been noted to have certain limitations. Though the investigators initially expected the effect of rituximab in more than half of the patients to be evident by 8 weeks, this was not seen until 20 weeks; this impacted upon the power calculations within the delayed-start design. It is also possible that the 8-week placebo phase (chosen due to ethical considerations) was too short resulting in no significant difference between the streams of the study. Selection of patients was done according to histological diagnosis with an adjudication committee reviewing medical records and biopsy results though it is not explicitly stated what criteria were used to differentiate PM from IBM [34]. Although the appearance of these subtypes can be similar on biopsy, IBM is generally held to be significantly more treatment refractory [35].
Use in specific sub-groups
Post hoc analysis of the RIM trial investigated whether antibody profile determined response to rituximab and found that the presence of anti-synthetase antibodies (most commonly anti-Jo1) and anti-Mi2 antibodies strongly predicted response [36]. A retrospective study published in 2019 specifically looking at anti-synthetase antibody-positive patients who received rituximab compared with anti-synthetase antibody-negative patients found that though both groups showed moderate/major improvement after rituximab, only the former experienced a significant glucocorticoid-sparing effect [37]. Barsotti et al. similarly found that patients with anti-synthetase antibodies (anti-Jo1, anti-Pl7 and anti-Pl12) showed more improvement with rituximab than those without these antibodies [38], whereas in a prospective study by Mahler et al. [39], rituximab response did not correlate with presence of anti-Jo1 antibodies.
Anti-Jo1 titres themselves do not significantly change after rituximab treatment and do not seem to correlate with disease activity [29, 40]. Though some studies report decrease in anti-Jo1 titres after rituximab [41, 42], patients can still have detectable anti-Jo1 despite being clinically in remission [43].
Interstitial lung disease (ILD) is an extra-musculoskeletal manifestation which carries significant morbidity and mortality in IIM [44]. The effect of rituximab on ILD is less well defined than its benefits on muscle and current investigation is ongoing with a multicentre randomised controlled trial (the RECITAL trial) in the UK comparing rituximab to cyclophosphamide in the treatment of ILD associated with connective tissue disease, including IIM [45]. In patients who are positive for anti-melanoma differentiation-associated gene 5 (anti-MDA5) antibody, associated with clinically amyopathic DM (CADM), a primary concern is rapidly progressive ILD (RP-ILD). In one retrospective study, four patients with CADM and ILD were given rituximab which resulted in improvement in pulmonary function tests (PFT), New York Heart Association (NYHA) functional grading and decreased glucocorticoid dose [46]. Conversely, when three patients with CADM and RP-ILD were given rituximab in another case series, the two which were anti-MDA5 positive died whereas the patient without this antibody improved [47].
Dermatomyositis is clinically defined by its characteristic skin manifestations. Few studies have examined the effect of rituximab on the dermatological signs of DM with results varying from no significant change in Dermatomyositis Skin Severity Index [48] to complete recovery [49].
Cardiac involvement in IIM, including myocarditis, fibrosis and arrhythmia is a negative prognostic factor [50]. There is only one case report on the use of rituximab in a woman with DM found to have elevated cardiac biomarkers (troponin T) as well as conduction abnormalities on Holter monitor (multifocal atrial tachycardia, ventricular premature beats and Mobitz type 1 AV block). Rituximab treatment led to reversal of all these abnormalities [51].
There is limited evidence on the use of rituximab in IIM other than PM and DM. In one retrospective analysis, 3/9 IMNM patients with anti-HMGCR antibodies responded to rituximab [52]. Similarly, an Australian study found that of eight IMNM patients receiving rituximab, only two had a positive response allowing weaning of glucocorticoids and intravenous immunoglobulin (IVIg) [53]. It is possible that certain subgroups of IMNM may have a better response to rituximab; a case series of eight patients describes dramatic improvement in refractory anti-signal recognition particle (anti-SRP) IMNM [54].
Two patients with IBM were included in a retrospective audit which predominantly examined patients with PM and DM treated with rituximab. Neither of them showed improvement in muscle strength or dysphagia post rituximab [38]. Similarly, one case report describes a patient with rheumatoid arthritis and IBM; rituximab treatment led to remission of arthritis but no improvement in muscle strength [55]. This is consistent with the generally held notion that IBM is refractory to immunosuppressive therapies.
Safety
The safety profile of rituximab has been reported extensively in rheumatoid arthritis [56]. In IIM trials, both bacterial infections (predominantly pneumonia, cellulitis and urinary tract infections) as well as herpes zoster reactivation have been documented. Opportunistic infection with pneumocystis jirovecii pneumonia has resulted in four deaths [41, 51]. Non-tuberculous mycobacterium infections have also been reported though whether these were unmasked by immunosuppression or newly contracted is unclear [57]. Malignancy is commonly screened for in the DM population and is of particular importance prior to commencing immunomodulatory therapy. There is a surprising paucity of reported malignant adverse effects in the IIM studies considered above though it is possible that this reflects a lack of longer term follow-up. One patient died from colorectal cancer post-rituximab [48] and another had a diagnosis of new breast cancer metastases on the background of previously diagnosed breast adenocarcinoma [58]. The feared complication of progressive multifocal leukoencephalopathy (PML) is rare but has been noted in two cases, both times fatal [59, 60].
TNF inhibitors
The tumour necrosis factor inhibitors (TNFi) are a group of drugs which target TNFα, a cytokine which plays an integral role in immune cell activation and proliferation as well as further cytokine and chemokine production. There are currently five TNFi approved for use—infliximab, adalimumab, etanercept, golilumab and certolizumab pegol though only the first three of these have been investigated in relation to IIM, with conflicting results.
Infliximab
Only one randomised, double blinded, controlled trial has been performed on infliximab. Twelve patients (11 PM, 1 DM) were enrolled. In phase 1, half of the patients were randomised to receive infliximab at 5 mg/kg and the other half to placebo. At 16 weeks, responders to infliximab continued on this agent whereas non-responders escalated to 7.5 mg/kg dosing. Patients receiving placebo crossed over to 5 mg/kg infliximab at this point in the trial. Overall, only 1/6 patients from the phase 1 infliximab group responded though 3/5 of the non-responders responded to the increased dose. Two of six patients who crossed over from placebo to infliximab responded. While this trial was under-powered, it did demonstrate that some patients respond to infliximab and response may be dose dependent [61].
In a group of 14 patients with DM and ILD detected by HRCT, Chen et al. found that ten responded to infliximab with improvement in muscle, skin and lung manifestations. The remaining four died from progressive respiratory failure. These patients had higher alveolar-arterial gradients at baseline, suggesting that infliximab may have a more favourable effect if given earlier in the disease process [62]. However, an open label trial performed uniquely on drug-naïve patients with PM/DM given methotrexate and infliximab, was terminated prematurely when 4/6 patients withdrew due to worsening weakness, progression of ILD and infusion reaction [63]. A negative result was also found when infliximab was given to 13 patients with IIM (5 PM, 4 DM and 4 IBM), none of whom improved in manual muscle test [64].
Etanercept
Etanercept has shown similarly mixed results. Again, only one randomised, double blinded, controlled trial has been performed on 16 patients with DM, 11 of which were randomised to receive etanercept and five to placebo. Five of 11 patients receiving etanercept were successfully weaned off prednisolone and overall, the treatment group had a significantly lower mean prednisolone dose as well as time to treatment failure than the placebo group [65].
Iannone et al. investigated five patients previously treated with cytotoxic therapy allowed to wash out before commencing etanercept (with prior prednisolone dosage maintained). All patients experienced increase in weakness and elevation of CK suggesting that etanercept is not effective without concurrent cytotoxic treatment [66]. A retrospective study of eight patients with refractory PM/DM who were commenced on TNFi (6 etanercept, 1 infliximab and 1 infliximab then etanercept) in addition to cytotoxics/intravenous immunoglobulin found that six patients (5/6 who received etanercept and the patient who had sequential therapy with both agents) had improved muscle strength and decreased subjective fatigue [67].
Adalimumab
Two case reports have been published on adalimumab use in IIM. Both patients were anti-Jo1 antibody-positive with ILD and refractory to multiple immunosuppressives. Adalimumab led to improvement in muscle strength but no change in ILD [68, 69].
Safety
From the literature on rheumatoid arthritis and spondyloarthropathies, TNFi are held to be generally safe and well tolerated with the main concern being an increased risk of infections and notably, tuberculosis [70]. Given only a small number of investigations with limited sample sizes it is difficult to comment on the safety of TNFi in IIM. Tuberculosis reactivation did not occur in any of these trials but serious infection with pneumonia has been reported to occur as a result of TNFi treatment [71].
TNFi causing IIM
A paradoxical relationship between TNFi and IIM has been observed in patients with other autoimmune diseases where new onset PM/DM has occurred following TNFi use. This has predominantly been reported in patients with rheumatoid arthritis who have been given infliximab, etanercept or adalimumab [72]. It is possible that these patients have an RA-IIM overlap syndrome (though this is rare [73]) or that they may have arthritis as part of anti-synthetase syndrome, the full manifestations of which are unmasked by TNFi treatment [74], but in almost all patients, withdrawal of TNFi resulted in improvement of myositis [72]. One suggested theory is that TNFα plays a role in inhibiting the production of type I interferons by plasmacytoid dendritic cells and the imbalance of these cytokines following TNFi treatment results in the development of PM/DM through the interferon pathways discussed above [17].
Notable myositis subgroups
Antisynthetase syndrome
Though the significance of antisynthetase antibodies in rituximab has been discussed above, there have also been reports and trials of biologic therapy in patients diagnosed with antisynthetase syndrome (ASS).
An open label trial published in 2015 by Allenbach et al. using rituximab in ASS patients refractory to conventional therapies found that only two of ten patients achieved the primary endpoint of objective muscle strength improvement in two different muscle groups. A non-significant increase in overall median strength and decrease in CK was noted [75].
Interstitial lung disease is a feature of ASS that is associated with significant morbidity and mortality. In a cohort of 24 patients with ILD as part of ASS, rituximab led to improved pulmonary function test indices and reduced lung inflammation as assessed by high-resolution CT (HRCT) chest with the most profound effect found in patients who had less than 12 months disease duration and/or current acute exacerbation of ILD [41]. This finding is consistent with other trials in ASS, suggesting that rituximab is effective in stabilising and/or improving ILD [42, 75, 76].
Interestingly, though ILD was the main focus of these trials, two of them also demonstrated significant improvement in manual muscle testing strength score and reductions in CK levels after treatment with rituximab [41, 42].
Juvenile dermatomyositis
Though the current literature predominantly focuses on biologic therapy in adult patients with IIM, there have been some significant publications in juvenile dermatomyositis (JDM). The opportunity to treat childhood myositis early and effectively could hold great promise for long-term outcomes in patients with JDM.
Though RIM was not powered towards analysis of myositis subtype, post hoc analysis has shown that patients with JDM were more likely than adult PM/DM patients to have a favourable response to rituximab. Furthermore, when analysed in isolation, JDM patients did actually meet the primary endpoint, with the rituximab early stream achieving DOI at 11.7 weeks compared to 20.2 weeks for the late stream, in keeping with the delayed start design of 8 weeks [36, 77].
A dedicated prospective study of JDM patients with active disease despite conventional therapies found that rituximab resulted in complete clinical response in the domains of muscle and skin manifestations in three of six patients. Calcinosis, a painful and debilitating complication of JDM, did not improve in any of the patients. Furthermore, non-responders were characterised by a longer disease duration than responders, highlighting the importance of early treatment [78].
In contrast, a trial of infliximab in five patients with refractory juvenile DM found improvement of muscle weakness and calcinosis though not skin rash. Significantly, in three of the patients, clinical improvement resulted in abrogation of the need for corticosteroids [79].
Rouster-Stevens et al. investigated the effect of etanercept in a prospective analysis of nine patients with JDM. Only two patients completed the 12-week treatment phase and overall, unlike the aforementioned infliximab trial in JDM, there was no change in serum muscle enzymes and no significant improvement in disease activity score or childhood myositis assessment scale [80].
Other biologic agents
Abatacept
Abatacept is a genetically engineered fusion protein synthesised from the Fc portion of IgG1 and the extracellular domain of cytotoxic T-cell lymphocyte-associated protein 4 (CTLA4). By binding to CD80 and CD86 on antigen-presenting cells, abatacept prevents T-cell activation [81].
The largest randomised trial on abatacept in IIM (ARTEMIS) involved 20 patients (11 PM, 9 DM) who had active disease despite glucocorticoids and at least one other immunosuppressant for at least 3 months. With a delayed treatment design, half of the patients commenced on abatacept at week 0 and the other half at week 10. The primary endpoint was the number of responders at 6 months regardless of treatment arm and nearly half the patients achieved this with improvement in disease activity. Comparisons between the arms of the study showed significant improvement in the treatment arm over the delayed arm at both 3 and 6 months. Notably, 75% of responders compared with 36% of non-responders were on concomitant methotrexate [82].
In addition to this, there are two case reports on abatacept use in patients with IIM refractory to multiple agents including other biologics—one with IMNM that relapsed after rituximab in a patient intolerant of tocilizumab [83] and the other in a patient with PM which worsened despite etanercept and IVIg [84]. Both these patients are reported improving in muscle strength and requiring lower doses of glucocorticoids after abatacept administration.
Alemtuzumab
Alemtuzumab is a biologic therapy targeting the widespread CD52 cell marker present on lymphocytes, eosinophils, monocytes/macrophages and peripheral blood dendritic cells. Binding of alemtuzumab to CD52 stimulates destruction of these cells resulting in depletion of both circulating B-cells and T-cells [85]. Immune reconstitution is theorised to favour immune regulation over autoimmunity and this effect has resulted in well-recognised use of alemtuzumab in the treatment of relapsing–remitting multiple sclerosis [86].
Two cases have been reported on alemtuzumab use in refractory PM [87, 88]. Both were associated with improvement in muscle strength and reduction in (but not normalisation of) CK levels. In one of these reports, despite muscle response, the patient had progression of respiratory disease on alemtuzumab and eventually died from interstitial fibrosis and pulmonary arterial hypertension [88].
Dalakas et al. published a proof of concept study in 2009 focusing on alemtuzumab in 13 patients with IBM who had not received any immunosuppressants in the preceding 12 months. Objective muscle testing scores were used to compare deterioration in this 12-month period (mean decrease of 14.9%) with the 6 months post alemtuzumab administration (mean decrease of 1.9%), suggesting that in this early period, lymphocyte depletion slowed the progression of disease with four patients demonstrating a gain in muscle strength of at least 10%. This was correlated with muscle biopsies at 6 months which showed a significant decrease in endomysial T-cells [89]. As a proof of concept study, there were significant limitations raised about comparisons between a 12-month and 6-month period, uncontrolled prophylactic antimicrobials used in addition to alemtuzumab as well as the possible placebo effect given the unblinded nature of the study [90].
Basiliximab
Basiliximab is a monoclonal antibody targeting the alpha subunit of the IL-2 receptor resulting in inhibition of IL-2 signalling, a key step in T-cell activation and proliferation.
There is one case report of a 42-year-old woman with PM which responded to prednisolone and MTX but flared when prednisolone was weaned. Basiliximab was given but ceased prior to the sixth infusion due to lack of improvement or tolerable reduction in glucocorticoid dose [91].
A small case series describes four patients with anti-MDA5 associated amyopathic DM with RP-ILD refractory to glucocorticoids, cyclosporine and IVIg. With addition of basiliximab, 3/4 patients had HRCT-confirmed improvement in pulmonary disease [92].
Sifalimumab
Sifalimumab, a monoclonal antibody targeting IFNα, is currently undergoing trials in systemic lupus erythematosus [93]. A randomised controlled, double blinded, crossover trial by Guo et al. published in 2014 examined 48 patients with DM/PM, half of whom received sifalimumab and the other half placebo for 3 months before crossing over. Biochemically, patients were assessed for dysregulated protein levels, some of which are known to be associated with interferon. The levels of these proteins as well as clinical muscle scores were measured before and after intervention (sifalimumab or placebo). Sifalimumab, but not placebo, was found to suppress multiple cytokines associated with interferon and of these, reduction in IL-2RA had the strongest correlation with improvement in muscle strength. Two patients with the greatest suppression of T cell-associated proteins had muscle biopsies after 3 months of sifalimumab. Compared with their baseline biopsies, there was a pronounced decrease in CD3 staining (a T-cell marker), suggesting that suppression of proteomic profile in the peripheral blood reflects reduced inflammatory infiltrate in the muscle [94].
Tocilizumab
Tocilizumab is a humanised monoclonal antibody which antagonises the IL-6 receptor [95]. Four case reports exist in the literature about tocilizumab use in refractory IIM patients. Three of these patients had previously tried other biologic therapy (rituximab [96, 97] and adalimumab [98]) without sustained improvement which they then achieved with tocilizumab manifesting as reduced muscle weakness and normalisation of CK.
Narazaki et al. reported two patients with an anti-Jo1 positive PM refractory to multiple immunosuppressants. In one of these patients, tocilizumab resulted in glucocorticoid dose reduction and normalisation of CK levels after two injections, whereas the second required increased frequency of administration (4 weekly to 3 weekly) and 12 doses before CK normalised and MRI of the thigh muscles showed resolution of muscle oedema [99].
Conclusion
There is growing evidence that biologic therapy in IIM has the potential to benefit patients with refractory disease, resulting in improved muscle strength, decreased biochemical markers of muscle inflammation and weaning of glucocorticoids. It is a significant barrier that IIM are rare diseases and this limits sample size in trials. Furthermore, though there have been significant steps in understanding the pathogenesis of IIM, there is still much unknown about the immune processes underlying these diseases.
Rituximab has been the most extensively investigated of the biologic therapies and appears to be effective for patients with PM, DM and JDM but less so for IBM and IMNM. It has a variable benefit for extra-muscular manifestations and subtleties of MSA profile may modulate its effectiveness.
Despite the theoretical role for TNF in the disease pathogenesis of IIM, the TNFi have a significantly less consistent effect on IIM in the literature than rituximab, sometimes even causing worsening of disease potentially through their ability to upregulate interferon pathways.
Other agents, though promising, are limited by minimal trial data and small sample sizes.
With a lack of large cohort randomised controlled trials, there is still no consensus on exactly what treatment algorithm should be used for patients with IIM, let alone how this might be modified for subtype or antibody profile. Dosage of all the biologics mentioned above is derived from trials in other autoimmune/oncology disease groups.
At our institution, it has been our practice to use steroids together with either methotrexate or azathioprine from the time of diagnosis. If a favourable response is not detected within 2–3 months, combination therapy with the two agents is employed, and thereafter if disease remains refractory, escalation of treatment to rituximab (using two intravenous doses of 1000 mg 2 weeks apart), or less commonly intravenous immunoglobulin (IVIg), is employed (Fig. 3).

Our approach to the management of inflammatory myositis
Overall, with a growing understanding of the pathogenic mechanisms of IIM and the specific differences between histological subtypes, the future of targeted therapy may involve IIM specific drugs rather than extrapolating from experience with other autoimmune diseases. However, presently, though it appears that biologics have the potential for a profound effect on the lives of patients afflicted with IIM, this is still a largely unexplored, untested and off-label area of ongoing investigation. Ultimately, translational research may enable personalised medicine in which treatments are precisely targeted to dominant cellular/cytokine pathways within an individual to optimise outcomes.
References
Marie I (2012) Morbidity and mortality in adult polymyositis and dermatomyositis. Curr Rheumatol Rep 14(3):275–285. https://doi.org/10.1007/s11926-012-0249-3
Wendling D (2007) Biologics in the treatment of primary inflammatory myositis. Jt Bone Spine 74(4):316–318. https://doi.org/10.1016/j.jbspin.2006.11.013
Lundberg IE, Vencovsky J, Alexanderson H (2014) Therapy of myositis: biological and physical. Curr Opin Rheumatol 26(6):704–711. https://doi.org/10.1097/BOR.0000000000000109
Dalakas MC, Hohlfeld R (2003) Polymyositis and dermatomyositis. Lancet 362(9388):971–982. https://doi.org/10.1016/S0140-6736(03)14368-1
Marie I, Hachulla E, Hatron PY, Hellot MF, Levesque H, Devulder B, Courtois H (2001) Polymyositis and dermatomyositis: short term and longterm outcome, and predictive factors of prognosis. J Rheumatol 28(10):2230–2237
Pipitone N (2016) Value of MRI in diagnostics and evaluation of myositis. Curr Opin Rheumatol 28(6):625–630. https://doi.org/10.1097/BOR.0000000000000326
Satoh M, Tanaka S, Ceribelli A, Calise SJ, Chan EK (2017) A comprehensive overview on myositis-specific antibodies: new and old biomarkers in idiopathic inflammatory myopathy. Clin Rev Allergy Immunol 52(1):1–19. https://doi.org/10.1007/s12016-015-8510-y
Luo YB, Mastaglia FL (2015) Dermatomyositis, polymyositis and immune-mediated necrotising myopathies. Biochim Biophys Acta 1852 4:622–632. https://doi.org/10.1016/j.bbadis.2014.05.034
Tieu J, Lundberg IE, Limaye V (2016) Idiopathic inflammatory myositis. Best Pract Res Clin Rheumatol 30(1):149–168. https://doi.org/10.1016/j.berh.2016.04.007
Katzap E, Barilla-LaBarca ML, Marder G (2011) Antisynthetase syndrome. Curr Rheumatol Rep 13(3):175–181. https://doi.org/10.1007/s11926-011-0176-8
Ghirardello A, Zampieri S, Tarricone E, Iaccarino L, Bendo R, Briani C, Rondinone R, Sarzi-Puttini P, Todesco S, Doria A (2006) Clinical implications of autoantibody screening in patients with autoimmune myositis. Autoimmunity 39(3):217–221. https://doi.org/10.1080/08916930600622645
Clark KEN, Isenberg DA (2018) A review of inflammatory idiopathic myopathy focusing on polymyositis. Eur J Neurol 25(1):13–23. https://doi.org/10.1111/ene.13357
Gherardi RK (2011) Pathogenic aspects of dermatomyositis, polymyositis and overlap myositis. Presse Med 40(4 Pt 2):e209–218. https://doi.org/10.1016/j.lpm.2010.12.013
Chahin N, Engel AG (2008) Correlation of muscle biopsy, clinical course, and outcome in PM and sporadic IBM. Neurology 70(6):418–424. https://doi.org/10.1212/01.wnl.0000277527.69388.fe
Greenberg SA (2019) Inclusion body myositis: clinical features and pathogenesis. Nat Rev Rheumatol 15(5):257–272. https://doi.org/10.1038/s41584-019-0186-x
Basharat P, Christopher-Stine L (2015) Immune-mediated necrotizing myopathy: update on diagnosis and management. Curr Rheumatol Rep 17(12):72. https://doi.org/10.1007/s11926-015-0548-6
Greenberg SA (2010) Type 1 interferons and myositis. Arthritis Res Ther 12(Suppl 1):S4. https://doi.org/10.1186/ar2885
Franzi S, Salajegheh M, Nazareno R, Greenberg SA (2013) Type 1 interferons inhibit myotube formation independently of upregulation of interferon-stimulated gene 15. PLoS ONE 8(6):e65362. https://doi.org/10.1371/journal.pone.0065362
Ladislau L, Suarez-Calvet X, Toquet S, Landon-Cardinal O, Amelin D, Depp M, Rodero MP, Hathazi D, Duffy D, Bondet V, Preusse C, Bienvenu B, Rozenberg F, Roos A, Benjamim CF, Gallardo E, Illa I, Mouly V, Stenzel W, Butler-Browne G, Benveniste O, Allenbach Y (2018) JAK inhibitor improves type I interferon induced damage: proof of concept in dermatomyositis. Brain 141(6):1609–1621. https://doi.org/10.1093/brain/awy105
Grundtman C, Bruton J, Yamada T, Ostberg T, Pisetsky DS, Harris HE, Andersson U, Lundberg IE, Westerblad H (2010) Effects of HMGB1 on in vitro responses of isolated muscle fibers and functional aspects in skeletal muscles of idiopathic inflammatory myopathies. FASEB J 24(2):570–578. https://doi.org/10.1096/fj.09-144782
Kuru S, Inukai A, Liang Y, Doyu M, Takano A, Sobue G (2000) Tumor necrosis factor-alpha expression in muscles of polymyositis and dermatomyositis. Acta Neuropathol 99(5):585–588
Lundberg I, Ulfgren AK, Nyberg P, Andersson U, Klareskog L (1997) Cytokine production in muscle tissue of patients with idiopathic inflammatory myopathies. Arthritis Rheum 40(5):865–874. https://doi.org/10.1002/art.1780400514
Efthimiou P (2006) Tumor necrosis factor-alpha in inflammatory myopathies: pathophysiology and therapeutic implications. Semin Arthritis Rheum 36(3):168–172. https://doi.org/10.1016/j.semarthrit.2006.07.003
Sultan SM, Ng KP, Edwards JC, Isenberg DA, Cambridge G (2008) Clinical outcome following B cell depletion therapy in eight patients with refractory idiopathic inflammatory myopathy. Clin Exp Rheumatol 26(5):887–893
Betteridge Z, McHugh N (2016) Myositis-specific autoantibodies: an important tool to support diagnosis of myositis. J Intern Med 280(1):8–23. https://doi.org/10.1111/joim.12451
Greenberg SA, Bradshaw EM, Pinkus JL, Pinkus GS, Burleson T, Due B, Bregoli L, O'Connor KC, Amato AA (2005) Plasma cells in muscle in inclusion body myositis and polymyositis. Neurology 65(11):1782–1787. https://doi.org/10.1212/01.wnl.0000187124.92826.20
Munoz-Beamud F, Isenberg DA (2013) Rituximab as an effective alternative therapy in refractory idiopathic inflammatory myopathies. Clin Exp Rheumatol 31(6):896–903
Levine TD (2005) Rituximab in the treatment of dermatomyositis: an open-label pilot study. Arthritis Rheum 52(2):601–607. https://doi.org/10.1002/art.20849
Limaye V, Hissaria P, Liew CL, Koszyka B (2012) Efficacy of rituximab in refractory antisynthetase syndrome. Intern Med J 42(3):e4–7. https://doi.org/10.1111/j.1445-5994.2011.02702.x
Noss EH, Hausner-Sypek DL, Weinblatt ME (2006) Rituximab as therapy for refractory polymyositis and dermatomyositis. J Rheumatol 33(5):1021–1026
Lopez J, Merino L, Piris L, Herrera FS, Llorente I, Humbria A, Ortiz AM, Velasco T, Garcia-Vicuna R, Castaneda S, Gonzalez Alvaro I, Munoz-Calleja C (2019) Rituximab induces a lasting, non-cumulative remodelling of the B-cell compartment. Clin Exp Rheumatol 37(4):615–622
Majmudar S, Hall HA, Zimmermann B (2009) Treatment of adult inflammatory myositis with rituximab: an emerging therapy for refractory patients. J Clin Rheumatol 15(7):338–340. https://doi.org/10.1097/RHU.0b013e3181bb8e70
Oddis CV, Reed AM, Aggarwal R, Rider LG, Ascherman DP, Levesque MC, Barohn RJ, Feldman BM, Harris-Love MO, Koontz DC, Fertig N, Kelley SS, Pryber SL, Miller FW, Rockette HE, Group RIMS (2013) Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: a randomized, placebo-phase trial. Arthritis Rheum 65(2):314–324. https://doi.org/10.1002/art.37754
de Visser M (2013) The efficacy of rituximab in refractory myositis: the jury is still out. Arthritis Rheum 65(2):303–306. https://doi.org/10.1002/art.37758
Aggarwal R, Oddis CV (2012) Inclusion body myositis: therapeutic approaches. Degener Neurol Neuromuscul Dis 2:43–52. https://doi.org/10.2147/DNND.S19899
Aggarwal R, Bandos A, Reed AM, Ascherman DP, Barohn RJ, Feldman BM, Miller FW, Rider LG, Harris-Love MO, Levesque MC, Group RIMS, Oddis CV (2014) Predictors of clinical improvement in rituximab-treated refractory adult and juvenile dermatomyositis and adult polymyositis. Arthritis Rheumatol 66(3):740–749. https://doi.org/10.1002/art.38270
Leclair V, Galindo-Feria AS, Dastmalchi M, Holmqvist M, Lundberg IE (2019) Efficacy and safety of rituximab in anti-synthetase antibody positive and negative subjects with idiopathic inflammatory myopathy: a registry-based study. Rheumatology (Oxford) 58(7):1214–1220. https://doi.org/10.1093/rheumatology/key450
Barsotti S, Cioffi E, Tripoli A, Tavoni A, D'Ascanio A, Mosca M, Neri R (2018) The use of rituximab in idiopathic inflammatory myopathies: description of a monocentric cohort and review of the literature. Reumatismo 70(2):78–84. https://doi.org/10.4081/reumatismo.2018.1011
Mahler EA, Blom M, Voermans NC, van Engelen BG, van Riel PL, Vonk MC (2011) Rituximab treatment in patients with refractory inflammatory myopathies. Rheumatology (Oxford) 50(12):2206–2213. https://doi.org/10.1093/rheumatology/ker088
Brulhart L, Waldburger JM, Gabay C (2006) Rituximab in the treatment of antisynthetase syndrome. Ann Rheum Dis 65(7):974–975. https://doi.org/10.1136/ard.2005.045898
Andersson H, Sem M, Lund MB, Aalokken TM, Gunther A, Walle-Hansen R, Garen T, Molberg O (2015) Long-term experience with rituximab in anti-synthetase syndrome-related interstitial lung disease. Rheumatology (Oxford) 54(8):1420–1428. https://doi.org/10.1093/rheumatology/kev004
Marie I, Dominique S, Janvresse A, Levesque H, Menard JF (2012) Rituximab therapy for refractory interstitial lung disease related to antisynthetase syndrome. Respir Med 106(4):581–587. https://doi.org/10.1016/j.rmed.2012.01.001
Zappa MC, Trequattrini T, Mattioli F, Rivitti R, Vigliarolo R, Marcoccia A, D'Arcangelo G (2011) Rituximab treatment in a case of antisynthetase syndrome with severe interstitial lung disease and acute respiratory failure. Multidiscip Respir Med 6(3):183–188. https://doi.org/10.1186/2049-6958-6-3-183
Torres C, Belmonte R, Carmona L, Gomez-Reino FJ, Galindo M, Ramos B, Cabello A, Carreira PE (2006) Survival, mortality and causes of death in inflammatory myopathies. Autoimmunity 39(3):205–215. https://doi.org/10.1080/08916930600622603
Saunders P, Tsipouri V, Keir GJ, Ashby D, Flather MD, Parfrey H, Babalis D, Renzoni EA, Denton CP, Wells AU, Maher TM (2017) Rituximab versus cyclophosphamide for the treatment of connective tissue disease-associated interstitial lung disease (RECITAL): study protocol for a randomised controlled trial. Trials 18(1):275. https://doi.org/10.1186/s13063-017-2016-2
So H, Wong VTL, Lao VWN, Pang HT, Yip RML (2018) Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol 37(7):1983–1989. https://doi.org/10.1007/s10067-018-4122-2
Tokunaga K, Hagino N (2017) Dermatomyositis with rapidly progressive interstitial lung disease treated with rituximab: a report of 3 cases in Japan. Intern Med 56(11):1399–1403. https://doi.org/10.2169/internalmedicine.56.7956
Chung L, Genovese MC, Fiorentino DF (2007) A pilot trial of rituximab in the treatment of patients with dermatomyositis. Arch Dermatol 143(6):763–767. https://doi.org/10.1001/archderm.143.6.763
Dinh HV, McCormack C, Hall S, Prince HM (2007) Rituximab for the treatment of the skin manifestations of dermatomyositis: a report of 3 cases. J Am Acad Dermatol 56(1):148–153. https://doi.org/10.1016/j.jaad.2006.05.068
Danko K, Ponyi A, Constantin T, Borgulya G, Szegedi G (2004) Long-term survival of patients with idiopathic inflammatory myopathies according to clinical features: a longitudinal study of 162 cases. Medicine (Baltimore) 83(1):35–42. https://doi.org/10.1097/01.md.0000109755.65914.5e
Touma Z, Arayssi T, Kibbi L, Masri AF (2008) Successful treatment of cardiac involvement in dermatomyositis with rituximab. Jt Bone Spine 75(3):334–337. https://doi.org/10.1016/j.jbspin.2007.05.011
Landon-Cardinal O, Allenbach Y, Soulages A, Rigolet A, Hervier B, Champtiaux N, Monzani Q, Sole G, Benveniste O (2019) Rituximab in the treatment of refractory anti-HMGCR immune-mediated necrotizing myopathy. J Rheumatol 46(6):623–627. https://doi.org/10.3899/jrheum.171495
Ashton C, Junckerstorff R, Bundell C, Hollingsworth P, Needham M (2016) Treatment and outcomes in necrotising autoimmune myopathy: an Australian perspective. Neuromuscul Disord 26(11):734–740. https://doi.org/10.1016/j.nmd.2016.08.013
Valiyil R, Casciola-Rosen L, Hong G, Mammen A, Christopher-Stine L (2010) Rituximab therapy for myopathy associated with anti-signal recognition particle antibodies: a case series. Arthritis Care Res (Hoboken) 62(9):1328–1334. https://doi.org/10.1002/acr.20219
Vordenbaumen S, Neuen-Jacob E, Richter J, Schneider M (2010) Inclusion body myositis in a patient with long standing rheumatoid arthritis treated with anti-TNFalpha and rituximab. Clin Rheumatol 29(5):555–558. https://doi.org/10.1007/s10067-009-1367-9
Rath E, Zwerina J, Oppl B, Nell-Duxneuner V (2015) Efficacy and safety of rituximab in rheumatic diseases. Wien Med Wochenschr 165(1–2):28–35. https://doi.org/10.1007/s10354-014-0331-8
Lutt JR, Pisculli ML, Weinblatt ME, Deodhar A, Winthrop KL (2008) Severe nontuberculous mycobacterial infection in 2 patients receiving rituximab for refractory myositis. J Rheumatol 35(8):1683–1685
Couderc M, Gottenberg JE, Mariette X, Hachulla E, Sibilia J, Fain O, Hot A, Dougados M, Euller-Ziegler L, Bourgeois P, Larroche C, Tournadre A, Amoura Z, Mazieres B, Arlet P, De Bandt M, Schaeverbeke T, Soubrier M (2011) Efficacy and safety of rituximab in the treatment of refractory inflammatory myopathies in adults: results from the AIR registry. Rheumatology (Oxford) 50(12):2283–2289. https://doi.org/10.1093/rheumatology/ker305
Belhassen-Garcia M, Rabano-Gutierrez A, Velasco-Tirado V, Romero-Alegria A, Perez-Garcia ML, Martin-Oterino JA (2015) Atypical progressive multifocal leukoencephalopathy in a patient with antisynthetase syndrome. Intern Med 54(5):519–524. https://doi.org/10.2169/internalmedicine.54.2748
Marie I, Guegan-Massardier E, Levesque H (2011) Progressive multifocal leukoencephalopathy in refractory polymyositis treated with rituximab. Eur J Intern Med 22(3):e13–14. https://doi.org/10.1016/j.ejim.2011.01.001
Schiffenbauer A, Garg M, Castro C, Pokrovnichka A, Joe G, Shrader J, Cabalar IV, Faghihi-Kashani S, Harris-Love MO, Plotz PH, Miller FW, Gourley M (2018) A randomized, double-blind, placebo-controlled trial of infliximab in refractory polymyositis and dermatomyositis. Semin Arthritis Rheum 47(6):858–864. https://doi.org/10.1016/j.semarthrit.2017.10.010
Chen D, Wang XB, Zhou Y, Zhu XC (2013) Efficacy of infliximab in the treatment for dermatomyositis with acute interstitial pneumonia: a study of fourteen cases and literature review. Rheumatol Int 33(10):2455–2458. https://doi.org/10.1007/s00296-012-2653-4
Hengstman GJ, De Bleecker JL, Feist E, Vissing J, Denton CP, Manoussakis MN, Slott Jensen H, van Engelen BG, van den Hoogen FH (2008) Open-label trial of anti-TNF-alpha in dermato- and polymyositis treated concomitantly with methotrexate. Eur Neurol 59(3–4):159–163. https://doi.org/10.1159/000114036
Dastmalchi M, Grundtman C, Alexanderson H, Mavragani CP, Einarsdottir H, Helmers SB, Elvin K, Crow MK, Nennesmo I, Lundberg IE (2008) A high incidence of disease flares in an open pilot study of infliximab in patients with refractory inflammatory myopathies. Ann Rheum Dis 67(12):1670–1677. https://doi.org/10.1136/ard.2007.077974
Muscle Study G (2011) A randomized, pilot trial of etanercept in dermatomyositis. Ann Neurol 70(3):427–436. https://doi.org/10.1002/ana.22477
Iannone F, Scioscia C, Falappone PCF, Covelli M, Lapadula G (2006) Use of etanercept in the treatment of dermatomyositis: a case series. J Rheumatol 33(9):1802–1804
Efthimiou P, Schwartzman S, Kagen LJ (2006) Possible role for tumour necrosis factor inhibitors in the treatment of resistant dermatomyositis and polymyositis: a retrospective study of eight patients. Ann Rheum Dis 65(9):1233–1236. https://doi.org/10.1136/ard.2005.048744
da Silva TC, Zon Pretti F, Shinjo SK (2013) Adalimumab in anti-synthetase syndrome. Jt Bone Spine 80(4):432. https://doi.org/10.1016/j.jbspin.2012.10.012
Park JK, Yoo HG, Ahn DS, Jeon HS, Yoo WH (2012) Successful treatment for conventional treatment-resistant dermatomyositis-associated interstitial lung disease with adalimumab. Rheumatol Int 32(11):3587–3590. https://doi.org/10.1007/s00296-011-2220-4
Wronski J, Fiedor P (2019) The safety profile of tumor necrosis factor inhibitors in ankylosing spondylitis: are TNF inhibitors safer than we thought? J Clin Pharmacol 59(4):445–462. https://doi.org/10.1002/jcph.1348
Dold S, Justiniano ME, Marquez J, Espinoza LR (2007) Treatment of early and refractory dermatomyositis with infliximab: a report of two cases. Clin Rheumatol 26(7):1186–1188. https://doi.org/10.1007/s10067-006-0325-z
Brunasso AM, Aberer W, Massone C (2014) New onset of dermatomyositis/polymyositis during anti-TNF-alpha therapies: a systematic literature review. Sci World J 2014:179180. https://doi.org/10.1155/2014/179180
Maundrell A, Proudman S, Limaye V (2019) Prevalence of other connective tissue diseases in idiopathic inflammatory myopathies. Rheumatol Int. https://doi.org/10.1007/s00296-019-04411-8
Ishikawa Y, Yukawa N, Kawabata D, Ohmura K, Fujii T, Usui T, Mimori T (2011) A case of antisynthetase syndrome in a rheumatoid arthritis patient with anti-PL-12 antibody following treatment with etanercept. Clin Rheumatol 30(3):429–432. https://doi.org/10.1007/s10067-010-1666-1
Allenbach Y, Guiguet M, Rigolet A, Marie I, Hachulla E, Drouot L, Jouen F, Jacquot S, Mariampillai K, Musset L, Grenier P, Devilliers H, Hij A, Boyer O, Herson S, Benveniste O (2015) Efficacy of Rituximab in refractory inflammatory myopathies associated with anti-synthetase auto-antibodies: an open-label phase II trial. PLoS ONE 10(11):e0133702. https://doi.org/10.1371/journal.pone.0133702
Sem M, Molberg O, Lund MB, Gran JT (2009) Rituximab treatment of the anti-synthetase syndrome: a retrospective case series. Rheumatology (Oxford) 48(8):968–971. https://doi.org/10.1093/rheumatology/kep157
Huber AM (2018) Update on the clinical management of juvenile dermatomyositis. Expert Rev Clin Immunol 14(12):1021–1028. https://doi.org/10.1080/1744666X.2018.1535901
Bader-Meunier B, Decaluwe H, Barnerias C, Gherardi R, Quartier P, Faye A, Guigonis V, Pagnier A, Brochard K, Sibilia J, Gottenberg JE, Bodemer C, Club Rhumatismes et I (2011) Safety and efficacy of rituximab in severe juvenile dermatomyositis: results from 9 patients from the French autoimmunity and rituximab registry. J Rheumatol 38(7):1436–1440. https://doi.org/10.3899/jrheum.101321
Riley P, McCann LJ, Maillard SM, Woo P, Murray KJ, Pilkington CA (2008) Effectiveness of infliximab in the treatment of refractory juvenile dermatomyositis with calcinosis. Rheumatology (Oxford) 47(6):877–880. https://doi.org/10.1093/rheumatology/ken074
Rouster-Stevens KA, Ferguson L, Morgan G, Huang CC, Pachman LM (2014) Pilot study of etanercept in patients with refractory juvenile dermatomyositis. Arthritis Care Res (Hoboken) 66(5):783–787. https://doi.org/10.1002/acr.22198
Lorenzetti R, Janowska I, Smulski CR, Frede N, Henneberger N, Walter L, Schleyer MT, Huppe JM, Staniek J, Salzer U, Venhoff A, Troilo A, Voll RE, Venhoff N, Thiel J, Rizzi M (2019) Abatacept modulates CD80 and CD86 expression and memory formation in human B-cells. J Autoimmun 101:145–152. https://doi.org/10.1016/j.jaut.2019.04.016
Tjarnlund A, Tang Q, Wick C, Dastmalchi M, Mann H, Tomasova Studynkova J, Chura R, Gullick NJ, Salerno R, Ronnelid J, Alexanderson H, Lindroos E, Aggarwal R, Gordon P, Vencovsky J, Lundberg IE (2018) Abatacept in the treatment of adult dermatomyositis and polymyositis: a randomised, phase IIb treatment delayed-start trial. Ann Rheum Dis 77(1):55–62. https://doi.org/10.1136/annrheumdis-2017-211751
Kerola AM, Kauppi MJ (2015) Abatacept as a successful therapy for myositis-a case-based review. Clin Rheumatol 34(3):609–612. https://doi.org/10.1007/s10067-014-2507-4
Musuruana JL, Cavallasca JA (2011) Abatacept for treatment of refractory polymyositis. Jt Bone Spine 78(4):431–432. https://doi.org/10.1016/j.jbspin.2011.03.022
Klotz L, Meuth SG, Wiendl H (2012) Immune mechanisms of new therapeutic strategies in multiple sclerosis-A focus on alemtuzumab. Clin Immunol 142(1):25–30. https://doi.org/10.1016/j.clim.2011.04.006
Coles AJ, Cox A, Le Page E, Jones J, Trip SA, Deans J, Seaman S, Miller DH, Hale G, Waldmann H, Compston DA (2006) The window of therapeutic opportunity in multiple sclerosis: evidence from monoclonal antibody therapy. J Neurol 253(1):98–108. https://doi.org/10.1007/s00415-005-0934-5
Ruck T, Bittner S, Kuhlmann T, Wiendl H, Meuth SG (2015) Long-term efficacy of alemtuzumab in polymyositis. Rheumatology (Oxford) 54(3):560–562. https://doi.org/10.1093/rheumatology/keu484
Thompson B, Corris P, Miller JA, Cooper RG, Halsey JP, Isaacs JD (2008) Alemtuzumab (Campath-1H) for treatment of refractory polymyositis. J Rheumatol 35(10):2080–2082
Dalakas MC, Rakocevic G, Schmidt J, Salajegheh M, McElroy B, Harris-Love MO, Shrader JA, Levy EW, Dambrosia J, Kampen RL, Bruno DA, Kirk AD (2009) Effect of alemtuzumab (CAMPATH 1-H) in patients with inclusion-body myositis. Brain 132(Pt 6):1536–1544. https://doi.org/10.1093/brain/awp104
Greenberg SA (2010) Comment on alemtuzumab and inclusion body myositis. Brain 133(Pt 5):e135. https://doi.org/10.1093/brain/awp275(author reply e136)
Pipitone N, Salvarani C (2013) CD25 blockade for refractory polymyositis. Clin Exp Rheumatol 31(3):474
Zou J, Li T, Huang X, Chen S, Guo Q, Bao C (2014) Basiliximab may improve the survival rate of rapidly progressive interstitial pneumonia in patients with clinically amyopathic dermatomyositis with anti-MDA5 antibody. Ann Rheum Dis 73(8):1591–1593. https://doi.org/10.1136/annrheumdis-2014-205278
Mathian A, Hie M, Cohen-Aubart F, Amoura Z (2015) Targeting interferons in systemic lupus erythematosus: current and future prospects. Drugs 75(8):835–846. https://doi.org/10.1007/s40265-015-0394-x
Guo X, Higgs BW, Rebelatto M, Zhu W, Greth W, Yao Y, Roskos LK, White WI (2014) Suppression of soluble T cell-associated proteins by an anti-interferon-alpha monoclonal antibody in adult patients with dermatomyositis or polymyositis. Rheumatology (Oxford) 53(4):686–695. https://doi.org/10.1093/rheumatology/ket413
Dhillon S (2014) Intravenous tocilizumab: a review of its use in adults with rheumatoid arthritis. BioDrugs 28(1):75–106. https://doi.org/10.1007/s40259-013-0076-8
Beaumel A, Muis-Pistor O, Tebib JG, Coury F (2016) Antisynthetase syndrome treated with tocilizumab. Jt Bone Spine 83(3):361–362. https://doi.org/10.1016/j.jbspin.2015.03.016
Murphy SM, Lilleker JB, Helliwell P, Chinoy H (2016) The successful use of tocilizumab as third-line biologic therapy in a case of refractory anti-synthetase syndrome. Rheumatology (Oxford) 55(12):2277–2278. https://doi.org/10.1093/rheumatology/kew296
Kondo M, Murakawa Y, Matsumura T, Matsumoto O, Taira M, Moriyama M, Sumita Y, Yamaguchi S (2014) A case of overlap syndrome successfully treated with tocilizumab: a hopeful treatment strategy for refractory dermatomyositis? Rheumatology (Oxford) 53(10):1907–1908. https://doi.org/10.1093/rheumatology/keu234
Narazaki M, Hagihara K, Shima Y, Ogata A, Kishimoto T, Tanaka T (2011) Therapeutic effect of tocilizumab on two patients with polymyositis. Rheumatology (Oxford) 50(7):1344–1346. https://doi.org/10.1093/rheumatology/ker152
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Thomas Khoo declares that he/she has no conflict of interest. Vidya Limaye declares that he/she has no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
No part of this article has been copied or published elsewhere in whole or in part.
Rights and permissions
About this article

Cite this article
Khoo, T., Limaye, V. Biologic therapy in the idiopathic inflammatory myopathies. Rheumatol Int 40, 191–205 (2020). https://doi.org/10.1007/s00296-019-04467-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00296-019-04467-6