
Abstract
Arenicin-1, a 21-mer antimicrobial peptide exerts significant broad-spectrum antimicrobial activity with membrane-active mechanisms. However, owing to multiple mechanisms of cell death, the antibacterial mechanism of arenicin-1 requires detailed analysis. In the present study, arenicin-1-treated bacteria underwent an apoptosis-like response, which was mechanistically and morphologically similar to eukaryotic apoptosis. Changes in the physiological status of arenicin-1-treated bacterial cells involved accumulation of reactive oxygen species, imbalance of intracellular calcium gradients, disruption of membrane potential, bacterial caspase-like protein activation, and DNA damage. In arenicin-1-induced apoptosis-like death, autocleavage of LexA was observed because of the activation of the caspase-like activity of RecA. Additionally, typical reactive oxygen species such as superoxide, hydrogen peroxide, and hydroxyl radicals, were scavenged in arenicin-1-treated cells to assess the role of specific reactive oxygen species. Scavenging of hydrogen peroxide interfered with the activity of arenicin-1 in Escherichia coli, whereas the superoxide and hydroxyl radicals level did not affect arenicin-1-induced apoptosis-like death activity. Furthermore, inhibition of Fenton reaction attenuated apoptosis-like response. In conclusion, arenicin-1-induced apoptosis like death requires SOS response proteins and is mediated by hydrogen peroxide and Fenton reaction in E. coli. Arenicin-1 may be a representative antimicrobial peptide with potent apoptotic response against E. coli.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The incidence of microbial infections has increased dramatically over the last few decades (Tanwar et al. 2014; Cansizoglu and Toprak 2017). Bacterial infections are extremely difficult to treat because of the general antibiotic resistance exhibited by subpopulations in the biofilm community, which are regarded as antibiotic-resistant bacteria (Xu et al. 2014). The continuous use of antimicrobial drugs in treating infections has led to the emergence of resistance among various microorganisms and hinders healthy host microbiota (Tanwar et al. 2014; Cansizoglu and Toprak 2017). Multidrug resistance is defined as the insensitivity or resistance of a microorganism to structurally unrelated antimicrobials with different molecular targets despite previously demonstrated sensitivity to those drugs (Tanwar et al. 2014). Bacteria acquired the resistance gene or mutation, can continue to grow in the presence of antibiotics, while antibiotic-sensitive bacteria have inhibited growth (Baym et al. 2016). As multidrug resistance among bacterial pathogens is becoming widespread, the need for new antibiotics is on the rise. Currently, the main approaches involve the production of second generation of existing antibiotics with improved activities and search strategies that can preserve the efficacy of existing antimicrobial agents (Cansizoglu and Toprak 2017; Kumar and Engelberg-Kulka 2014).
Under certain hostile conditions, bacteria undergo programmed cell death (PCD), which involves death mediated by an intracellular program (Nagamalleswari et al. 2017). The prokaryotic death shows mechanistic similarities to eukaryotic cell apoptosis (Hakansson et al. 2011). Although bacterial PCD is a relatively new area of research, it can lead to the development of innovative antibacterial compounds that can artificially induce PCD in bacteria (Dewachter et al. 2016). Different mechanisms of bacterial PCD have been associated with various cellular phenomena (Nagamalleswari et al. 2017). The three major PCD pathways are mazEF-mediated death, thymine-less death (TLD), and apoptotic-like death (ALD) (Nagamalleswari et al. 2017; Erental et al. 2014). The mazEF, a well-studied toxin–antitoxin system, induces PCD in most of the population by increasing the synthesis of specific protein related to cell death (Erental et al. 2014; Choi et al. 2016). ALD is an extreme DNA damage-induced death pathway, and is mainly dependent on RecA as a caspase substrate binding partner (Erental et al. 2014). The TLD pathway is triggered by thymine starvation leading to the DNA replication fork stalling (Nagamalleswari et al. 2017; Matic 2017).
Antimicrobial peptides (AMPs), composed of less than 50 amino acid residues, have been isolated from plants, insects, and vertebrates (including humans), which constitute host defense systems against invading pathogenic microorganisms. These peptides not only directly kill pathogens (Gram-positive and -negative bacteria, fungi, parasites, enveloped viruses, and multidrug-resistant microorganisms), but also modulate innate immunity and even bridge the innate and adaptive immune responses. Several attempts have been made to utilize AMPs as novel antibiotics because they exhibit a broad spectrum of antimicrobial activities and do not easily induce resistance compared to conventional antibiotics, although they do eventually evoke resistance (Matsuzaki 2009). They interact with microbial membranes through electrostatic interactions. Unregulated ion efflux/influx result in bacterial morphological damage (Lam et al. 2016; Panteleev et al. 2015; Lee and Lee 2014). Arenicin-1 (RWCVYAYVRVRGVLVRYRRCW), a family of 21-residue β-hairpin AMPs, was identified in the coelomocytes of the marine polychaeta lugworm Arenicola marina (Panteleev et al. 2017). This peptide has been extensively investigated because of its high cytotoxicity and potent antimicrobial activity (Panteleev et al. 2017; Park et al. 2011). Their mechanistic studies indicate cytoplasmic membrane permeation, reactive oxygen species (ROS) accumulation and changes in physiological status responded intracellular stress in microbial cells (Choi and Lee 2012; Panteleev et al. 2015). In this study, we investigated the potential of bacterial ALD by arenicin-1.
Materials and methods
Reagent preparation, strains, and cell growth conditions
Peptide synthesis was performed by ANYGEN Co. Ltd. (Gwangju, Korea) and dissolved in milliQ water (a stock solution of 10 mg/mL). Norfloxacin was purchased from Sigma-Aldrich (St. Louis, MO, USA). E. coli MG1655 cells were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA). We also studied MG1655ΔRecA and K996 cells expressing the mutant repressor LexA3(Ind-) (Shah et al. 2013). MG1655ΔRecA is MG1655 with ΔRecA allele originated from JW2669 by P1 transduction. E. coli K996 cells is lexA3(ind-), malE300::Tn10 from E. coli Genetic Stock Center (Shah et al. 2013). For all experiments, cells were grown in Luria–Bertani (LB) (BD Pharmingen, San Diego, CA, USA) agar plates at 37 °C and inoculated in LB broth under aerobic conditions at 37 °C and 120 rpm. The bacterial strains in the exponential phase were harvested and then resuspended in phosphate-buffered saline (PBS). Arenicin-1 (12.5 µM) and norfloxacin (2.3 µM) were treated in the follow-up assays and the cells were incubated under aerobic conditions for 4 h at 37 °C and 120 rpm. Sodium pyruvate, tiron (4,5-dihydroxy-1,3-benzenedisulfonic acid), thiourea, and dipyridyl were purchased from Sigma-Aldrich. Dipyridyl prevents hydroxyl radical production by Fenton reaction (Dewachter et al. 2016).
Assessment of intracellular ROS levels
Intracellular ROS levels were assessed using 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA; Molecular Probes, Eugene, OR, USA), a cell-permeant ROS indicator. After incubation, the cells were suspended in PBS, and 10 µM H2DCFDA was added to the suspension. After incubation at 37 °C for 1 h, the cells were washed with PBS, and the fluorescent cells were detected using a FACSVerse™ flow cytometer (Becton–Dickinson, San Jose, CA, USA).
Assessment of oxidative damage
Reduced glutathione (GSH) levels were evaluated as described by Kumar et al. (2011) with slight modifications. Cells were washed with PBS, lysed via sonication, and centrifuged at 12,000 rpm for 20 min. The supernatant was added to 20 µL 100% trichloroacetic acid (TCA) solution. The mixture was then incubated on ice for 10 min, centrifuged at 12,000 rpm for 10 min, and the supernatants were collected. GSH was quantified by measuring the absorbance at 415 nm using a microtiter enzyme-linked immunosorbent assay (ELISA) reader (Molecular Devices Emax) following a reaction containing 100 µL of 0.01% dithionitrobenzoate (DTNB) and 20 µL of the supernatant for 15 min. The Bradford method was used to determine total protein levels in treated cultures (Bio-Rad, Hercules, CA, USA). GSH levels were estimated and expressed as GSH µg/mg of protein.
Lipid peroxidation was estimated by quantifying malonyl dialdehyde (MDA) levels via the (2-thiobarbituric acid reactive substance) TBARS assay. The incubated cells were washed with PBS and sonicated for 1 min in lysis buffer. Trichloroacetic acid (TCA, 5%) was added to precipitate proteins. After centrifugation at 12,000 rpm for 5 min, the supernatant was incubated with thiobarbituric acid (TBA) solution and subjected to heat shock at 95 °C for 40 min. The extent of lipid peroxidation was analyzed using a spectrophotometer (DU530; Beckman, Fullerton, CA, USA) at 570 nm (Nordberg and Arner 2001).
Analysis of intracellular calcium levels
Intracellular calcium levels were measured using Fura-2 AM, a cell-permeant intracellular calcium indicator. When Fura-2 AM enters the cell, its acetoxymethyl groups are removed by cellular esterases and the calcium-sensitive indicator (Fura-2) is generated. After incubation, the cells were washed thrice with Krebs buffer [4 mM KCl, 132 mM NaCl, 1.4 mM MgCl2, 1 mM CaCl2, 10 mM NaHCO3, 10 mM HEPES, and 6 mM glucose (pH 7.4)], and treated with 1% bovine serum albumin and 0.01% Pluronic® F-127 (Molecular Probes). Next, the cells were treated with Fura-2 AM (5 µM; Molecular Probes) and incubated at 37 °C for 40 min. After incubation, the cells were washed thrice with calcium-free Krebs buffer and analyzed using a spectrofluorophotometer (Shimadzu RF-5301PC) at wavelengths of 340 nm (excitation) and 510 nm (emission).
Assessment of changes in membrane potential and cell size
Plasma membrane potential was analyzed using bis-(1,3-dibutylbarbituric acid) trimethine oxonol [DiBAC4(3)] staining. DiBAC4(3) exhibits increased fluorescence by binding to the membranes or intracellular proteins of depolarized cells. After incubation, the cells were washed and resuspended in PBS before staining with 10 µg/mL DiBAC4(3). Flow cytometry was performed using a FACSVerse flow cytometer (Nagamalleswari et al. 2017; Cho and Lee 2011). To investigate morphological change, the incubated cells were harvested by centrifugation and resuspended in PBS. The morphological changes were analyzed using a FACSVerse flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA).
Assessment of nucleoid damage
Nucleoid damage was analyzed using 4′,6-diamidino-2-phenylindole (DAPI) staining. For nucleoid staining, cells were washed twice with PBS, treated with a permeabilization solution (0.1% Triton X-100 and 0.1% sodium citrate), and incubated with DAPI (1 µg/mL) in the dark for 20 min (Cho and Lee 2011). The fluorescence intensity was examined using a spectrofluorophotometer (Shimadzu RF-5301PC, Shimadzu, Kyoto, Japan).
Assessment of DNA fragmentation
DNA fragmentation was quantified by the terminal deoxynucleotidyl transferase (TdT) dUTP nick-end labeling (TUNEL) assay. Briefly, compound-treated cells and untreated cells were incubated and suspended in permeabilization solution consisting of 0.1% Triton X-100 and 0.1% sodium citrate for 2 min on ice. The cell suspensions were fixed in 2% paraformaldehyde for 1 h. Cells were then washed and labeled using an in situ cell death detection kit for 1 h at 37 °C. The stained cells were analyzed with a FACSverse flow cytometer (Cho and Lee 2011).
Assessment of activation caspase-like protein
Caspase activity was measured using the CaspACE™ FITC-VAD-FMK in situ marker (Promega). Bacterial cells were resuspended in PBS containing peptides. After incubation, cells were washed with PBS and stained with 2.5 µM of CaspACE™ FITC-VAD-FMK for 30 min at 28 °C. Fluorescence intensity was assessed using a FACSVerse flow cytometer (Cho and Lee 2011).
Western blotting for RecA and LexA
Cells were collected and resuspended in PBS. The suspensions were then lysed using a sonicator (10 pulses of 2 min each at an amplitude of 38) (Sonics, Newtown, CT, USA), and centrifuged to remove the intact cells and cell debris. The supernatants were collected, and proteins were precipitated with 5% TCA at 4 °C for 10 min. The TCA-precipitated proteins were washed twice with cold acetone, harvested, and dissolved in H2O. Quantitation of solubilized proteins was performed using Bradford’s assay. Each 50 µg protein sample was resolved using 7.5% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE), followed by transfer to a nitrocellulose membrane. The membrane was blocked in 3% skimmed milk for 1 h at room temperature, and incubated with rabbit polyclonal anti-RecA (1:3000; Abcam, Cambridge, UK) and LexA antibodies (Thermo fisher) for 12 h at 4 °C. Subsequently, the membrane was incubated with horseradish peroxidase conjugated goat anti-rabbit IgG (1:2000; Biovision, Milpitas, CA, USA) as the secondary antibody for 6 h at 4 °C. Pierce ECL Plus western blotting substrate (Thermo Scientific, Waltham, MA, USA) was added, and the membranes were exposed to an X-ray film (Dwyer et al. 2012).
Statistical analysis
All experiments were performed for three independent times. The data represent mean ± standard deviation. Statistical significance was determined via Student’s t test. p < 0.05 was considered to indicate statistical significance.
Result
ROS induced by arenicin-1 and its oxidative damage
To evaluate the oxidative effect of arenicin-1, intracellular ROS levels were determined using H2DCFDA. Norfloxacin was used as the positive control due to its oxidative effect on E. coli without the induction of membrane damage (Dwyer et al. 2012). As shown in Fig. 1a, arenicin-1 treatment increased in H2DCFDA fluorescence. This suggested that arenicin-1 produced excessive ROS and could induce oxidative stress. Bacterial cells treated with arenicin-1 and norfloxacin exhibited significantly lower GSH levels than untreated cells. Thus, the GSH level was reduced regardless of the elevated intracellular ROS levels (Fig. 1b). Furthermore, the TBARS assay was used to detect the extent of lipid peroxidation. MDA, an end product of lipid peroxidation, reacts with TBA (Nie et al. 2009). Notably, a considerable increase in the MDA level was observed after treatment with arenicin-1 (Fig. 1c). Therefore, we concluded that arenicin-1 induced an imbalance in the redox equilibrium and oxidative damage via ROS generation.

Oxidative stress induced by arenicin-1. E. coli cells were treated with arenicin-1 (12.5 µM) or norfloxacin (2.3 µM). a Accumulation of ROS was determined by flow cytometric analysis using H2DCFDA. b Decrease in GSH levels indicates oxidative damage. 1 Untreated; 2: arenicin-1; 3: norfloxacin. c Increase in MDA levels indicates lipid peroxidation. 1 Untreated; 2: arenicin-1; 3: norfloxacin. Data represents the average, standard deviation, and p values from three independent experiments (*p < 0.1; **p < 0.05; ***p < 0.01 vs. the untreated; Student’s t test)
Changes in intracellular calcium and nucleoid fragmentation
To investigate the effect of arenicin-1 on cytosolic calcium levels, a membrane-permeable derivative of a ratiometric calcium indicator was used. In addition, the intracellular physiological changes were observed via elimination of typical ROS. Sodium pyruvate is oxidatively decarboxylated by hydrogen peroxide to produce carbon dioxide and acetate, which subsequently scavenges hydrogen peroxide (Troxell et al. 2014). Tiron is a cell-permeable superoxide scavenger (Hernansanz-Agustin et al. 2014) and thiourea is a potent hydroxyl radical scavenger that mitigates the effects of hydroxyl radical damage in both eukaryotes and prokaryotes (Hwang et al. 2012). Cells exposed to arenicin-1 showed increased cytosolic calcium levels. While sodium pyruvate-treated cells exhibited more fluorescence than arenicin-1-treated cells, tiron- and thiourea-treated cells individually exhibited fluorescence levels similar to those of arenicin-1-treated cells (Fig. 2a). These results indicated that hydrogen peroxide may exert different effects on arenicin-1-induced bacterial cell death, compared to other typical ROS. Moreover, DAPI staining was performed to monitor changes in the nucleoid. Spectrofluorophotometric analyses revealed that cells treated with arenicin-1 or norfloxacin displayed more concentrated fluorescence compared to untreated cells, which is indicative of nucleoid condensation. Nucleoid fragmentation was weaker in sodium pyruvate-co-treated cells than in arenicin-1-treated cells. Tiron and thiourea-co-treated cells exhibited nucleoid damage similar to those of arenicin-1-treated cells (Fig. 2b). Monitoring of DNA fragmentation via the TUNEL assay showed that fluorescence increased in arenicin-1-treated cells. Tiron- and thiourea-co-treated samples showed a similar increase in fluorescence, whereas sodium pyruvate-treated cells exhibited a decrease in fluorescence intensity (Fig. 2c). These results demonstrated that arenicin-1 induces nucleoid fragmentation, and that each ROS scavenger exhibit different effects, especially hydrogen peroxide.

Changes in physiological state in the presence of arenicin-1. Analysis of calcium influx and DNA damage in E. coli following treatment with arenicin-1 (12.5 µM) and norfloxacin (2.3 µM). a Spectrofluorophotometric analysis of calcium influx using Fura-2 AM. The data represents the average, standard deviation, and p values from three independent experiments (*p < 0.1; **p < 0.05; ***p < 0.01 vs. the untreated; #p < 0.1; ##p < 0.05; ###p < 0.01 vs. arenicin-1-treated cells; Student’s t test;). 1 Untreated; 2: Arenicin-1; 3: Arenicin-1 + sodium pyruvate; 4: Arenicin-1 + tiron; 5: Arenicin-1 + thiourea; 6: Norfloxacin. b DNA condensation was detected by DAPI staining using a spectrofluorophotometer. 1: Arenicin-1 + sodium pyruvate; 4: Arenicin-1 + tiron; 5: Arenicin-1 + thiourea; 6. Norfloxacin; 6. c DNA fragmentation was assessed by TUNEL staining
Confirmation of bacterial PCD
The fluorescent dye DiBAC4(3) was used to determine whether arenicin-1 triggered an analogous PCD mechanism in E. coli. DiBAC4(3) binds to intracellular proteins or membranes to exhibit enhanced fluorescence, indicating membrane depolarization (Nagamalleswari et al. 2017; Li et al. 2008; Cochrane et al. 2016). As shown in Fig. 3a, arenicin-1-treated cells showed higher DiBAC4(3) fluorescence intensity than the untreated cells. Unlike the arenicin-1-treated cells co-treated with tiron and thiourea, the pyruvate/arenicin-1-treated cells showed significant reduction in DiBAC4(3) fluorescence. We also observed filamentation of cells treated with arenicin-1 or norfloxacin while monitoring morphological changes. The cell size of arenicin-1-treated cells was reduced by the sodium pyruvate-mediated scavenging of hydrogen peroxide. Tiron and thiourea did not affect arenicin-1-induced cell elongation (Fig. 3b). Furthermore, VAD-FMK was transported inside cells, where it binds to the caspase-active site. The fluorescence in arenicin-1-treated cells was higher than that in untreated cells, indicating an active expression of proteins that specifically bind to the caspase substrate. Flow cytometric analysis showed that cells treated with arenicin-1 and norfloxacin were fluorescent following incubation with VAD-FMK (Fig. 3c). Compared to the arenicin-1-treated cells, the sodium pyruvate-treated samples showed reduction in green fluorescence, whereas the tiron- and thiourea-treated samples showed similar fluorescence.

Features of apoptotic-like death induced by arenicin-1. Analysis of membrane depolarization, cell filamentation and caspase activation in E. coli following treatment with arenicni-1 (12.5 µM) and norfloxacin (2.3 µM). a Flow cytometric analysis of membrane depolarization using DiBAC4(3). b Induction-mediated cell size increase. An increase in FS- and SS-indicated cell filamentation. c Flow cytometric analysis of caspase-like response using FITC-VAD-FMK. d Analysis of RecA/LexA expression levels by western blotting
We performed western blotting to determine whether arenicin-1 induced the bacterial SOS response. RecA plays a significant role in DNA damage via the SOS response and stimulates the self-cleavage of LexA, leading to the derepression of SOS genes (Zgur-Bertok 2013). As shown in Fig. 3d, the banding pattern of RecA showed similarities with caspase activation. An opposite pattern was observed for the LexA bands. The bands obtained from arenicin-1-treated cells were fainter than the bands from untreated cells, whereas the bands from sodium pyruvate-treated cells were thinner than those from arenicin-1-treated cells. The bands from tiron- and thiourea-treated cells were of an intensity similar to those of arenicin-1-treated cells. Therefore, we assumed that arenicin-1-induced bacterial cell death involved in DNA damage and that the DNA repair system was also activated by the SOS regulon. Furthermore, these observations were diminished when hydrogen peroxide was scavenged. These features indicated that arenicin-1 could induce apoptosis-like death, which was suppressed by sodium pyruvate, a hydrogen peroxide scavenger.
Oxidative stress and features of ALD in ΔRecA and K996 cells
We used the ΔRecA strain and K996 cells expressing the mutant repressor, LexA3(Ind-), for determining the relationship between arenicin-1-induced ALD and SOS response (Dudas et al. 2003). ΔRecA and K996 cells treated with arenicin-1 or norfloxacin exhibited an increase in fluorescence intensity compared to untreated cells, indicating that arenicin-1 induces ROS accumulation (Fig. S1). Furthermore, GSH depletion and lipid peroxidation indicated that arenicin-1 induced oxidative stress regardless of the presence of RecA and LexA (Fig. S1). Thus, oxidative stress can occur without SOS response in E. coli and oxidative damage and imbalance of redox equilibrium can occur regardless of the presence of RecA and LexA.
Hallmarks of apoptosis were observed in the same strains. Above all, we detected changes in membrane potential in ΔRecA and K996 cells. The membrane potential was unchanged in all compound-treated ΔRecA cells, whereas changes in fluorescent intensity were observed slightly in K996 cells. K996 cells were depolarized by arenicin-1, which was inhibited by sodium pyruvate (Fig. S2). Changes in cell morphology were also observed. The ΔRecA cells did not exhibit significant morphological differences. K996 cells showed slight morphological changes, and hydrogen peroxide scavenging inhibited morphological changes by arenicin-1 (Fig. S3). Moreover, caspase activation did not occur in the absence of RecA, whereas it increased in the presence of the modified LexA3 (Fig. 4). Additionally, ROS scavenging did not affect the action of arenicin-1 in the absence of RecA. Caspase-like protein was slightly stimulated by arenicin-1 in K996 cells, and hydrogen peroxide scavenging inhibited arenicin-1 action. Tiron and thiourea did not interfere significantly with arenicin-1 action. RecA was necessary for arenicin-1-mediated ALD, which was attenuated by modifying LexA3.

Features of apoptotic-like death in ΔRecA and K996 cells induced by arenicin-1. Analysis of caspase activation in ΔRecA and K996 cells in the presence/absence of arenicin-1 and ROS scavenger. Flow cytometric analysis of caspase-like response using FITC-VAD-FMK
Inhibition of Fenton reaction was attenuated by arenicin-1-induced bacterial apoptosis
Arenicin-1-induced ALD was affected by intracellular hydrogen peroxide level. E. coli synthesizes catalase and NADH peroxidase to minimize hydrogen peroxide levels (Khademian and Imlay 2017). This oxidant also oxidizes the intracellular pool of unincorporated iron via the Fenton reaction, thereby generating DNA-damaging hydroxyl radicals (Khademian and Imlay 2017). In contrast, hydroxyl radical level did not affect by ALD in the presence of thiourea. Fenton reaction was studied using the iron chelator, dipyridyl (Dewachter et al. 2016). Figure 5 shows that dipyridyl lowers arenicin-1-induced ALD. Overall, these results demonstrated that Fenton reaction may reinforce arenicin-1-mediated bacterial ALD.

Decrease in arenicin-1-induced ALD features in the presence of dipyridyl. Analysis of membrane depolarization, caspase activation and DNA fragmentation. K996 cells were treated with arenicin-1 and dipyridyl. a Flow cytometric analysis of membrane depolarization using DiBAC4(3). b Flow cytometric analysis of caspase-like response using FITC-VAD-FMK. c DNA fragmentation was assessed by TUNEL staining
Discussion
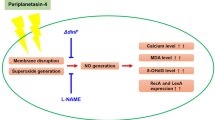
Arenicin-1 consists of two antiparallel β-sheets linked by a hydrophilic β-turn. The Arg and Trp residues in the N-terminal region and the Arg residue in the β-turn region were suggested as key roles in antibacterial activity and membrane targeting (Cho and Lee 2011a, b; Panteleev et al. 2017). Moreover, in artificial membrane condition, arenicin-1 implies the formation of mixed lipid/peptide pores (Panteleev et al. 2015; Shenkarev et al. 2011) but the mechanisms of the peptide–lipid interactions, disruption and translocation across target membranes are still obscure against bacterial cells. AMPs induce bacterial cell death by multifaceted mechanisms involving loss of membrane integrity, upregulation of ion gradient and induction ALD (Choi et al. 2016; Rios Colombo et al. 2018; Lam et al. 2016; Lee and Lee 2014). Based on the observations of this study, we propose bacterial apoptosis-like cell death to be a possible antimicrobial mechanism. A series of experiments were performed to confirm the antibacterial function of arenicin-1 based on the hypothesis that the underlying mechanism is bacterial ALD.
Reactive oxygen species attack intracellular contents and alter redox-related physiological conditions; however, before exerting their deleterious effects, ROS are usually scavenged by intracellular enzymatic antioxidants (Zhao and Drlica 2014; Evans et al. 2004; Dwyer et al. 2014). Among ROS, three naturally occurring species, namely superoxide, hydrogen peroxide, and hydroxyl radicals, have received the most attention. Superoxide and hydrogen peroxide are formed when molecular oxygen adventitiously oxidizes redox enzymes that normally transfer electrons to other substrates (Zhao and Drlica 2014). Hydrogen peroxide, which can also be produced by the dismutation of superoxide, acts as a substrate for hydroxyl radical formation via Fenton reaction. This oxidative process can be fatal if hydroxyl radical accumulation is not controlled, since hydroxyl radicals disrupt nucleic acids, carbonylated proteins, and peroxide lipids (Zhao and Drlica 2014; Green and Paget 2004). In summary, arenicin-1-mediated bacterial cell death generated ROS, which resulted in lipid peroxidation and an imbalance of the antioxidant system.
Different classes of bactericidal antibiotics, regardless of their drug–target interactions, have been proposed to generate varying levels of deleterious ROS that contribute to cell death (Dwyer et al. 2014). ROS play a central role in regulating cell fate by altering the cellular redox balance (Bidle 2016). Therefore, we used a representative ROS scavenger to observe the changes in the antibacterial mechanism of arenicin-1 for each type of ROS molecule. The calcium gradient is constantly maintained by a system of channels and transporters, and it contributes to oxidative stress signaling in bacterial cells (Dominguez 2004). Arenicin-1 disrupted intracellular calcium homeostasis. Furthermore, the intracellular calcium gradient by arenicin-1 was enhanced in the absence condition of hydrogen peroxide. However, the intracellular calcium gradient was not significantly changed when superoxide and hydroxyl radical were scavenged. The mutual interplay between ROS and calcium signaling systems appears to affect the fine tuning of cellular signaling networks (Gorlach et al. 2015). The intracellular response induced by arenicin-1 stimulates calcium signaling, which is associated with the ROS network.
Oxidized DNA is repaired by bacterial repair systems. Before the critical phases of DNA replication, cell division is arrested to provide more time for repair (Kaufmann and Paules 1996). Calcium fluxes activate a series of events leading to phosphorylation reactions, activation of regulatory proteins, and cell cycle regulation (Dominguez 2004; Ferullo and Lovett 2008). Arenicin-1-mediated nucleoid and DNA fragmentation did not change significantly in the absence of superoxide and hydroxyl radicals, whereas DNA damage was reduced in the absence of the hydrogen peroxide. The DNA damage caused by arenicin-1 is possibly because of the electrical interactions of the peptide rather than the ROS-mediated oxidation. DNA damage and elevation of calcium level underline the differences in physiological status induced by arenicin-1 in the absence of hydrogen peroxide.
Reactive oxygen species generation, calcium influx, and severe DNA damage are observed in bacterial PCD. Particularly, DNA fragmentation, membrane depolarization, and hydroxyl radical formation are bacterial PCD hallmarks that accompany caspase-like protein and RecA activation and cell filamentation in response to severe DNA damage (Erental et al. 2014; Nagamalleswari et al. 2017; Dewachter et al. 2016; Dwyer et al. 2012). Bacterial cells exposed to arenicin-1 exhibit apoptotic features and RecA activation simultaneously with LexA autocleavage. This indicates that arenicin-1 triggers SOS response to cause LexA autocleavage, and that RecA was activated beyond the threshold to serve as a caspase.
Stress-induced DNA cleavage induces the SOS response, which subsequently triggers the expression of genes involved in DNA repair, and causes RecA activation and LexA proteolysis (Nagamalleswari et al. 2017). RecA and LexA play critical roles as regulators of genes comprising the SOS response network. LexA prevents the expression of more than 40 genes required for DNA damage repair and binds to a specific DNA sequence (the SOS box) (Shkilnyj et al. 2013). Under stressful conditions, the active form of RecA, which is stimulated by single-stranded DNA, induces the SOS response by promoting LexA cleavage (Sharma et al. 2013; Shkilnyj et al. 2013; Adikesavan et al. 2011). However, severely damaged cells cannot be repaired, in which case RecA triggers ALD pathway instead of the SOS response (Cox 2007). RecA-deficient or LexA3-modified-cells scarcely appeared ALD in despite of ROS accumulation. Typical ALD characteristics were not observed in the absence of RecA. ALD characteristics were mild in the strain harboring denatured LexA3, possibly because of the effect of existing RecA (Dwyer et al. 2012). In the absence of RecA, cleavage of LexA protein is not sufficient to stimulate the apoptotic pathway. Thus, arenicin-1-induced ALD requires RecA overexpression and LexA autocleavage, which is similar to the features of an SOS response and have no relevance with ROS levels.
The apoptotic characteristics of cells exposed to arenicin-1 were attenuated in the absence of hydrogen peroxide, whereas scavenging of other typical ROS did not affect the apoptotic effect, indicating that they do not play any role in the regulation of this type of cell death. Hydroxyl radical formation is one of the major features of apoptosis. ALD may be triggered by hydroxyl radical formation from hydrogen peroxide via Fenton reaction. Catalases and NADH peroxidase degrade hydrogen peroxide. In addition, hydrogen peroxide permeates inside cells, initiating the Fenton reaction by oxidation of iron (Mishra and Imlay 2012). Hydroxyl radical is formed through the reduction of hydrogen peroxide (Mishra and Imlay 2012; Lee and Tan 2015). However, lack of hydroxyl radical did not affect features of ALD induced by arenicin-1. Hence, we focused on the relationship between Fenton reaction and bacterial ALD and used an iron chelator for blocking Fenton reaction by sequestering unbound intracellular iron (Lee and Tan 2015). Scavenging of hydrogen peroxide and inhibition of Fenton reaction machinery reduced arenicin-1-mediated ALD, which indicates a relationship between Fenton reaction and bacterial ALD. Removal of hydrogen peroxide in RecA-deleted and LexA-modified cells did not show any unusual fluctuation of the hallmarks of arenicin-1-induced ALD. The cell death action of arenicin-1 showed that proteins related to SOS response require the activation of ALD by arenicin-1 in the presence of hydrogen peroxide and the inhibition of Fenton reaction attenuates bacterial ALD.
In summary, we suggest the possibility that arenicin-1 induces bacterial ALD response. Cell filamentation, DNA damage, ROS accumulation, caspase activation, RecA protein activation, and LexA protein autocleavage were typical ALD characteristics. Arenicin-1-mediated ALD requires RecA and stimulation of cleavage LexA. Moreover, the absence of hydrogen peroxide suggests that a network of SOS responses is highly associated with arenicin-1-induced ALD. The conversion of hydrogen peroxide to hydroxyl radical via Fenton reaction is related to ALD. These finding suggest that ALD induced by AMPs is associated with SOS repair gene, RecA and LexA, and specific ROS, hydrogen peroxide is also affected. Recently, several AMPs have been suggested the evidence of programmed cell death in prokaryotes. These AMPs proceed a multimodal mechanisms of bacterial cell death by membrane destabilization, disruption of ion homeostasis across the cytoplasmic membrane and induction of the PCD pathway (Choi et al. 2016; Lee and Lee 2014, 2016; Lam et al. 2016; Hakansson et al. 2011). These findings are the initial step to figure out ALD as a novel mechanism for applying antimicrobial agents. AMPs and arenicin-1 can be a role as a representative in elucidating the molecular detail of bacterial ALD.
Abbreviations
- PCD:
-
Programmed cell death
- TLD:
-
Thymine-less death
- ALD:
-
Apoptotic-like death
- AMP:
-
Antimicrobial peptides
- PBS:
-
Phosphate-buffered saline
- GSH:
-
Reduced glutathione
- DiBAC4(3):
-
Bis-(1,3-dibutylbarbituric acid) trimethine oxonol
- H2DCFDA:
-
2′,7′-Dichlorodihydrofluorescein diacetate
References
Adikesavan AK, Katsonis P, Marciano DC, Lua R, Herman C, Lichtarge O (2011) Separation of recombination and SOS response in Escherichia coli RecA suggests LexA interaction sites. PLoS Genet 7(9):e1002244. https://doi.org/10.1371/journal.pgen.1002244
Baym M, Stone LK, Kishony R (2016) Multidrug evolutionary strategies to reverse antibiotic resistance. Science 351(6268):aad3292. https://doi.org/10.1126/science.aad3292
Bidle KD (2016) Programmed cell death in unicellular phytoplankton. Curr Biol 26(13):R594–R607. https://doi.org/10.1016/j.cub.2016.05.056
Cansizoglu MF, Toprak E (2017) Fighting against evolution of antibiotic resistance by utilizing evolvable antimicrobial drugs. Curr Genet 63(6):973–976. https://doi.org/10.1007/s00294-017-0703-x
Cho J, Lee DG (2011a) The characteristic region of arenicin-1 involved with a bacterial membrane targeting mechanism. Biochem Biophys Res Commun 405(3):422–427. https://doi.org/10.1016/j.bbrc.2011.01.046
Cho J, Lee DG (2011b) The antimicrobial peptide arenicin-1 promotes generation of reactive oxygen species and induction of apoptosis. Biochim Biophys Acta 1810(12):1246–1251. https://doi.org/10.1016/j.bbagen.2011.08.011
Choi H, Lee DG (2012) Synergistic effect of antimicrobial peptide arenicin-1 in combination with antibiotics against pathogenic bacteria. Res Microbiol 163(6–7):479–486. https://doi.org/10.1016/j.resmic.2012.06.001
Choi H, Hwang JS, Lee DG (2016) Coprisin exerts antibacterial effects by inducing apoptosis-like death in Escherichia coli. IUBMB Life 68(1):72–78. https://doi.org/10.1002/iub.1463
Cochrane SA, Findlay B, Bakhtiary A, Acedo JZ, Rodriguez-Lopez EM, Mercier P, Vederas JC (2016) Antimicrobial lipopeptide tridecaptin A1 selectively binds to Gram-negative lipid II. Proc Natl Acad Sci USA 113(41):11561–11566. https://doi.org/10.1073/pnas.1608623113
Cox MM (2007) Regulation of bacterial RecA protein function. Crit Rev Biochem Mol Biol 42(1):41–63. https://doi.org/10.1080/10409230701260258
Dewachter L, Herpels P, Verstraeten N, Fauvart M, Michiels J (2016) Reactive oxygen species do not contribute to ObgE*-mediated programmed cell death. Sci Rep 6:33723. https://doi.org/10.1038/srep33723
Dominguez DC (2004) Calcium signalling in bacteria. Mol Microbiol 54(2):291–297. https://doi.org/10.1111/j.1365-2958.2004.04276.x
Dudas A, Markova E, Vlasakova D, Kolman A, Bartosova Z, Brozmanova J, Chovanec M (2003) The Escherichia coli RecA protein complements recombination defective phenotype of the Saccharomyces cerevisiae rad52 mutant cells. Yeast 20(5):389–396. https://doi.org/10.1002/yea.971
Dwyer DJ, Camacho DM, Kohanski MA, Callura JM, Collins JJ (2012) Antibiotic-induced bacterial cell death exhibits physiological and biochemical hallmarks of apoptosis. Mol Cell 46(5):561–572. https://doi.org/10.1016/j.molcel.2012.04.027
Dwyer DJ, Belenky PA, Yang JH, MacDonald IC, Martell JD, Takahashi N, Chan CT, Lobritz MA, Braff D, Schwarz EG, Ye JD, Pati M, Vercruysse M, Ralifo PS, Allison KR, Khalil AS, Ting AY, Walker GC, Collins JJ (2014) Antibiotics induce redox-related physiological alterations as part of their lethality. Proc Natl Acad Sci USA 111(20):E2100–E2109. https://doi.org/10.1073/pnas.1401876111
Erental A, Kalderon Z, Saada A, Smith Y, Engelberg-Kulka H (2014) Apoptosis-like death, an extreme SOS response in Escherichia coli. MBio 5(4):e01426–e01414. https://doi.org/10.1128/mBio.01426-14
Evans MD, Dizdaroglu M, Cooke MS (2004) Oxidative DNA damage and disease: induction, repair and significance. Mutat Res 567(1):1–61. https://doi.org/10.1016/j.mrrev.2003.11.001
Ferullo DJ, Lovett ST (2008) The stringent response and cell cycle arrest in Escherichia coli. PLoS Genet 4(12):e1000300. https://doi.org/10.1371/journal.pgen.1000300
Gorlach A, Bertram K, Hudecova S, Krizanova O (2015) Calcium and ROS: a mutual interplay. Redox Biol 6:260–271. https://doi.org/10.1016/j.redox.2015.08.010
Green J, Paget MS (2004) Bacterial redox sensors. Nat Rev Microbiol 2(12):954–966. https://doi.org/10.1038/nrmicro1022
Hakansson AP, Roche-Hakansson H, Mossberg AK, Svanborg C (2011) Apoptosis-like death in bacteria induced by HAMLET, a human milk lipid-protein complex. PLoS One 6(3):e17717. https://doi.org/10.1371/journal.pone.0017717
Hernansanz-Agustin P, Izquierdo-Alvarez A, Sanchez-Gomez FJ, Ramos E, Villa-Pina T, Lamas S, Bogdanova A, Martinez-Ruiz A (2014) Acute hypoxia produces a superoxide burst in cells. Free Radic Biol Med 71:146–156. https://doi.org/10.1016/j.freeradbiomed.2014.03.011
Hwang IS, Lee J, Hwang JH, Kim KJ, Lee DG (2012) Silver nanoparticles induce apoptotic cell death in Candida albicans through the increase of hydroxyl radicals. FEBS J 279(7):1327–1338. https://doi.org/10.1111/j.1742-4658.2012.08527.x
Kaufmann WK, Paules RS (1996) DNA damage and cell cycle checkpoints. FASEB J 10(2):238–247
Khademian M, Imlay JA (2017) Escherichia coli cytochrome c peroxidase is a respiratory oxidase that enables the use of hydrogen peroxide as a terminal electron acceptor. Proc Natl Acad Sci USA 114(33):E6922–E6931. https://doi.org/10.1073/pnas.1701587114
Kumar S, Engelberg-Kulka H (2014) Quorum sensing peptides mediating interspecies bacterial cell death as a novel class of antimicrobial agents. Curr Opin Microbiol 21:22–27. https://doi.org/10.1016/j.mib.2014.09.001
Kumar A, Pandey AK, Singh SS, Shanker R, Dhawan A (2011) Engineered ZnO and TiO(2) nanoparticles induce oxidative stress and DNA damage leading to reduced viability of Escherichia coli. Free Radic Biol Med 51(10):1872–1881. https://doi.org/10.1016/j.freeradbiomed.2011.08.025
Lam SJ, O’Brien-Simpson NM, Pantarat N, Sulistio A, Wong EH, Chen YY, Lenzo JC, Holden JA, Blencowe A, Reynolds EC, Qiao GG (2016) Combating multidrug-resistant Gram-negative bacteria with structurally nanoengineered antimicrobial peptide polymers. Nat Microbiol 1(11):16162. https://doi.org/10.1038/nmicrobiol.2016.162
Lee W, Lee DG (2014) Magainin 2 induces bacterial cell death showing apoptotic properties. Curr Microbiol 69(6):794–801. https://doi.org/10.1007/s00284-014-0657-x
Lee J, Lee DG (2016) Concentration-dependent mechanism alteration of pleurocidin peptide in Escherichia coli. Curr Microbiol 72(2):159–164. https://doi.org/10.1007/s00284-015-0937-0
Lee P, Tan KS (2015) Effects of Epigallocatechin gallate against Enterococcus faecalis biofilm and virulence. Arch Oral Biol 60(3):393–399. https://doi.org/10.1016/j.archoralbio.2014.11.014
Li T, Jiang L, Chen H, Zhang X (2008) Characterization of excitability and voltage-gated ion channels of neural progenitor cells in rat hippocampus. J Mol Neurosci 35(3):289–295. https://doi.org/10.1007/s12031-008-9065-7
Matic I (2017) The major contribution of the DNA damage-triggered reactive oxygen species production to cell death: implications for antimicrobial and cancer therapy. Curr Genet. https://doi.org/10.1007/s00294-017-0787-3
Matsuzaki K (2009) Control of cell selectivity of antimicrobial peptides. Biochim Biophys Acta 1788(8):1687–1692. https://doi.org/10.1016/j.bbamem.2008.09.013
Mishra S, Imlay J (2012) Why do bacteria use so many enzymes to scavenge hydrogen peroxide? Arch Biochem Biophys 525(2):145–160. https://doi.org/10.1016/j.abb.2012.04.014
Nagamalleswari E, Rao S, Vasu K, Nagaraja V (2017) Restriction endonuclease triggered bacterial apoptosis as a mechanism for long time survival. Nucleic Acids Res 45(14):8423–8434. https://doi.org/10.1093/nar/gkx576
Nie X, Gu J, Lu J, Pan W, Yang Y (2009) Effects of norfloxacin and butylated hydroxyanisole on the freshwater microalga Scenedesmus obliquus. Ecotoxicology 18(6):677–684. https://doi.org/10.1007/s10646-009-0334-1
Nordberg J, Arner ES (2001) Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic Biol Med 31(11):1287–1312
Panteleev PV, Bolosov IA, Balandin SV, Ovchinnikova TV (2015) Design of antimicrobial peptide arenicin analogs with improved therapeutic indices. J Pept Sci 21(2):105–113. https://doi.org/10.1002/psc.2732
Panteleev PV, Myshkin MY, Shenkarev ZO, Ovchinnikova TV (2017) Dimerization of the antimicrobial peptide arenicin plays a key role in the cytotoxicity but not in the antibacterial activity. Biochem Biophys Res Commun 482(4):1320–1326. https://doi.org/10.1016/j.bbrc.2016.12.035
Park C, Cho J, Lee J, Lee DG (2011) Membranolytic antifungal activity of arenicin-1 requires the N-terminal tryptophan and the beta-turn arginine. Biotechnol Lett 33(1):185–189. https://doi.org/10.1007/s10529-010-0402-x
Rios Colombo NS, Chalon MC, Navarro SA, Bellomio A (2018) Pediocin-like bacteriocins: new perspectives on mechanism of action and immunity. Curr Genet 64(2):345–351. https://doi.org/10.1007/s00294-017-0757-9
Shah AA, Wang C, Yoon SH, Kim JY, Choi ES, Kim SW (2013) RecA-mediated SOS response provides a geraniol tolerance in Escherichia coli. J Biotechnol 167(4):357–364. https://doi.org/10.1016/j.jbiotec.2013.07.023
Sharma V, Sakai Y, Smythe KA, Yokobayashi Y (2013) Knockdown of recA gene expression by artificial small RNAs in Escherichia coli. Biochem Biophys Res Commun 430(1):256–259. https://doi.org/10.1016/j.bbrc.2012.10.141
Shenkarev ZO, Balandin SV, Trunov KI, Paramonov AS, Sukhanov SV, Barsukov LI, Arseniev AS, Ovchinnikova TV (2011) Molecular mechanism of action of beta-hairpin antimicrobial peptide arenicin: oligomeric structure in dodecylphosphocholine micelles and pore formation in planar lipid bilayers. Biochemistry 50(28):6255–6265. https://doi.org/10.1021/bi200746t
Shkilnyj P, Colon MP, Koudelka GB (2013) Bacteriophage 434 Hex protein prevents recA-mediated repressor autocleavage. Viruses 5(1):111–126. https://doi.org/10.3390/v5010111
Tanwar J, Das S, Fatima Z, Hameed S (2014) Multidrug resistance: an emerging crisis. Interdiscip Perspect Infect Dis 2014:541340. https://doi.org/10.1155/2014/541340
Troxell B, Zhang JJ, Bourret TJ, Zeng MY, Blum J, Gherardini F, Hassan HM, Yang XF (2014) Pyruvate protects pathogenic spirochetes from H2O2 killing. PLoS One 9(1):e84625. https://doi.org/10.1371/journal.pone.0084625
Xu W, Zhu X, Tan T, Li W, Shan A (2014) Design of embedded-hybrid antimicrobial peptides with enhanced cell selectivity and anti-biofilm activity. PLoS One 9(6):e98935. https://doi.org/10.1371/journal.pone.0098935
Zgur-Bertok D (2013) DNA damage repair and bacterial pathogens. PLoS Pathog 9(11):e1003711. https://doi.org/10.1371/journal.ppat.1003711
Zhao X, Drlica K (2014) Reactive oxygen species and the bacterial response to lethal stress. Curr Opin Microbiol 21:1–6. https://doi.org/10.1016/j.mib.2014.06.008
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) Grant funded by the Korean government (MSIP) (No. 2017R1A2B4005811).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interests.
Additional information
Communicated by M. Kupiec.
Electronic supplementary material
Below is the link to the electronic supplementary material.
294_2018_855_MOESM1_ESM.tif
Fig. S1. Oxidative stress in ΔRecA and K996 cells treated with arenicin-1. ΔRecA cells were treated with arenicin-1 (12.5 µM) or norfloxacin (2.3 µM). K996 cells were treated with arenicin-1 (12.5 µM) or norfloxacin (2.3 µM). (A) ROS production was detected via H2DCFDA staining (B) Decrease in GSH levels indicate oxidative damage. (C) Increase in MDA levels indicate lipid peroxidation. The data represents the average, standard deviation, and p values from three independent experiments (*p < 0.1; **p < 0.05; ***p < 0.01 vs the untreated; Student’s t test) (TIF 10063 KB)
294_2018_855_MOESM2_ESM.tif
Fig. S2. Features of apoptotic-like death in ΔRecA and K996 cells induced by arenicin-1. Analysis of membrane depolarization in ΔRecA and K996 cells in the presence/absence of arenicin-1 and ROS scavenger. Flow cytometric analysis of membrane depolarization using DiBAC4(3) (TIF 12006 KB)
294_2018_855_MOESM3_ESM.tif
Fig. S3 Features of apoptotic-like death in ΔRecA and K996 cells induced by arenicin-1. Analysis of cell filamentation in ΔRecA and K996 cells treated with/without arenicin-1 and ROS scavenger. Induction-mediated cell size changes. An increase in FS and SS indicated cell filamentation (TIF 13952 KB)
Rights and permissions
About this article

Cite this article
Lee, H., Lee, D.G. Arenicin-1-induced apoptosis-like response requires RecA activation and hydrogen peroxide against Escherichia coli. Curr Genet 65, 167–177 (2019). https://doi.org/10.1007/s00294-018-0855-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-018-0855-3