
Abstract
Cellulase production in filamentous fungi is repressed by various carbon sources. In our preliminary survey in Aspergillus nidulans, degree of de-repression differed depending on carbon sources in a mutant of creA, encoding the transcriptional repressor for carbon catabolite repression (CCR). To further understand mechanisms of CCR of cellulase production, we compared the effects of creA deletion with deletion of protein kinase A (pkaA) and G (ganB) genes, which constitute a nutrient sensing and signaling pathway. In plate culture with carboxymethyl cellulose and d-glucose, deletion of pkaA and ganB, but not creA, led to significant de-repression of cellulase production. In submerged culture with cellobiose and d-glucose or 2-deoxyglucose, both creA or pkaA single deletion led to partial de-repression of cellulase genes with the highest level by their double deletion, while ganB deletion caused de-repression comparable to that of the creA/pkaA double deletion. With ball-milled cellulose and d-glucose, partial de-repression was detected by deletion of creA but not of pkaA or ganB. The creA/pkaA or creA/ganB double deletion led to earlier expression than the creA deletion. Furthermore, the effect of each deletion with d-xylose or L-arabinose as the repressing carbon source was significantly different from that with d-glucose, d-fructose, and d-mannose. Consequently, this study revealed that PkaA and GanB participate in CreA-independent CCR and that contribution of CreA, PkaA, and GanB in CCR differs depending on the inducers, repressing carbon sources, and culture conditions (plate or submerged). Further study of CreA-independent mechanisms is needed to fully understand CCR in filamentous fungi.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Microorganisms possess a carbon catabolite repression (CCR) system, which ensures preferential utilization of easily metabolizable carbon sources such as d-glucose by preventing the utilization of difficult to metabolize carbon sources. As a result, the CCR system determines the hierarchy of carbon substrate utilization in filamentous fungi, which are known to utilize a huge variety of carbon substrates. A number of genes involved in the carbon catabolism, including industrially important carbohydrate-degrading-enzymes such as cellulase, amylase, and xylanase are regulated by CCR. The transcriptional repressor that mediates CCR in Aspergillus nidulans is CreA (Dowzer and Kelly 1989), with orthologues such as CRE-1 in Neurospora crassa (de la Serna et al. 1999) and CRE1 in Trichoderma reesei (Strauss et al. 1995). CreA has a C2H2-type DNA-binding motif and directly binds to 5′-SYGGRG-3′ on the promoters of target genes to prevent transcription (Cubero and Scazzocchio 1994). CreA activity is regulated according to nuclear/cytoplasmic trafficking and stability. That is, CreA is excluded from the nucleus under de-repressing conditions, and is degraded in the cytoplasm (Brown et al. 2013; de Assis et al. 2018; Tanaka et al. 2018). The protein kinases SnfA and SchA are involved in the exclusion of CreA from the nucleus (Brown et al. 2013), while the cAMP-dependent protein kinase (PKA) encoded by pkaA counteracts SnfA function (de Assis et al. 2015).
CreA directly and indirectly regulates gene expression. It represses the expression of the xlnR gene encoding the major transcriptional activator of the xylanolytic enzyme genes in A. niger, such that the genes regulated by XlnR, including the xylanase genes xlnA and xlnB, are downregulated (Orejas et al. 2001; Tamayo et al. 2008). In the case of proline utilization, CreA directly represses prnB expression, which encodes the proline permease. This causes inducer exclusion, such that the expression of prnC and prnD genes remains at a low level, even in the presence of proline (Cubero et al. 2000).
The CreA-dependent system described above is not the sole system governing CCR. The transcription of A. nidulans α-L-rhamnosidase genes rhaA and rhaE is repressed and the uptake of the inducer molecule L-rhamnose is inhibited by d-glucose even in the creAd30 mutant (Tamayo-Ramos et al. 2012). Uptake of phenylacetate has also been reported to be inhibited by d-glucose in a CreA-independent manner (Fernändez-Canón and Luengo 1997). A recent report found that an Arrestin-like protein CreD and a ubiquitin ligase HulA, identified via the characterization of the suppressor mutants of the creB and creC mutations (Boase and Kelly 2004), regulate d-glucose-induced endocytosis of the maltose transporter MalP, thus affecting CCR of α-amylase genes in A. oryzae (Hiramoto et al. 2015). CreB encodes a putative deubiquitinase and CreC encodes a WD40 protein, which interact with each other (Boase et al. 2003; Lockington and Kelly 2002). Although CreA was initially suggested to be a possible target of CreB, a recent study denied such possibility based on the result that CreA was not ubiquitinated (Alam el al. 2017). In fact, mutation or deletion of creB does not affect CreA stability in A. nidulans or A. oryzae (Ries et al. 2016; Tanaka et al. 2018), while a mutation in CreC has been found to lead to the destabilization of CreA (Ries et al. 2016). The creA and creB double deletion mutant has been found to lead to a higher production of α-amylase, xylanase, and β-glucosidase activity than single creA or creB deletion mutants in A. oryzae (Ichinose et al. 2014, 2018). These reports point towards the presence of a CreB-dependent but CreA-independent CCR. Alternatively, CreB might target a CreA-interacting protein that is required for function of CreA (Alam and Kelly 2017).
Although many of the abovementioned studies used d-glucose as a representative repressing substrate, many other carbon substrates also show repressing activity at varying strengths. Moreover, the level of repression exerted by certain repressing substrates can differ from gene to gene. Currently, no concrete regulatory mechanisms are known to determine the hierarchy of carbon source utilization, indicating that our understanding of CCR is still not satisfactory. In this study, we compared the effects on CCR of cellulase genes caused by the deletion of creA and pkaA, as well as of ganA, ganB, and fadA, which encode Gα, a subunit of the trimeric G protein, in the presence of various carbon substrates. Our results demonstrated that CreA, PkaA, and GanB play distinct roles in CCR depending on the repressing carbon sources as well as the growth conditions.
Materials and methods
Strains and media
The A. nidulans strains used in this study are listed in Table S1. ABPU1 was the parent strain of all the deletion mutants. ABU was generated by introduction of the wild-type pyroA gene into ABPU1. The creA and pkaA single deletion strains (ΔcreA and ΔpkaA, respectively) were constructed by replacing the genes with A. nidulans pyroA (Fig. S1A and B). The creA and pkaA double deletion strain ΔcreA/ΔpkaA was constructed using the marker recycling strategy (Akada et al. 2006); first, the pkaA gene in the ΔcreA strain was deleted using a deletion cassette specially designed for marker recycling, with A. oryzae pyrG as a selectable marker, then the pyrG gene was eliminated by selecting 5-fluoroorotic acid (FOA)-resistant strains (Fig. S2A). The ganA, ganB, and fadA single deletion strains (ΔganA, ΔganB, and ΔfadA, respectively) were also constructed from ABPU1 using the marker recycling strategy (Fig. S2B, C, D). The ΔcreA/ΔganB double deletion strain was constructed from the ΔganB strain by the marker recycling strategy using A. oryzae pyrG as a selectable marker (Fig. S2E).
Plasmids carrying the deletion cassettes were constructed by joining PCR-generated DNA fragments via the conventional method using restriction enzymes and DNA ligase for the pkaA single deletion and via the seamless cloning technique for the rest using the GeneArt™ Seamless Cloning and Assembly kit (Thermo fisher Scientific, Waltham, MA, USA) or the In-Fusion® HD Cloning Kit (Takara, Shiga, Japan). The primers used for the construction of the plasmids are listed in Table S2. Plasmid construction was performed using Escherichia coli DH5α and XL1-Blue strains. Plasmids for complementation of the deletions were also constructed via the seamless cloning technique.
Transformation of A. nidulans was carried out by protoplast transformation (balance and Turner 1985), with slight modifications, as previously described (Makita et al. 2009). Deletion of the target genes was first examined by PCR (data not shown), and then confirmed by Southern blot analysis (Figs. S1 and S2). The presence of the re-introduced genes in the complementary strains was confirmed by PCR. A. nidulans strains were grown at 37 °C in the standard minimal medium (MM) with the appropriate supplements to fulfill auxotrophy, unless otherwise stated (Rowlands and Turner 1973).
Plate assay of amylase and cellulase activity
An aliquot of suspension containing 104 conidia of each A. nidulans strain was spotted onto agar plates of the standard minimal medium containing 0.1% TritonX-100 with 1% starch or 0.5% carboxymethylcellulose (CMC). In addition, each repressing monosaccharide was added at a final concentration of 1%. After incubation at 37 °C for 3 days, amylase activity was visualized using an iodine test. Cellulase activity was visualized by staining with 0.1% Congo red followed by de-staining with 0.7 M NaCl.
Transcriptional analysis
A. nidulans strains were grown in MM containing 1% Bacto™ Peptone (Becton Dickinson, Franklin Lakes, NJ, USA) and 0.1% Bacto™ Yeast Extract (Becton Dickinson) instead of d-glucose for 22 h at 37 °C. The mycelia were collected and washed with MM without carbon sources. The mycelia were transferred into fresh MM containing 3 mM cellobiose and 50 µM 1-deoxynojirimycin (DNJ) with or without repressing carbon substrates at 2.5 mM for 2-deoxyglucose (2DG) and 30 mM for hexoses and pentoses, then cultivated at 37 °C for 1.5 h. DNJ was added to prevent the hydrolysis of cellobiose. When ball-milled cellulose (BMC) was used, the mycelia were transferred into fresh MM containing 0.5% BMC with or without d-glucose at 1%, and cultivated for 6 and 12 h. The BMC used in this study was prepared from KC flock W-300G (Nippon Paper Industries, Tokyo, Japan) by wet ball milling. The average degree of polymerization was 190, which was calculated by dividing the total sugars by the reducing sugars. Total and reducing sugar concentrations were determined by the phenol–sulfuric acid method and Somogyi–Nelson method, respectively (Dubois et al. 1951; Somogyi 1952). The mycelia were harvested, frozen in liquid nitrogen, and ground into a fine powder using an SK mill SK-100 (Tokken, Chiba, Japan). RNA extraction was carried out as previously described (Kunitake et al. 2016). Genomic DNA removal and cDNA synthesis were carried out using ReverTra Ace® qPCR RT Master Mix with gDNA Remover (Toyobo, Osaka, Japan). Quantitative PCR was performed as previously described (Kunitake et al. 2016) using the THUNDERBIRD SYBR qPCR Mix (Toyobo) with the StepOnePlus™ Real-time PCR system (Thermo Fisher Scientific). The primers used for qPCR are listed in Table S3.
Zymography of cellulase activity
After cultivation with BMC as the inducer, 15 µl of culture supernatant was subjected to 12% native-PAGE containing 0.1% CMC. The gels were then rinsed with 50 mM sodium phosphate buffer (pH 6.5) three times and incubated in the same buffer for 3 h at 37 °C. The protein bands containing cellulase activity were visualized by staining with 0.1% Congo-Red for 30 min, followed by de-staining with 1 M NaCl.
Results
PkaA and Gα-mediated but CreA-independent CCR
Screening of the protein kinase deletion library of A. nidulans, generated by De Souza et al. (De Souza et al. 2013a), by plate assay for cellulase production revealed that the deletion of pkaA, encoding protein kinase A, led to a significant de-repression of cellulase production with carboxymethylcellulose (CMC) plus d-glucose as the carbon sources. PkaA is partially involved in CCR by counteracting SnfA that promotes exclusion of CreA from the nucleus (de Assis et al. 2015). However, the creA204 mutation, which possesses an amino acid substitution in the DNA-binding domain of CreA (Shroff et al. 1996), did not cause such de-repression of cellulase production in our preliminary studies (data not shown). This suggested that PkaA is involved in CreA-independent CCR. To obtain further evidence of a PkaA-dependent and CreA-independent CCR, amylase and cellulase production in the creA, pkaA, and creA/pkaA knockouts, derived from the same parent strain ABPU1, was compared by the plate assay with d-glucose as the repressing carbon source. In addition, the knockouts of the genes ganA, ganB, and fadA, encoding the trimeric G-protein α subunit (Gα), were examined considering that protein kinase A is generally under the control of Gα in eukaryotes. The ΔcreA and ΔpkaA strains grew poorly on starch, d-glucose, CMC, starch plus d-glucose, and CMC plus d-glucose, where the double knockout ΔcreA/ΔpkaA exhibited the slowest growth (Fig. 1a).

Growth (a) and amylase and cellulase production (b) of the deletion strains on agar plates containing starch or CMC with d-glucose. Positions of strains are shown in (c)
Despite the growth defect, the ΔcreA, ΔcreA/ΔpkaA, and ΔganB/ΔcreA strains were found to have extremely higher amylase activity, based on the sizes of the clear zones (halos) around their colonies, than the other strains on starch and starch plus d-glucose, while such an elevated production was not observed in the ΔpkaA strain (Fig. 1b). Consequently, PkaA does not participate in the CCR of amylase, or if it does, only plays a minor role. In contrast, the effects of the creA, pkaA, and ganB deletions on cellulase production were completely different from those on amylase production. High cellulase production in the presence of d-glucose was observed in the ΔpkaA, ΔganB, ΔcreA/ΔpkaA, and ΔcreA/ΔganB strains, but not in the ΔcreA strain (Fig. 1b). The ΔcreA/ΔpkaA and ΔcreA/ΔganB strains produced slightly higher cellulase activity than the ΔpkaA and ΔganB strains, despite that the double deletants grew more poorly than the single deletants. Defects in CCR of amylase or cellulase production were recovered by reintroduction of the creA, pkaA, and ganB genes into the deletants (Fig. S3). As a single exception, trial to reintroduce creA into the ΔcreA/ΔganB strain failed because protoplasts were not formed.
Consequently, CCR of amylase production is CreA dependent, PkaA independent, and partially GanB dependent while CCR of cellulase production is PkaA and GanB dependent, but surprisingly almost CreA independent. De-repression of the cellulase activity by the ganB deletion suggests that the extracellular d-glucose signal sensed by G-protein-coupled receptors (GPCRs) is mainly transmitted to GanB, but not to GanA or FadA.
Effects of deletion of creA, pkaA, and Gα genes on CCR of cellulase genes in submerged culture
Although cellulase production in the presence of d-glucose was not de-repressed in the ΔcreA strain, the ΔcreA/ΔpkaA and ΔcreA/ΔganB strains produced slightly higher cellulase activity than the ΔpkaA and ΔganB strains (Fig. 1b). This suggests that CreA plays a minor role in CCR of cellulase production, but we could not conclude this because plate assay is not quantitative enough. To obtain quantitative data, we investigated expression of cellulase genes by RT-qPCR in submerged culture with cellobiose as an inducer with 2-deoxyglucose (2DG) as a repressing carbon source.
In the pilot experiments, we determined experimental conditions for cellobiose induction and 2DG repression. When induced with 3 mM cellobiose for 1.5 h, the expression of eglA and cbhA, encoding an endoglucanase and a cellobiohydrolase, was found to be several fold higher in the ΔcreA and ΔpkaA strains compared to that of the reference strain ABU (creA+pkaA+), even without d-glucose addition (Fig. S4). The low-level expression in ABU was recovered by the addition of 50 µM 1-deoxynojirimycin (DNJ), an inhibitor of β-glucosidase, indicating that d-glucose released from cellobiose represses cellulase gene expression (Fig. S4). Therefore, DNJ was added in all subsequent transcriptional analyses using cellobiose as the inducer. In addition, a non-metabolizable d-glucose analog, 2DG, was used as a repressing carbon substrate. When cellobiose, DNJ, and various concentrations of 2DG were added, repression levels of eglA and cbhA were not significantly different between 0.5 mM and 2.5 mM 2DG in the ABU strain, as well as in the ΔcreA and ΔpkaA strains (Fig. S5). Based on these results, further experiments were performed using 3 mM cellobiose, 50 µM DNJ, and 2.5 mM 2DG.
The addition of 2DG caused a remarkable decrease in the expression levels of eglA and cbhA not only in ABU but also in the ΔcreA, ΔpkaA, ΔcreA/ΔpkaA, and ΔganB strains to different degrees (Fig. 2a, b). The differences in their expression levels without 2DG were not negligible; the expression of cbhA in the ΔcreA/ΔpkaA strain was 3.3-fold higher than in the reference strain (Fig. 2b). Considering such a difference in the absence of 2DG, the expression of eglA and cbhA in the presence of 2DG relative to that in its absence in each strain was compared to evaluate the degree of de-repression caused by each deletion (Fig. 2c, d). The expression of eglA and cbhA in ABU in the presence of 2DG was only 0.6% and 0.7%, respectively, compared to that without 2DG. The relative expression of the genes in the presence of 2DG increased to 11% and 8% in the ΔcreA strain, 17% and 20% in the ΔpkaA strain, and 20% and 19% in the ΔcreA/ΔpkaA strain. As for the Gα genes, the expression of eglA and cbhA in the presence of 2DG was 1% and 2% in both the ΔganA and ΔfadA strains, 25% and 34% in the ΔganB strain, and 11% and 14% in the ΔcreA/ΔganB strain. Additionally, the increase in the de-repression levels in the ΔcreA/ΔpkaA strain compared to the ΔcreA strain (p = 0.0698 for eglA, and 0.0009 for cbhA by the Welch’s t test) was consistent with the observation in the plate assay (Fig. 1b). De-repression level of the genes was the highest in the ΔganB strain, and unexpectedly, the level decreased in the ΔcreA/ΔganB strain. As to creA, pkaA, and ganB, the deletions did not significantly affect expression of the undeleted genes (Fig. S6).

Expression of eglA (a, c) and cbhA (b, d), evaluated by RT-qPCR, under 3 mM cellobiose-induced conditions with or without 2.5 mM 2-DG. a, b Relative expression levels of the genes were normalized with actA and shown by a logarithmic scale. White bar, cellobiose; black bar, cellobiose with 2DG. c, d Expression ratio in the presence of 2DG compared to in its absence. Error bars indicate the standard errors of more than three biological independent experiments. Asterisks indicate statistically significant differences (*P < 0.05, **P < 0.01, ***P < 0.001, Welch’s t test)
Although it was not obvious whether CreA is involved in CCR of cellulase production in the plate assay (Fig. 1b), transcriptional analysis clearly revealed its involvement. However, considering that deletion of pkaA or ganB caused stronger de-repression than that of creA, our results here again confirmed the presence of CreA-independent CCR of the cellulase genes.
Effects of the creA, pkaA, and ganB deletions on CCR under ball-milled cellulose-induced conditions
In the above transcriptional analyses, the inducer of the expression of the cellulase genes used was cellobiose, which is produced by endoglucanases and cellobiohydrolases from cellulose. When only cellulose is present, the enzymes produced at a basal level and/or induced by starvation are thought to produce cellobiose to boost cellulase expression. Thus, the effects of creA and/or pkaA deletion on cellulose induction may differ from cellobiose induction. To assess this issue, the effects of the deletions on the production of endoglucanases (EglA and EglB) and the expression of eglA and cbhA were examined by zymography and transcriptional analysis using 0.5% ball-milled cellulose (BMC) as the inducer and 1% d-glucose as the repressing carbon substrate. BMC was prepared from crystalline cellulose by grinding with ceramic ball, resulting in segmentation of the polymers. Its average degree of polymerization was 190. As shown in Fig. 3a, cellulase production in the absence of d-glucose did not significantly differ among the ABU, ΔcreA, ΔpkaA, and ΔganB strains, while that in the ΔcreA/ΔpkaA strain was clearly elevated at 6 h of induction. The lower activity band represents EglA and the upper two bands represent EglB in differently glycosylated forms (Chikamatsu et al. 1999; Endo et al. 2008). In the presence of d-glucose, endoglucase activity was not detected in the ABU, ΔpkaA, and ΔganB strains, but was detected in the ΔcreA strain at 12 h. In the ΔcreA/ΔpkaA and ΔcreA/ΔganB strains, the activity was detected at 6 h and further increased at 12 h.

Cellulase production and eglA and cbhA expression under 0.5% BMC-induced conditions with or without 1% d-glucose. Each strain was cultivated in MM plus BMC with or without d-glucose for 6 h and 12 h. Culture supernatant (15 µl) of each deletion strain was subjected to zymography (a), and total RNA extracted from the mycelia was subjected to RT-qPCR analysis (b). White bar, BMC for 6 h; light gray bar, BMC for 12 h, dark gray bar, BMC + d-glucose for 6 h; black bar, BMC + d-glucose for 12 h
The expression of eglA and cbhA exhibited very similar results; their expression was significantly repressed by d-glucose in the ABU, ΔpkaA, and ΔganB strains, while it was detected at 12 h in the ΔcreA strain and at 6 h with an increased production at 12 h in the ΔcreA/ΔpkaA and ΔcreA/ΔganB strains (Fig. 3b). Although de-repression from CCR was not observed in the ΔpkaA and ΔganB strains, the CreA-independent participation of PkaA and GanB in CCR is evident considering the earlier expression of eglA and cbhA, as well as the earlier production of endoglucanases in the ΔcreA/ΔpkaA and ΔcreA/ΔganB strains compared to the ΔcreA strain. It should be noted that the effects of the creA, pkaA, and ganB deletion on the cellulase expression in the submerged culture were the opposite of those in the plate assay (Figs. 1b, 3a).
Effects of the creA, pkaA, and ganB deletions on CCR with various carbon sources
Not only d-glucose but also other carbon sources repress the expression of cellulase genes. Among the monosaccharides examined in this study, d-fructose, d-mannose, and d-xylose were found to cause a strong repression of cellulase production in the reference strain ABU, and d-galactose and L-arabinose were found to be weakly repressing carbon sources (Fig. 4b). The effects of the deletion of the creA, pkaA, creA/pkaA, and Gα genes on the repression of cellulase production and cellulase gene expression were examined, since these genes may be involved in CCR via different mechanisms depending on the carbon sources used. Growth of the deletion mutants on various carbon sources was similar to that on d-glucose, as shown in Fig. 1a. A single exception was the ΔpkaA and ΔganB strains in which little growth was observed on both d-galactose and CMC plus d-galactose. Interestingly, the growth defect on d-galactose was recovered to a level comparable to that of other carbon sources in the ΔcreA/ΔpkaA strain. Furthermore, the growth defect of the ΔcreA/ΔganB strain was hardly observed on both d-galactose and CMC plus d-galactose. These results suggest that the cAMP signaling pathway plays an essential role in the d-galactose catabolism.

Growth on (a) and repression of cellulase production by (b) various monosaccharides. Strains were cultured on MM agar plates containing indicated sugars as carbon sources. Positions of strains are shown in c
The cellulase production of the deletion mutants on the plates containing various monosaccharides revealed a striking difference from the data with d-glucose shown in Fig. 1b, where de-repression was observed on all the monosaccharides by the creA deletion. Additive de-repression by double deletion of creA and pkaA as well as creA and ganB was evident on d-fructose and d-mannose, while cellulase production in ΔcreA, ΔcreA/ΔpkaA, and ΔcreA/ΔganB was comparable to each other on d-xylose and L-arabinose (Fig. 4b). These observations imply that CCR caused by pentoses might be different from that by hexoses, and that contribution of PkaA is very minor or negligible on pentoses. The pkaA deletion led to a weakly de-repressed production of cellulase on all the monosaccharides, while the ganB deletion led to stronger de-repression than the pkaA deletion. In addition, the fadA deletion led to a weak de-repression on d-xylose. Due to the extremely impaired growth of the ΔpkaA and ΔganB strains, de-repression on d-galactose could not be evaluated.
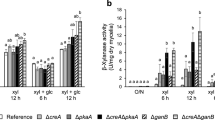
The effects of various repressing monosaccharides on the expression of eglA and cbhA were examined in the submerged culture with cellobiose as the inducer (Fig. 5a, b). d-glucose, d-fructose, and d-mannose appeared to be strong repressing carbon sources in the reference strain ABU, and the ΔcreA strain was found to exhibit a higher expression of the cellulase genes than the ΔpkaA strain in the presence of hexoses. Compared to the hexoses, the pentoses d-xylose and L-arabinose were weaker repressing carbon sources in ABU. The deletion of creA led to the highest levels of expression, while the effects of the pkaA deletion were very minor on d-xylose, with no changes observed on L-arabinose.

Transcriptional analysis of eglA (a, c) and cbhA (b, d) on 3 mM cellobiose-induced conditions in the presence of various repressing monosaccharides (30 mM). The data without the repressing carbon sources are the same as those used in Fig. 2. a, b Relative expression levels normalized with actA. White bar, cellobiose; black bar, cellobiose + d-glucose; dark gray bar, cellobiose + d-fructose; light gray bar, cellobiose + d-mannose; shaded bar, cellobiose + d-xylose; horizontal striped bar, cellobiose + L-arabinose. c, d Expression ratio in the presence of a repressing monosaccharide compared to in its absence. White bar, ABU; black bar, ΔcreA; dark gray bar, ΔpkaA; light gray bar, ΔcreAΔpkaA; shaded bar, ΔganB; horizontal striped bar, ΔfadA; dotted bar, ΔcreAΔganB. Error bars indicate the standard errors of three biological independent experiments. Asterisks indicate statistically significant differences (*P < 0.05, **P < 0.01, ***P < 0.001, Welch’s t test)
The expression levels in the presence of the monosaccharides relative to those in their absence were compared (Fig. 5c, d), which revealed significant differences between CCR by the hexoses and by the pentoses. In the case of CCR by the hexoses, the effects of the creA and pkaA deletions were additive, leading to higher expression levels of eglA and cbhA in the ΔcreA/ΔpkaA strain than in the ΔcreA and ΔpkaA strains, although this additive action was not very evident in eglA expression upon d-mannose addition, which was the only exception. The ΔganB and ΔcreA/ΔganB strains also exhibited the expression of the genes to a greater extent than the ΔcreA and ΔpkaA strains. On the other hand, the deletion of creA led to an extremely high expression of the genes in the presence of the pentoses, which exceeded the expression level in the absence of the repressing carbon sources. The ganB deletion was found to cause de-repression: however, the degree was much lower than in the creA deletion. Consequently, CCR by the hexoses appeared to be mediated by the additive actions of CreA and PkaA, while that by the pentoses was mainly CreA-mediated. Furthermore, a higher de-repression was found in the ΔganB strain than the ΔpkaA strain on the pentoses, implying that GanB has a role in addition to PkaA regulation in cellulase gene expression.
Discussion
The expression of genes related to the utilization of carbon in filamentous fungi is induced in the presence of an inducer, while this expression is repressed by CCR if easily metabolizable carbon substrates co-exist with the inducer. The repressing activity differs depending on the carbon substrates, among which d-glucose and d-xylose are known to be the strongest. This suggests that CCR plays an important role in determining the order of carbon sources to be utilized in the conditions where various carbon substrates are present, as is often the case in natural environments. CCR is regulated by CreA-dependent as well as CreA-independent mechanisms in Aspergillus, as described in the introduction. In this study, we provided evidence of the involvement of PkaA and GanB in CreA-independent CCR by observing the effects of deletions of genes encoding the proteins on the production and gene expression of cellulases.
PkaA partially regulates nuclear-cytoplasmic shuttling of CreA by counteracting SnfA, which directly phosphorylates CreA in A. nidulans (Brown et al. 2013; de Assis et al. 2018). If the role of PkaA in CCR is limited to this function, de-repression caused by the pkaA deletion should be weaker than that by the creA deletion. However, the production of cellulase on the plate culture was significantly de-repressed from d-glucose repression as a result of pkaA and ganB deletions, but not by creA deletion (Fig. 1), indicating that PkaA and GanB participate CreA-independently in CCR in the case of the cellulase production. GanB is the Gα subunit of trimeric G protein, which generally responds to extracellular signals sensed by GPCRs and regulates adenylate cyclase, which produces cAMP. In contrast, the CreA-dependent system is thought to respond to intracellular d-glucose as the CreA orthologue Mig1 in S. cerevisiae is regulated by hexokinase 2, which is reported to be an intracellular d-glucose sensor (Vega et al. 2016). CCR of cellulase genes in A. nidulans may be regulated by a double lock system caused by both extracellular and intracellular d-glucose. It should be noted that the effects of the creA, pkaA, and ganB deletions were the opposite in the production of amylase. This suggests that GanB/PkaA-mediated CCR specifically functions for the regulation of cellulase genes.
The expression of eglA and cbhA in the submerged culture also confirmed the differential contribution of CreA and PkaA in CCR, considering that the highest level of de-repression of eglA and cbhA expression as well as cellulase production was observed in the ΔcreA/ΔpkaA strain compared to the single deletion strains (Figs. 2, 3, 5). However, while de-repression as a result of the creA deletion was not observed in the plate culture (Fig. 1), it was detected in the submerged culture with cellobiose plus 2DG (Fig. 2), with BMC plus d-glucose (Fig. 3), and with cellobiose plus d-glucose (Fig. 5). These findings suggest that the degree of contribution of CreA and PkaA changes depending on the cultural conditions. De-repression due to pkaA and ganB deletions was detected with cellobiose plus d-glucose (Fig. 5) but not with BMC plus d-glucose (Fig. 2). De-repression of eglA and cbhA in the ΔganB strain was the most marked. While full and 72% de-repression of eglA and cbhA, respectively, occurred with cellobiose plus d-glucose (Fig. 5), the expression was found to be completely repressed with BMC plus d-glucose (Fig. 3). Although we were unable to identify the possible causes of this difference in the literature, the creA but not ganB deletion might have led to an increased basal-level production of cellulases required for the production of the physiological inducer cellobiose from BMC.
The ΔganB strain exhibited approximately 6.1-fold and 3.4-fold higher de-repression of eglA and cbhA (p = 0.003 and 0.005, respectively, by Welch’s t test) compared to the ΔpkaA strain in the cellobiose plus d-glucose culture (Fig. 5). The higher level of de-repression implies that the role of GanB in CCR is not limited to the regulation of PkaA activity. The ganB deletion may affect the phospholipase C-dependent signaling pathway, which is also known to be under the regulation of trimeric G protein in eukaryotes, although the involvement of this phospholipase in cellulase regulation has not been elucidated in filamentous fungi. Another possible pathway may be GanB to FbxA via cAMP. FbxA is an F-box protein, which are known to be involved in ubiquitination, and has cAMP-binding motifs. This protein is required for the full expression of xylanase genes in A. nidulans and has been previously suggested to be involved in CCR (Colabardini et al. 2012).
Catabolite repression is caused not only by d-glucose but also by various other carbon substrates. As shown in Fig. 5, CreA, PkaA, and GanB are clearly involved in the CCR caused by hexoses. On the other hand, full de-repression was observed in the ΔcreA strain by pentoses. The contribution by PkaA was either very weak or nonexistent. The significant effect caused by the creA deletion may be at least partially due to the XlnR-dependent activation of cellulase gene expression, since XlnR participates in cellulase gene expression in Aspergillus species (Gielkens et al. 1999; Marui et al. 2002; Noguchi et al. 2009; van Peij et al. 1998), and is activated not only by d-xylose, but also by commercially available L-arabinose (de Souza et al. 2013b; Ishikawa et al. 2018; Noguchi et al. 2011). Interestingly, GanB was found to be involved in CCR caused by all the monosaccharides examined (Fig. 5). However, the contribution of PkaA was very small on pentoses. This is consistent with previous findings that conidial germination was impaired on various carbon sources in a ganB mutant (Lafon et al. 2005), implying that hexoses and pentoses were sensed by the GPCRs which exist upstream of GanB. There are 16 genes encoding GPCRs in A. nidulans. However, monosaccharide-sensing by GPCRs are not well characterized, except for some reports which state that the GprH of A. nidulans is a possible d-glucose sensor (Brown et al. 2015; Lafon et al. 2005). Further studies on the ligand–receptor relationship in GPCRs may increase our understanding of CCR. Possible signaling pathways in CCR, based on this study and literatures, are illustrated in Fig. 6.

Model for CCR of cellulase genes mediated by CreA, PkaA, and GanB. In the presence of a hexose such as d-glucose (left), sugar recognition by GPCR causes activation of cAMP synthesis via GanB. PkaA activated by cAMP indirectly represses cellulase gene expression by regulating an unknown factor and SnfA. In the presence of a pentose such as d-xylose (right), CreA mainly functions in CCR, but GanB-dependent, PkaA-independent system is also involved. Possible pathway for the GanB-dependent system may include FbxA, a protein with a cAMP-binding motif. The dashed lines represent hypothetical pathways
In conclusion, this study revealed that CreA, PkaA, and GanB have distinct functions in CCR. This finding provides an insight into the mechanisms determining the hierarchy of carbon source utilization in filamentous fungi, where the identification of the factors upstream and downstream of GanB and PkaA is crucial to improve our understanding of the tactics employed by A. nidulans for its survival in nature. From a biotechnological point of view, the double knockout of creA and pkaA exhibited the best performance in cellulase gene expression; however, the resulting significantly impaired growth of the strain would be a limitation for industrial application. Further research on CCR would be needed to construct a CCR-free strain with better growth for industrial use.
References
Akada R, Kitagawa T, Kaneko S, Toyonaga D, Ito S, Kakihara Y, Hoshida H, Morimura S, Kondo A, Kida K (2006) PCR-mediated seamless gene deletion and marker recycling in Saccharomyces cerevisiae. Yeast 23:399–405. https://doi.org/10.1002/yea.1365
Alam MA, Kelly JM (2017) Proteins interacting with CreA and CreB in the carbon catabolite repression network in Aspergillus nidulans. Curr Genet 63:669–683. https://doi.org/10.1007/s00294-016-0667-2
Alam MA, Kamlangdee N, Kelly JM (2017) The CreB deubiquitinating enzyme does not directly target the CreA repressor protein in Aspergillus nidulans. Curr Genet 63:647–667. https://doi.org/10.1007/s00294-016-0666-3
Boase NA, Kelly JM (2004) A role for creD, a carbon catabolite repression gene from Aspergillus nidulans, in ubiquitination. Mol Microbiol 53:929–940. https://doi.org/10.1111/j.1365-2958.2004.04172.x
Boase NA, Lockington RA, Adams JR, Rodbourn L, Kelly JM (2003) Molecular characterization and analysis of the acrB gene of Aspergillus nidulans: a gene identified by genetic interaction as a component of the regulatory network that includes the CreB deubiquitination enzyme. Genetics 164:95–104
Brown NA, de Gouvea PF, Krohn NG, Savoldi M, Goldman GH (2013) Functional characterisation of the non-essential protein kinases and phosphatases regulating Aspergillus nidulans hydrolytic enzyme production. Biotechnol Biofuels 6:91. https://doi.org/10.1186/1754-6834-6-91
Brown NA, Dos Reis TF, Ries LN, Caldana C, Mah JH, Yu JH, Macdonald JM, Goldman GH (2015) G-protein coupled receptor-mediated nutrient sensing and developmental control in Aspergillus nidulans. Mol Microbiol 98:420–439. https://doi.org/10.1111/mmi.13135
Chikamatsu G, Shirai K, Kato M, Kobayashi T, Tsukagoshi N (1999) Structure and expression properties of the endo-βAspergillus nidulans. FEMS Microbiol Lett 179:239–245
Colabardini AC, Humanes AC, Gouvea PF, Savoldi M, Goldman MH, Kress MR, Bayram Ö, Oliveira JV, Gomes MD, Braus GH, Goldman GH (2012) Molecular characterization of the Aspergillus nidulans fbxA encoding an F-box protein involved in xylanase induction. Fungal Genet Biol 49:130–140. https://doi.org/10.1016/j.fgb.2011.11.004
Cubero B, Scazzocchio C (1994) Two different, adjacent and divergent zinc finger binding sites are necessary for CREA-mediated carbon catabolite repression in the proline gene cluster of Aspergillus nidulans. EMBO J 13:407–415
Cubero B, Gómez D, Scazzocchio C (2000) Metabolite repression and inducer exclusion in the proline utilization gene cluster of Aspergillus nidulans. J Bacteriol 182:233–235. https://doi.org/10.1128/JB.182.1.233-235.2000
de Souza CP, Hashmi SB, Osmani AH, Andrews P, Ringelberg CS, Dunlap JC, Osmani SA (2013a) Functional analysis of the Aspergillus nidulans kinome. PLoS One 8:e58008. https://doi.org/10.1371/journal.pone.0058008
de Souza WR, Maitan-Alfenas GP, de Gouvêa PF, Brown NA, Savoldi M, Battaglia E, Goldman MH, de Vries RP, Goldman GH (2013b) The influence of Aspergillus niger transcription factors AraR and XlnR in the gene expression during growth in d-xylose, L-arabinose and steam-exploded sugarcane bagasse. Fungal Genet Biol 60:29–45. https://doi.org/10.1016/j.fgb.2013.07.007
de Assis LJ, Ries LN, Savoldi M, Dos Reis TF, Brown NA, Goldman GH (2015) Aspergillus nidulans protein kinase A plays an important role in cellulase production. Biotechnol Biofuels 8:213. https://doi.org/10.1186/s13068-015-0401-1
de Assis LJ, Ulas M, Ries LNA, El Ramli NAM, Sarikaya-Bayram O, Braus GH, Bayram O, Goldman GH (2018) Regulation of Aspergillus nidulans CreA-mediated catabolite repression by the F-Box proteins Fbx23 and Fbx47. MBio 9:e00840–e00818. https://doi.org/10.1128/mBio.00840-18
de la Serna I, Ng D, Tyler BM (1999) Carbon regulation of ribosomal genes in Neurospora crassa occurs by a mechanism which does not require Cre-1, the homologue of the Aspergillus carbon catabolite repressor, CreA. Fungal Genet Biol 26:253–269. https://doi.org/10.1006/fgbi.1999.1121
Dowzer CE, Kelly JM (1989) Cloning of the creA gene from Aspergillus nidulans: a gene involved in carbon catabolite repression. Curr Genet 15:457–459
Dubois M, Gilles K, Hamilton JK, Rebers PA, Smith F (1951) A colorimetric method for the determination of sugars. Nature 168:167. https://doi.org/10.1038/168167a0
Endo Y, Yokoyama M, Morimoto M, Shirai K, Chikamatsu G, Kato N, Tsukagoshi N, Kato M, Kobayashi T (2008) Novel promoter sequence required for inductive expression of the Aspergillus nidulans endoglucanase gene eglA. Biosci Biotechnol Biochem 72:312–320. https://doi.org/10.1271/bbb.70278
Fernändez-Canón JM, Luengo JM (1997) The phenylacetic acid uptake system of Aspergillus nidulans is under a creA-independent model of catabolic repression which seems to be mediated by acetyl-CoA. J Antibiot (Tokyo) 50:45–52. https://doi.org/10.7164/antibiotics.50.45
Gielkens MMC, Dekkers E, Visser J, de Graaff LH (1999) Two cellobiohydrolase-encoding genes from Aspergillus niger require d-xylose and the xylanolytic transcriptional activator XlnR for their expression. Appl Environ Microbiol 65:4340–4345
Hiramoto T, Tanaka M, Ichikawa T, Matsuura Y, Hasegawa-Shiro S, Shintani T, Gomi K (2015) Endocytosis of a maltose permease is induced when amylolytic enzyme production is repressed in Aspergillus oryzae. Fungal Genet Biol 82:136–144. https://doi.org/10.1016/j.fgb.2015.05.015
Ichinose S, Tanaka M, Shintani T, Gomi K (2014) Improved αAspergillus oryzae after a double deletion of genes involved in carbon catabolite repression. Appl Microbiol Biotechnol 98:335–343. https://doi.org/10.1007/s00253-013-5353-4
Ichinose S, Tanaka M, Shintani T, Gomi K (2018) Increased production of biomass-degrading enzymes by double deletion of creA and creB genes involved in carbon catabolite repression in Aspergillus oryzae. J Biosci Bioeng 125:141–147. https://doi.org/10.1016/j.jbiosc.2017.08.019
Ishikawa K, Kunitake E, Kawase T, Atsumi M, Noguchi Y, Ishikawa S, Ogawa M, Koyama Y, Kimura M, Kanamaru K, Kato M, Kobayashi T (2018) Comparison of the paralogous transcription factors AraR and XlnR in Aspergillus oryzae. Curr Genet. https://doi.org/10.1007/s00294-018-0837-5
Kunitake E, Hagiwara D, Miyamoto K, Kanamaru K, Kimura M, Kobayashi T (2016) Regulation of genes encoding cellulolytic enzymes by Pal-PacC signaling in Aspergillus nidulans. Appl Microbiol Biotechnol 100:3621–3635. https://doi.org/10.1007/s00253-016-7409-8
Lafon A, Seo JA, Han KH, Yu JH, d’Enfert C (2005) The heterotrimeric G-protein GanB(alpha)-SfaD(beta)-GpgA(gamma) is a carbon source sensor involved in early cAMP-dependent germination in Aspergillus nidulans. Genetics 171:71–80. https://doi.org/10.1534/genetics.105.040584
Lockington RA, Kelly JM (2002) The WD40-repeat protein CreC interacts with and stabilizes the deubiquitinating enzyme CreB in vivo in Aspergillus nidulans. Mol Microbiol 43:1173–1182
Makita T, Katsuyama Y, Tani S, Suzuki H, Kato N, Todd RB, Hynes MJ, Tsukagoshi N, Kato M, Kobayashi T (2009) Inducer-dependent nuclear localization of a Zn(II)2Cys6 transcriptional activator, AmyR, in Aspergillus nidulans. Biosci Biotechnol Biochem 73:391–399. https://doi.org/10.1271/bbb.80654
Marui J, Kitamoto N, Kato M, Kobayashi T, Tsukagoshi N (2002) Transcriptional activator, AoXlnR, mediates cellulose-inductive expression of the xylanolytic and cellulolytic genes in Aspergillus oryzae. FEBS Lett 528:279–282
Noguchi Y, Sano M, Kanamaru K, Ko T, Takeuchi M, Kato M, Kobayashi T (2009) Genes regulated by AoXlnR, the xylanolytic and cellulolytic transcriptional regulator, in Aspergillus oryzae. Appl Microbiol Biotechnol 85:141–154. https://doi.org/10.1007/s00253-009-2236-9
Noguchi Y, Tanaka H, Kanamaru K, Kato M, Kobayashi T (2011) Xylose triggers reversible phosphorylation of XlnR, the fungal transcriptional activator of xylanolytic and cellulolytic genes in Aspergillus oryzae. Biosci Biotechnol Biochem 75:953–959. https://doi.org/10.1271/bbb.100923
Orejas M, MacCabe AP, Pérez-González JA, Kumar S, Ramón D (2001) The wide-domain carbon catabolite repressor CreA indirectly controls expression of the Aspergillus nidulans xlnB gene, encoding the acidic endo-β
Ries LN, Beattie SR, Espeso EA, Cramer RA, Goldman GH (2016) Diverse regulation of the CreA carbon catabolite repressor in Aspergillus nidulans. Genetics 203:335–352. https://doi.org/10.1534/genetics.116.187872
Rowlands RT, Turner G (1973) Nuclear and extranuclear inheritance of oligomycin resistance in Aspergillus nidulans. Mol Gen Genet 126:201–216
Shroff RA, Lockington RA, Kelly JM (1996) Analysis of mutations in the creA gene involved in carbon catabolite repression in Aspergillus nidulans. Can J Microbiol 42:950–959
Somogyi M (1952) Notes on sugar determination. J Biol Chem 195:19–23
Strauss J, Mach RL, Zeilinger S, Hartler G, Stöffler G, Wolschek M, Kubicek CP (1995) Cre1, the carbon catabolite repressor protein from Trichoderma reesei. FEBS Lett 376:103–107
Tamayo EN, Villanueva A, Hasper AA, de Graaff LH, Ramón D, Orejas M (2008) CreA mediates repression of the regulatory gene xlnR which controls the production of xylanolytic enzymes in Aspergillus nidulans. Fungal Genet Biol 45:984–993. https://doi.org/10.1016/j.fgb.2008.03.002
Tamayo-Ramos JA, Flipphi M, Pardo E, Manzanares P, Orejas M (2012) L-rhamnose induction of Aspergillus nidulansα
Tanaka M, Ichinose S, Shintani T, Gomi K (2018) Nuclear export-dependent degradation of the carbon catabolite repressor CreA is regulated by a region located near the C-terminus in Aspergillus oryzae. Mol Microbiol. https://doi.org/10.1111/mmi.14072
van Peij NN, Gielkens MM, de Vries RP, Visser J, de Graaff LH (1998) The transcriptional activator XlnR regulates both xylanolytic and endoglucanase gene expression in Aspergillus niger. Appl Environ Microbiol 64:3615–3619
Vega M, Riera A, Fernández-Cid A, Herrero P, Moreno F (2016) Hexokinase 2 is an intracellular glucose sensor of yeast cells that maintains the structure and activity of Mig1 protein repressor complex. J Biol Chem 291:7267–7285. https://doi.org/10.1074/jbc.M115.711408
Acknowledgements
This work was partially supported by the Program for Promotion of Basic and Applied Research for Innovations in Bio-oriented Industry and by the Science and Technology Research Promotion Program for Agriculture, Forestry, Fisheries, and Food Industry. It was also supported by JSPS KAKENHI (Grant Numbers JP 18H02125 and JP 17H06763) and funding from Institute for Fermentation, Osaka (IFO) and Noda Institute for Scientific Research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Communicated by M. Kupiec.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kunitake, E., Li, Y., Uchida, R. et al. CreA-independent carbon catabolite repression of cellulase genes by trimeric G-protein and protein kinase A in Aspergillus nidulans. Curr Genet 65, 941–952 (2019). https://doi.org/10.1007/s00294-019-00944-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-019-00944-4