
Abstract
In the current study, a porous composite material, hydroxyapatite/chitosan/microcrystalline-cellulose (CaHAp–CS–MCC), was successfully prepared and used for the adsorption of Eriochrome Black T (EBT) from water. X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FTIR), scanning electron microscopy (SEM), differential thermal analysis (DTA), thermogravimetric analysis (TGA), specific surface area (SSA), and atomic force microscope (AFM) were used to evaluate the physicochemical properties of the prepared adsorbents. CaHAp–CS–MCC exhibited a decrease in crystallinity and reduction in crystallite size. CaHAp–CS–MCC surface exhibited cracks and pores. AFM displayed changes in surface micro-texture of CaHAp, formation of new surface structures, and variations in particle size and distribution. Upon grafting CaHAp with CS and MCC, its specific surface area decreased from 139.224 to 11.775 m2/g. BET suggested that filling CaHAp pores by CS and MCC contributed to the reduction in surface area. The pore volume values decreased from 2.56 × 10–1 (CaHAp) to 3.91 × 10–2 cm3/g (CaHAp–CS–MCC). Adsorption equilibrium was reached at about 40 min. The adsorption capacities values of CaHAp–CS–MCC and CaHAp were equal to 248 mg/g and 92 mg/g, respectively. The pseudo-second order and the Langmuir equations fitted well the adsorption phenomenon. The calculated thermodynamic parameters showed an exothermic and non-spontaneous system.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Dyes are widely used in industries including paper and textiles. As a result, the world produces more than one million tons of dyes annually. This contributes to the serious environmental problem of dye pollution [1, 2]. Even minimal traces of dyes in water sources not only cause visual displeasure but also pose potential health risks [3, 4]. The complexity of dye molecules, which show exceptional resistance to deterioration when exposed to light, temperature, and chemicals, makes these challenges much tougher. Consequently, the complete removal of dyes through the common conventional methods becomes a daunting task [5]. The lack of effective dye treatment prior to their discharge into the environment can result in the generation of toxic and carcinogenic byproducts, highlighting the urgent need for control and containment [4,5,6].
In response to these pressing concerns, adsorption has emerged as a highly promising solution for environmental remediation. Adsorption, which is a process involving the adherence of molecules to solid surfaces, could offer an efficient and straightforward method to improve the water quality [7,8,9]. In particular, hydroxyapatite (CaHAp) has been used as an effective adsorbent due to its environmental friendliness, non-toxicity, non-inflammatory, non-immunogenicity, low cost, and crystallinity [10,11,12,13,14]. To improve its function as an adsorbent, the surface of CaHAp can be modified by certain organic and inorganic substance [15]. According to literature, a number of composites based on CaHAp have been effectively used to eliminate dangerous dyes from water. For instance, hydroxyapatite–chitosan–montmorillonite was used to eliminate Indigo Carmine [16]. In other study, a composite based on hydroxyapatite biocrystal and poly (aniline-o-phenylenediamine) copolymer was employed to adsorb orange G and thymol blue [17]. Also, graphene oxide-doped nano-hydroxyapatite was used to remove Congo Red and Trypan Blue [18]. In earlier research, we looked at how sodium alginate [19], λ-carrageenan [20], and sodium silicate [21] can modify hydroxyapatite using the co-precipitation method. High adsorption capabilities toward cationic dye molecules were shown by the produced hybrid composites.
Eriochrome Black T, containing a mono azo group (N = N), is a largely used dye in textile and carpet manufacturing industries [22]. It is a toxic and potential environmental and public health concern [23]. It has been shown that direct contact of Eriochrome with skin leads to alterations in metabolic and antioxidant enzymes activities [24].
Biopolymers including chitosan and cellulose are getting more attention because they are non-toxic, renewable, and biocompatible [16]. The presence of amino and hydroxyl groups in their structures makes these materials very reactive. Consequently, their blend with inorganic particles may offer good properties. Hydroxyapatite surface exhibits poor adsorption properties toward anionic pollutants. Considering the biocompatibility, hydrophilicity, and porosity of hydroxyapatite, chitosan, and microcrystalline cellulose, the current work is devoted to prepare a porous composite that combine the properties of hydroxyapatite, chitosan, and microcrystalline cellulose and evaluate the effectiveness of the resulting composite to eliminate Eriochrome Black T from water. XRD, FT-IR, SEM, TGA, DTA, SSA, and AFM techniques were used to analyze the prepared materials. In order to better understand how various parameters can affect the adsorption process, different experimental parameters were investigated which include pH, contact time, initial concentration, and temperature. Pseudo-first order, pseudo-second order, Elovich, and intraparticle diffusion were used to analyze the kinetic data. However, the isotherms were investigated using Langmuir, Freundlich and Temkin equations.
Experimental
Materials and methods
Ca(NO3)2.4H2O, (> 99%) and (NH4)2HPO4 (> 99%) were purchased from Sigma-Aldrich. Chitosan (DD = 72.5%) and microcrystalline cellulose were used as biopolymers to modify hydroxyapatite. Eriochrome Black T (an anionic dye) was used as adsorbate. Their physicochemical characteristics are summarized in Table 1. Sodium hydroxide (NaOH), hydrochloric acid (HCl, 0.1 M), acetic acid (CH3COOH), and ammonia solution (NH4OH) were used to adjust the pH of solution.
Synthesis of CaHAp and CaHAp–CS–MCC
Hydroxyapatite was prepared through a co-precipitation technique [20, 21]. To summarize the process, 250 mL of a solution containing Ca(NO3)2·4H2O was slowly dripped into a solution of (NH4)2HPO4, with a Ca/P ratio of 1.67. This was carried out while maintaining the mixture in a nitrogen environment to prevent any potential contamination. In order to ensure that the pH of the reaction mixture stayed above 10, a gradual addition of NH4OH solution was implemented. After the introduction of Ca(NO3)2·4H2O, the mixture underwent additional stirring for a period of 4 h and was then left to mature for 72 h at room temperature, with continuous magnetic stirring. The resultant precipitate was then filtered, undergoing thorough washing with distilled water until the pH of the filtrate reached a value close to 7. Finally, it was subjected to overnight drying at 80 °C.
The following protocol was used to prepare the CaHAp–CS–MCC solution. Firstly, an amount of 3 g of chitosan powder was mixed evenly with 3 g of microcrystalline cellulose. This preparation was designated as A. The solution B: the suspension of hydroxyapatite was prepared by mixing 4 g of CaHAp with 150 mL distilled water. The solution A was added drop by drop into the solution B. To create a homogenous mixture, the solution was continually stirred for 2 h at room temperature. Then, 3 mL of glacial acetic acid was added, and the mixture was allowed to sit under agitation until it solidified. After that, the solvent was frozen by placing the mixture solidified in a -30 °C freezer for an overnight. Finally, the sample obtained was dried and lyophilized in a lyophilizer to produce scaffolding. After immersing the resultant composite in a NaOH (0.2 M) solution for several hours, it was cleaned with deionized water to remove any traces of acetic acid. Finally, samples were dried in an oven set at 60 °C. Figure 1 describes the main synthesis steps of CaHAp–CS–MCC.

Synthesis steps of CaHAp–CS–MCC
Characterization techniques
At room temperature, X-ray powder diffractograms were produced using a PANalytical X’Pert PRO MPD fitted with a copper anticathode tube. The UATR-unit (diamond) in the 4000–400 cm−1 range of a PerkinElmer Spectrum Two ATR–FTIR was used to analyze the chemical structure of the resulting hybrid compound and calcium phosphate. Information regarding the specific surface area (SSA), pore volume pore size and particle size measurements was obtained by Brunauer–Emmett–Teller (BET) method (adsorptive/desorptive gas N2) using an ASAP 2020 (Micromeritics Instrument Corp, Norcross, GA, USA) instrument.
Using an NT-MDT NTEGRA Probe Nano Laboratory instrument (NT-MDT, Moscow, Russia), atomic force microscopy (AFM) was used to determine the surface morphology of the samples. The samples’ AFM topographies were captured in non-contact mode in atmospheric conditions (at 25 °C and less than 60% relative humidity). The scanning area was 10 µm × 10 µm with a scan speed of 10–20 μm/s, resulting in 512 × 512 pixel images. The AFM instrument was equipped with a silicon NT-MDT NSG01 cantilever (NT-MDT, Moscow, Russia) measuring 125 µm in length, 30 µm in width, and 1.5–2.5 µm in thickness, with a tip curvature radius of 10 nm. The cantilever's resonant frequency ranged from 87 to 230 kHz, and its force constant was between 1.45 and 15.1 N/m. SEM analysis was performed in secondary electron (SE) and backscattered electrons (BSE) modes with × 400–600 K magnification, acceleration voltage 100 kV, working distance 10 mm, and image resolution of 1,536 × 1,094 px.
Batch adsorption experiments
The adsorption experiments were carried out in a batch mode. 0.012 g of adsorbent and 20 mL of EBT were continuously stirred in Erlenmeyer flasks. The mixture was stirred for different periods of time at 150 rpm in order to calculate how long it would take to reach equilibrium. To ascertain the maximum adsorption capacity with respect to temperature change in the 22–60 °C range, a wide range of initial dye concentrations (from 0 to 400 mg/L) was examined. The pH values were varied from 3 to 10. Following the shaking of the dye solution, the liquid supernatant was filtered, and an UV–Vis spectrophotometer was used to measure the absorbance at 550 nm, corresponding to the maximum wavelength of absorption of EBT. The adsorbed amount of dye molecules (mg/g) was determined using the following formula:
The initial and residual dye concentrations are denoted by C0 and Ce, respectively. V is the volume of the used dye solution, and m is the mass of the used adsorbent.
The adsorption kinetics were investigated using pseudo-first order [25], pseudo-second-order [26], Elovich [27], and intraparticle diffusion [28] models. Their linear forms were given in the following equations:
qe is the adsorbed capacity (mg g−1) at equilibrium, qt, is the adsorbed capacity (mg g−1) at time t, k1 is the rate constant for pseudo-first order (L min−1). k2 is the rate constant for pseudo-second order (g mg−1 min−1), α (mg g−1 min−1) and β (mg g−1 min−1) are the constants of Elovich, and ki is the rate constant for intraparticle diffusion.
The isotherms were studied using Langmuir [29], Freundlich [30], and Temkin [31]. Their linear forms were described as:
\(q_{e}\) is the adsorbed amount of the adsorbate (mg·g−1),\(c_{e}\) is the remaining concentration (mg·L−1), Kl is Langmuir constant (L·mg−1), n is the heterogeneous factor of Freundlich, \(k_{F}\) is the capacity of adsorption of Langmuir [mg·g−1 (L·mg−1)1/n], and Bt and At are Temkin constants.
Zero-point contaminated pH
The adsorption process in a water-based solution depends mainly on the adsorbent material's point of zero charge (ZPC) [32, 33]. One of the most important factors in figuring out the total surface charge of the ions in the solution is the ZPC of an adsorbent. The number of positively charged surface sites is equal to the number of negatively charged sites at the point of zero charge (pHpzc), where [MO−] = [MOH2+]. Because it indicates the adsorbent's acidity or basicity as well as its net surface charge, the pHpzc of the adsorbent’s surface is very important. This value essentially indicates the pH of the solution at which acidic or basic functional groups cease to contribute [34]. This parameter is particularly important for comprehending adsorption phenomena, particularly when electrostatic forces are involved in the underlying mechanisms, which is frequently the case in biomass-related applications.
Results and discussion
FT-IR spectroscopy
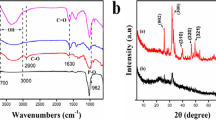
Figure 2 displays the FT-IR spectra of CS, MCC, CaHAp, and CaHAp–CS–MCC samples. The FTIR spectra of the CS sample highlight the chitosan saccharide structure by the presence of peaks in the 520–560 cm−1 spectral domain [35]. These peaks appear due to variations in the CO groups. Also, it is observed that the CS spectra are dominated by the intense bands centered at 894, 1027, 1065 and 1150 cm−1 that are specific to the saccharide structure from CS [36]. Moreover, in the CS spectra, a vibration band appears at ~ 1420 cm−1 that belongs to ν(amide III) from CS [37]. Usually, at ~ 1557 cm−1 and at ~ 1652 cm−1 could be noticed peaks that arise due to the N–H bending (amide II) and C–O stretching (amide I), in CS [35,36,37,38]. The wide peaks observed between 2874 and 3360 cm−1 could be ascribed to stretching vibration of hydroxyl groups, to the vibrational extension of the –NH2, and to the internal hydrogen bonds of the polysaccharide groups (v (OH and NH)) from CS structure [37,38,39].

FT-IR spectra of CS, MCC, CaHAp and CaHAp–CS–MCC
Furthermore, FTIR spectra of MCC exhibit various bands that are characteristic of MCC structure. The peaks characteristic of the O–H out of plane bending in C–OH alcoholic groups from MCC could be easily observed at ~ 664 cm−1 (Fig. 2) [40]. The most intense adsorption bands of MCC are centered at 892, 1052, and 1160 cm−1 and are specific for the skeletal vibrations, C–O stretching, and β-glycosidic linkages between MCC sugar units [40]. The adsorption bands from 1315 to 1408 cm−1 could be assigned to the C–H bending and to symmetrical stretching [41]. The peaks seen between 2900 and 2980 cm−1 are usually attributed to the C–H stretching vibrations in the methyl and methylene groups of MCC structure [39, 40, 42, 43]. In the meantime, O–H stretching vibrations may be the cause of the peak seen at approximately 3334 cm−1 [40]. The presence of the peak at ~ 3672 cm−1 in the FTIR spectra of MCC is commonly associated with the hydroxyl groups stretching vibrations and indicate their presence within the MCC structure [39].
The FTIR spectra of the CaHAp sample show a number of different absorption bands, such as those at 1091, 1021, 962, 599, and 561 cm−1, which are suggestive of the (PO43−) groups in the hydroxyapatite [35, 44,45,46,47,48]. Notably, two more bands are also seen in the spectrum, one at 3386 cm−1 and the other at roughly 1644 cm−1, which both point to the existence of water molecules adsorbed onto the hydroxyapatite surface [35, 44,45,46,47,48]. Subsequent examination reveals two unique bands at 874, 1336 and 1409 cm−1, corresponding to the (HPO42−) and (CO32−) groups [35, 44,45,46,47,48]. When surface modifications are applied, minor changes of peak heights and band positions are observed when the CaHAp–CS–MCC spectrum is compared with the original materials spectra. This behavior suggests that there are interactions between CaHAp–CS–MCC composite’s constituent parts (CaHAp, CS and MCC), but the distinctive peaks of CaHAp, CS and MCC can still be seen in the composite spectra. Remarkably, the strength of vibration bands corresponding to OH ions progressively decreases, probably because of the reduced degree of crystallinity brought about by these changes. Additionally, new bands emerge, corresponding to the presence of CS and MCC on the hydroxyapatite surface. For example, new vibration bands corresponding to (O–H) and (N–H) bonds appear at 3336 cm−1 and 3677 cm−1, respectively, while an additional band at 893 cm−1 indicates aromatic hydrogen that has been isolated from MCC. These findings imply that there is little interaction between HPO groups and the biopolymer. Rather, interactions between the functional groups within each biopolymer and the active Ca–OH sites on the apatitic surface result in the fixation of the biopolymer.
Therefore, in order to obtain more information regarding the subtle structure of molecular vibrations of the analyzed samples, the FTIR second derivative spectra’s (Fig. 3) were presented for the following spectral domains: 450–650 cm−1; 850–1200 cm−1 and 2800–3100 cm−1. For CS and CaHAp samples, the FTIR second derivative spectra for 2800–3100 cm−1 spectral domain are not presented due to their low contribution in this region.

FT-IR second derivative spectra of CS, MCC, CaHAp and CaHAp–CS–MCC for 450–650 cm−1 a 850–1200 cm−1 b and 2800–3100 cm−1 c spectral domains
The FTIR second derivative spectrum of CS sample obtained in the 450–650 cm−1 spectral domain highlights the presence of the polysaccharides structure vibrations such as C–O stretching vibrations in the saccharide ring, skeletal vibrations of the glucosamine units, and specific vibrations associated with C–N deformation and stretching of the amine group from CS structure [35, 46]. The range from 500 to 900 cm−1 is sensitive for the polysaccharides structure vibrations [46], according to the study published by Burkhanova, et al. [47]. On the other hand, between 850 and 1200 cm−1 (the fingerprint region), CS sample exhibits vibrational bands that arise due to the vibration of β-glycosidic bonds (at 894 cm−1), ν(C = C) at 948 cm−1, vibration of CS skeletal ν (C = O) at 1027 cm−1 and ν(C–H) at 1150 cm−1 [43]. The MCC common spectral features noticed in the 450–650 cm−1 spectral domain include peaks associated with C–O–C stretching vibrations and glycosidic bonds in the MCC structure, along with peaks that are characteristics for bending vibrations of CH2 and CH3 groups [40,41,42]. More than that, all the vibration bands observed between 850 and 1200 cm−1 (Fig. 3) for MCC sample are specific structural features for the skeletal vibrations, C–O stretching, and β-glycosidic linkages [40,41,42]. The methyl and methylene groups of the MCC structure exhibited peaks between 2833 and 2980 cm−1 that were linked to the C–H stretching vibrations [40,41,42]. The FTIR second derivative spectra of the CaHAp sample, which were obtained in the 450–650 cm−1 region, highlight the existence of vibrational bands linked to the ν2 and ν4 of the (PO4)3− groups from the CaHAp structure. The bands seen between 850 and 1200 cm−1 are specific to the ν1 and ν3 of phosphate group from CaHAp. In the case of the CaHAp–CS–MCC composite sample, the FTIR second derivative spectra confirm and sustain the results of the FTIR spectroscopy studies regarding the contribution of hydroxyapatite, microcrystalline cellulose and chitosan vibration bands to the spectrum (for all the studied spectral domains). In conclusion, the FTIR and FTIR second derivative data unequivocally demonstrate that the CaHAp–CS–MCC composite was formed.
X-Ray diffraction
Figure 4 exhibits the XRD patterns for MCC, CS, CaHAp and CaHAp–CS–MCC composite. All diffraction peaks of CaHAp were associated with a pure apatitic phase of hexagonal symmetry and P63/m space group, and they were in good agreement with the JCPDS n° 09–432 card [49]. The CaHAp–CS–MCC composite preserves the characteristic HAp structure in accordance with JCPDS n° 09–432 card [49]. On the other hand, characteristic maxima of MCC and CS were observed in the diffraction pattern of the CaHAp–CS–MCC composite. Five new peaks were identified at 2θ = 10.95°, 14.48°, 20.73°, 22.84° and 34.06°. The peaks observed at 2θ = 14.48°, 22.84°, 34.06° were associated with MCC diffraction planes (110), (002) and (310) in agreement with JCPDS n° 003–0289 card [50]. In addition, the two peaks identified at 2θ = 10.95° and 20.73° were attributed to CS in agreement with JCPDS n°067–1540 card [51]. Moreover, CaHAp–CS–MCC exhibits a decrease in intensity and a peaks broadening. The lower intensity of peaks may point to the decrease in crystallinity as a result of the presence of MCC and CS. Also, the broadening of XRD peaks is commonly linked to the reduction in crystallite size. This phenomenon occurs due to the presence of deteriorated ligands at the surface of nanoparticles. When particle size decreases, the proportion of disordered atoms at the particle surface becomes higher compared to those in the center of the bulk. XRD patterns exhibit peak broadening, and the intensities of all peaks decrease [48]. This behavior is a consequence of the nanometric dimensions and structural disorder inherent to nanoparticles.

XRD patterns of the CaHAp–CS–MCC composite, CaHAp, MCC, CS and MCC
Surface morphology characterization: AFM and SEM features
AFM was employed to analyze the surface of CaHAp. To ensure comprehensive analysis, measurements were taken at five different locations across various regions of each sample, providing a representative overview of the surface characteristics. The findings from AFM analysis were visually depicted through a selection of 2D and 3D AFM images, as shown in Fig. 5. These images illustrated the surface features of CaHAp before and after modification. Upon comparing the AFM images of CaHAp before and after modification, distinct differences in surface topography were observed. The images revealed alterations in the surface morphology, suggesting modifications induced by the presence of CS–MCC. These modifications include changes in surface micro-texture, formation of new surface structures, or variations in particle size and distribution.

a AFM of CaHAp, and b AFM of CaHAp–CS–MCC
Fractal dimension (D) represents the degree of complexity or irregularity of a structure, with higher values indicating greater complexity [52,53,54]. It is known that the box-counting dimension of a subset, denoted by D, can be computed according to the following formula [52]:
where Nδ(F) can be defined in any of the following ways: (i) as the minimum number of closed balls of radius δ > 0 needed to cover F; (ii) as the minimum number of cubes with side length δ needed to cover F; (iii) as the count of δ-mesh cubes that intersect F; (iv) as the minimum number of sets with a diameter no greater than δ needed to cover F; (v) as the maximum number of disjoint balls with radius δ and centers within F.
The basic parameters of the height values distribution for the surface micro-texture provide quantitative measures and essential insights into the surface morphology and roughness characteristics [55,56,57,58]. A summary of the basic parameters of the height values distribution of the surface samples, including fractal dimensions, with coefficients of correlation (R2), determined by the cube counting method [52], based on the linear interpolation type, is shown in Table 2. The correlation coefficients (R2) for all linear fits were 0.995, indicating a stronger correlation.
The statistical analyses were conducted utilizing the GraphPad InStat version 3.20 software package (GraphPad, San Diego, CA, USA). The normal distribution of quantitative variables was assessed using the Kolmogorov–Smirnov test.
The independent samples T test was used to compare several areas within the same sample. When statistical significance was found, the Mann–Whitney U test was used to compare two groups more thoroughly. P values of 0.05 or less were used to define statistical significance for differences. The mean values and standard deviations for the average results were provided.
The (Sa) parameter represents the arithmetic mean height, indicating the average roughness of the surface. For CaHAp, it is 0.038 μm, while for CaHAp–CS–MCC, it is considerably higher at 0.335 μm, suggesting a rougher surface texture in the latter. (Sq) measures the root mean square height of the surface. Similar to (Sa), the values for CaHAp–CS–MCC are notably larger than those for CaHAp, indicating a higher overall surface roughness. Skewness (Ssk) represents the third statistical moment, providing a measure of the symmetry of the height distribution. Kurtosis (Sku) represents the fourth statistical moment and reflects the flatness of the height distribution. Both parameters suggest slightly different distributions between the two surfaces, with CaHAp–CS–MCC having a slightly higher skewness and kurtosis compared to CaHAp. Inclination parameters (θ and φ) indicate the tilt or orientation of surface features. CaHAp–CS–MCC exhibits slightly higher inclinations in both θ and φ angles compared to CaHAp. Lastly, the parameter D represents the fractal dimension, providing insights into the surface complexity [59]. CaHAp–CS–MCC shows a slightly higher fractal dimension compared to CaHAp, indicating a more complex surface structure. These topographies can be attributed to the interaction between CaHAp and CS-MCC in the CaHAp–CS–MCC hybrid compound. These observations are consistent with the decrease in crystallinity degree and crystallite size observed in the XRD patterns.
Furthermore, SEM analysis was utilized to delve deeper into the surface characteristics of CaHAp. The SEM images provided detailed insights into the morphology and structural properties of both CaHAp particles and CaHAp–CS–MCC. When observing the SEM image of unmodified CaHAp particles (Fig. 6a), several notable features were evident. Firstly, irregularities in size and shape were observed, indicating variations in particle morphology. Additionally, there was a distinct tendency for the particles to aggregate, suggesting a propensity for particle–particle interactions leading to clustering or agglomeration. In contrast, SEM image of CaHAp–CS–MCC (Fig. 6b) reveals a different surface landscape. Here, the surface exhibits various features such as cracks and pores, which were not as prominently observed in the unmodified CaHAp particles. These features suggest interactions between the hydroxyapatite and the chitosan-based biopolymers used for modification. The presence of cracks and pores can be attributed to the grafting of biopolymers onto the surface of CaHAp, indicating a modified surface structure resulting from the interaction between the two materials. These observations deepen our understanding of the structural changes and interactions occurring at the surface level of the samples.

SEM images of CaHAp, and CaHAp–CS–MCC composite
Thermal analysis
The decomposition behavior of CaHAp–CS–MCC composite is depicted in Fig. 7. In the TGA curves, four distinct steps are observed. The first phase, with weight loss occurring between 25 °C and 177 °C (approximately 5% mass loss), is attributed to the desorption of water and acetic acid from the surface. The thermal degradation of chitosan is linked to the second phase, which is detected between 177 and 360 °C and exhibits a 40% mass loss. The final weight loss, approximately 30%, is detected between 360 and 650 °C, and it is believed to result from the breakdown of organic matter, specifically MCC. These four mass losses correspond to four endothermic peaks observed in the ATD at 80 °C, 344 °C, 489 °C, and 569 °C. The last three losses are absent from the DTA plot of CaHAp and are attributed to the breakdown of chitosan and MCC.

TGA and DTA curves of CaHAp, and b CaHAp–CS–MCC powders
Textural properties
The surface characteristics of CaHAp and CaHAp–CS–MCC samples acquired by the BET technique are shown in Table 3. Upon grafting CaHAp with CS and MCC biopolymers, its specific surface area decreases from 139.224 to 11.775 m2/g compared to unmodified CaHAp. The BET results suggest that the filling of CaHAp surface pores by CS and MCC contributes to the reduced surface area of CaHAp–CS–MCC compared to CaHAp. Additionally, the pore volume also decreases, from 2.56·10–1 to 3.91·10–2 cm3/g.
Adsorption of Eriochrome Black T onto CaHAp and CaHAp–CS–MCC
Effect of experimental conditions
Figure 8a illustrates the pHpzc (point of zero charge) for CaHAp and CaHAp–CS–MCC. The curves exhibit similar shapes. Initially, as the pH increases to 6, the adsorbent surface absorbs the added H+ ions from the solution. The final pH values remain relatively stable within the pH range of 6–9, indicating buffering properties of the samples. On the other hand, when the starting pH is higher than 8, the end pH falls because the material’s surface sites deprotonate and consume OH− ions. The pHpzc values of 6.25 and 6.89 were observed on the surface of CaHAp and CaHAp–CS–MCC composite, respectively. The difference between the initial and final pH values, along with the buffering lengths of the pH area, were used to extrapolate the behavior of synthetic CaHAp (with a pHpzc = 5.6). This suggests that the surface charge of the adsorbents is positive below pH = pHpzc, favoring the adsorption of anionic species, while it becomes negative above pHpzc, facilitating the adsorption of cationic species.

a Plots of pHpzc of CaHAp and CaHAp–CS–MCC, b Effect of pH on the adsorption of EBT using CaHAp and CaHAp–CS–MCC, c Evolution of the EBT adsorbed quantity as a function of time, d Effect of initial concentration on EBT adsorption, and e, f effect of temperature using CaHAp and CaHAp–CS–MCC
Managing the adsorption processes involves considering the pH level of the dye solution, as it directly affects the adsorption capacity. The effectiveness of adsorption relies on the pH of the solution because changes in pH influence the surface characteristics of the adsorbent and the degree of ionization of the adsorptive molecule. In this section, experiments were conducted across pH values ranging from 3 to 11 to explore the adsorption of EBT on CaHAp and the CaHAp–CS–MCC composite (Fig. 8b) (C0 = 10 mg/L, time = 120 min, and T = 22 °C). At pH = 5, the maximum adsorption capacities of CaHAp and CaHAp–CS–MCC were found to be 14.8 mg/g and 13.1 mg/g, respectively. Indeed, EBT, being an anionic dye with negatively charged sulfonate groups (-SO3−) in aqueous solution, exhibits anionic behavior. The adsorption capacity of EBT for both CaHAp and CaHAp–CS–MCC noticeably increases as the solution pH is raised from 3 to 5. This observation can be attributed to changes in the surface charge of the adsorbent, which enhances the adsorption capacity in more acidic solutions. The surface charge of the adsorbents becomes more positive at pH values lower than the pHpzc, facilitating electrostatic attraction and EBT adsorption. Conversely, electrostatic repulsions between the negatively charged adsorbent and anionic dye increase in the basic pH range. Since the pH is above the pHpzc, the surfaces of CaHAp and CaHAp–CS–MCC are negatively charged, resulting in a decrease in available sites for EBT removal and a higher likelihood of the dye being repelled. Both CaHAp and CaHAp–CS–MCC exhibit a preference for lower pH values in EBT adsorption. Therefore, the adsorption of EBT is highly influenced by pH.
The evolution of the adsorbed amount of EBT with the reaction time is assessed in Fig. 8c (C0 = 10 mg/L, pH = 5, and T = 22 °C). The adsorption of EBT onto CaHAp and CaHAp–CS–MCC composite materials was studied at different time intervals (0 and 240 min). The adsorption process exhibits an initial rapid phase, predominantly within the first 120 min, followed by a gradual slowdown. This phenomenon can be explained by the sorption sites on the sample surfaces being depleted. The slight decrease in adsorption rate beyond 120 min is due to EBT molecules occupying most of the adsorption sites within the adsorbents’ pores.
The variation of the initial EBT concentration is investigated in Fig. 8d while maintaining constant values for pH, temperature, and contact time (time = 120 min, pH = 5)). Results show that when the initial concentration of EBT rose from 10 mg/L to 400 mg/L, CaHAp’s adsorption ability improved considerably. For CaHAp and CaHAp–CS–MCC, the highest adsorption capabilities are 92 mg/g and 248 mg/g, respectively. As the adsorption process proceeds, EBT molecules bonded to the material’s surface layer through interaction with surface-active sites. The higher adsorption capacity observed for the prepared porous composite was attributed to the increased availability of surface area in the adsorbents, enabling them to compete effectively with dye ions at higher initial dye concentrations. At a specific concentration threshold, the saturation of adsorption sites was noted, suggesting that the adsorbent’s surface had the same active centers. As the initial adsorbate concentration increased, these active centers became saturated over time, making it increasingly difficult for additional surface sites to be occupied due to repulsive forces between the EBT molecules on the surface of CaHAp and CaHAp-MCC and the solution phase.
Finally, the removal of EBT using CaHAp and CaHAp–CS–MCC as adsorbents at three distinct temperature ranges, 20 °C, 40 °C, and 60 °C, is described in Figs. 8 e, f. Results indicate that as the solution temperature increased from 22 to 60 °C, the removal of EBT dye decreased. These results suggest that the adsorption of EBT dye onto CaHAp–CS–MCC is an exothermic process. The decrease in EBT dye removal with increasing temperature can be attributed to the weakening of attractive forces between the EBT dye molecules and the active sites of CaHAp–CS–MCC. The concept of physisorption is further supported by the fact that interactions between adjacent molecules weaken as temperature increases in the adsorbed phase.
Adsorption kinetics
The adsorption of EBT onto the surfaces of CaHAp and CaHAp–CS–MCC was explored using various kinetic models, including Elovich, pseudo-first-order, pseudo-second-order, and intraparticle diffusion models (Fig. 9; Table 4). Based on the examination of linear regression correlation coefficients (R2 values and adsorption capabilities, qe), the best model was chosen. As shown in Table 4, the pseudo-second-order model among the models under investigation showed significantly higher R2 values, almost reaching unity. Furthermore, the closely matched calculated qe values provided further support for the applicability of this adsorption model. This finding suggests that the adsorption of EBT onto CaHAp and CaHAp–CS–MCC is chemical [60]. According to the plots of the intraparticle diffusion, the plots are noticeably diverged from the origin. This demonstrates that diffusion step and more than kinetic processes may possibly governor the adsorption phenomenon of EBT molecules [26].

Kinetic plots of: a pseudo-first order, b pseudo-second order, c Elovich, and d intraparticle diffusion
Adsorption isotherms
The relationship between the equilibrium concentration of the adsorbate material and the amount adsorbed at a constant temperature is captured by the adsorption isotherms. Its significance extends beyond academia to real-world applications, serving as a crucial tool for understanding the intricate interactions between adsorbate and adsorbent within diverse systems. Derived parameters from various models offer valuable insights into the adsorbent’s affinities, surface properties, and the underlying mechanisms of adsorption. Within our study, we employed the theoretical isotherms such as the Freundlich, Temkin, and Langmuir models. The corresponding plots and the computed parameters are depicted in Figs. 10 and 11 and Table 5, respectively. The regression coefficients observed for Langmuir are higher than 0.98 which are more important compared to Freundlich (0.93 < R2 < 0.98) and Temkin (0.89 < R2 < 0.96). This trend indicates that the adsorption phenomenon of EBT, using CaHAp and CaHAp–CS–MCC, complies well with Langmuir model. This postulates the existence of uniform adsorption sites favorable to monolayer formation by anion species [30]. The favorability of the adsorption phenomenon could be, moreover, considered regarding the parameter “n” calculated from Freundlich equation. The calculated values show 0 < n suggesting a favorable phenomenon [31].

Plots of the isotherms of EBT for CaHAp: a Langmuir, b Freundlich, and c Temkin

Plots of the isotherms of EBT for CaHAp–CS–MCC: a Langmuir, b Freundlich, and c Temkin
The thermodynamic parameters such as the enthalpy (ΔH°), Gibbs energy (ΔG°), and the entropy (ΔS°) values were determined using the following equations [61]:
where Ce is the equilibrium concentration (mg/L), qe is the adsorbed amount of dye (mg/g) at equilibrium, R is the gas constant (J/K/mol), and T is the temperature (K).
Plots of ln Kd versus 1/T (Fig. 12) were used to calculate the changes in free energy (ΔG°), enthalpy (ΔH°), and entropy (ΔS°). Table 6 summarizes the calculated thermodynamic parameters. Notably, the Gibbs free energy values were determined to be positive. These positive ΔG° values show that the adsorption system is non-spontaneous. The adsorption of EBT was identified as exothermic, as evidenced by the negative ΔH° values observed at different temperatures, suggesting the predominance of physical adsorption in this process. Additionally, the entropy values, which reflect the disorderliness of a system, was observed to be negative. The decrease in entropy arises from the reduced randomness resulting from the interaction between the adsorbent and adsorbate. In a solid–liquid interface, the negative ΔS° values indicate a decrease in the degrees of freedom for the adsorbed EBT dye molecules, suggesting an increase in EBT dye concentration on the CaHAp–CS–MCC surface. ease in disorder and randomness at the solid–liquid interface during the adsorption process.

Plots of Ln Kd versus the inverse of temperature
Adsorption mechanism
The adsorption mechanism of EBT using CaHAp–CS–MCC can be interpreted by the electrostatic interactions between the negatively charged molecules of EBT and positively charged surface of the composite due to the presence of reactive amino groups of chitosan. In addition, hydrogen bonds can also occur between hydroxyl groups of either chitosan or cellulose and (–P–OH) group of CaHAp. However, the adsorption of EBT using CaHAp can be conducted due to the electrostatic interactions between Ca2+ of CaHAp and the sulfonate groups of EBT.
Conclusion
In this study, CaHAp–CS–MCC was successfully prepared, characterized, and employed as an efficient adsorbent of EBT dye from water. XRD results indicated that the crystallinity and the crystallite size were reduced after the addition of chitosan and cellulose to CaHAp. The morphological characteristics of the prepared composite were changed showing cracks and pores. The AFM revealed changes in surface micro-texture of CaHAp and variations in particle size and distribution. The specific surface area decreased from 139.224 (CaHAp) to 11.775 m2/g (CaHAp–CS–MCC). The pore volume decreased from 2.56 × 10–1 (CaHAp) to 3.91 × 10–2 cm3/g (CaHAp–CS–MCC). The adsorption of Eriochrome, using CaHAp and CaHAp–CS–MCC, depended on several experimental parameters including pH, time, temperature, and initial dye concentration. Notably, the kinetics of EBT adsorption exhibited a rapid adsorption process, achieving equilibrium in 40 min. Remarkably, the adsorption data complied well with the pseudo-second order and Langmuir equations. Thermodynamic analysis exhibited the non-spontaneity of the EBT adsorption process and an exothermic nature across 22 °C to 60 °C temperature range. Given its affordability and biocompatibility, the utilization of CaHAp–CS–MCC for EBT adsorption presents a promising and environmentally responsible approach to pollution control. These attributes underscore the potential for adopting a more sustainable stance in addressing water pollution challenges. In order to lessen negative effects on the environment and public health, it is critical to address these concerns efficiently. Therefore, prioritizing the implementation of environmentally friendly technologies for managing solid waste and wastewater is essential to ensure a cleaner and more sustainable future.
Data availability
No datasets were generated or analyzed during the current study.
References
Al-Tohamy R, Ali SS, Li F, Okasha KM, Mahmoud YA-G, Elsamahy T, Jiao H, Yinyi F, Sun J (2022) A critical review on the treatment of dye-containing wastewater: ecotoxicological and health concerns of textile dyes and possible remediation approaches for environmental safety. Ecotoxicol Environ Safety 231:113160. https://doi.org/10.1016/j.ecoenv.2021.113160
Vassalini I, Maddaloni M, Depedro M, De Villi A, Ferroni M, Alessandri I (2024) From water for water: PEDOT:PSS-chitosan beads for sustainable dyes adsorption. Gels 10:37
Akartasse N, Azzaoui K, Mejdoubi E, Hammouti B, Elansari LL, Abou-salama M, Aaddouz M, Sabbahi R, Rhazi L, Siaj M (2022) Environmental-friendly adsorbent composite based on hydroxyapatite/hydroxypropyl methyl-cellulose for removal of cationic dyes from an aqueous solution. Polymers 14:2147
Martínez-Rico Ó, Blanco L, Domínguez Á, González B (2023) Removal of azo dyes orange II and reactive black 5 from aqueous solutions by adsorption on chitosan beads modified with choline chloride: urea deep eutectic solvent and FeO. Separations 10:426
Barka N, Abdennouri M, Makhfouk ME (2010) Removal of methylene blue and eriochrome black t from aqueous solutions by biosorption on Scolymus hispanicus L.: kinetics, equilibrium and thermo dynamics. J Taiwan Inst Chem Eng 42:320–326
Rajabi M, Mahanpoor K, Moradi O (2017) Removal of dye molecules from aqueous solution by carbon nanotubes and carbon nanotube functional groups: critical review. RSC Adv 7:47083–47090
Parimelazhagan V, Chinta A, Shetty GG, Maddasani S, Tseng W-L, Ethiraj J, Sundaram GA, Kumar ASK (2024) Process optimization and equilibrium, thermodynamic, and kinetic modeling of toxic congo red dye adsorption from aqueous solutions using a copper ferrite nanocomposite adsorbent. Molecules 29(2):418. https://doi.org/10.3390/molecules29020418
Daniela P, Iconaru SL, Predoi M, Motelica-Heino M (2020) Removal and oxidation of As(III) from water using iron oxide coated CTAB as adsorbent. Polymers 12:1687
Najah M, Araissi M, Mhamdi M, Elaloui E (2022) Valorisation, characterisation and application of natural materials (Zeolite and Chert) as adsorbents for the removal of chromium (III) from an aqueous solution. Water Air Soil Pollut 233:246
Predoi SA, Ciobanu SC, Chifiriuc MC, Motelica-Heino M, Predoi D, Iconaru SL (2023) Hydroxyapatite nanopowders for effective removal of strontium ions from aqueous solutions. Materials 16:229
Zhou H, Yang M, Zhang M, Hou S, Kong S, Yang L, Deng L (2016) Preparation of Chinese mystery snail shells derived hydroxyapatite with different morphology using condensed phosphate sources. Ceram Int 42:16671–16676
Udoetok IA, Karoyo AH, Mohamed MH, Wilson LD (2024) Chitosan biocomposites with variable cross-linking and copper-doping for enhanced phosphate removal. Molecules 29:445
Alfuraydi RT, Al-Harby NF, Alminderej FM, Elmehbad NY, Mohamed NA (2023) Poly (Vinyl alcohol) hydrogels boosted with cross-linked chitosan and silver nanoparticles for efficient adsorption of congo red and crystal violet dyes. Gels 9:882
Deng L, Li Y, Zhang A, Zhang H (2020) Nano-hydroxyapatite incorporated gelatin/zein nanofibrous membranes: fabrication, characterization and copper adsorption. Int J Biol Macromol 154:1478–1489
Teng S-H, Lee E-J, Wang P, Kim H-E (2008) Collagen/hydroxyapatite composite nanofibers by electrospinning. Mater Lett 62:3055–3058
Joudi M, Nasserlah H, Hafdi H et al (2020) Synthesis of an efficient hydroxyapatite–chitosan–montmorillonite thin film for the adsorption of anionic and cationic dyes: adsorption isotherm, kinetic and thermodynamic study. SN Appli Sci 2:1078
Aziz K, El Achaby M, Jacquemin J, Kurniawan TA, Aziz F (2023) A composite based on hydroxyapatite biocrystal and poly(aniline-o-phenylenediamine) copolymer for efficient removal of anionic dyes from wastewater. J Taiwan Ins Chem Eng. https://doi.org/10.1016/j.jtice.2023.105256
Prabhu SM, Abuzar Khan M, Farzana H, Hwang GC, Lee W, Lee G (2018) Synthesis and characterization of graphene oxide-doped nano-hydroxyapatite and its adsorption performance of toxic diazo dyes from aqueous solution. J Mol Liquids 269:746–754. https://doi.org/10.1016/j.molliq.2018.08.044
Youssef G, Hassen A, Ridha L, Mahjoub J, Amor H (2018) Synthesis of hydroxyapatite-sodium alginate via a co-precipitation technique for efficient adsorption of Methylene Blue dye. J Mol Liq 249:912–920
Hassen A, Mahjoub J, Hatem M (2017) Synthesis, characterization of hydroxyapatite-lambda carrageenan, and evaluation of its performance for the adsorption of methylene blue from aqueous suspension. Appl Polym Sci 134(40):45385
Hassen A, Nouha S, Jabli M, Yassine E-G, khaled B (2021) Synthesis of hydroxyapatite-sodium metasilicate via double decomposition method: Characterization and application to the removal of methylene blue. Inorgc Chem Commun 133:108986
Bootharaju MS, Pradeep T (2013) Facile and rapid synthesis of a dithiol-protected Ag7 quantum cluster for selective adsorption of cationic dyes. Langmuir 29(25):8125–8132. https://doi.org/10.1021/la401180r
Morris AD, Léonce S, Guilbaud N, Tucker GC, Pérez V (1997) Eriochrome Black T, structurally related to suramin, inhibits angiogenesis and tumor growth in vivo. Anticancer Drugs 8:746–755
Srivastava A, Verma N, Mistri A, Ranjan B, Nigam AK (2017) Alterations in the skin of Labeo rohita exposed to an azo dye, Eriochrome black T: a histopathological and enzyme biochemical investigationEnviron. Sci Pollut Res 24:8671–8681
Lagergren S (1898) About the theory of so-called adsorption of soluble substances, Kungliga Svenska Vetenskapsakademiens Handlingar. Water Resource and Protection 24:1–39
Ho YS, McKay G (1999) Pseudo-second order model for sorption processes. Process Biochem 34:451–465
Cheung CW, Porter JF, McKay G (2000) Elovich equation and modified second-order equation for sorption of cadmium ions onto bone char. Chem Technol Biotechnol 75:963–970
Ahmad AA, Hameed BH (2010) Fixed-bed adsorption of reactive azo dye onto granular activated carbon prepared from waste. J Hazard Mater 175:298–303
Freundlich HMF (1906) Over the adsorption in solution. J Phys Chem 57:385–471
Langmuir I (1918) The adsorption of gases on plane surfaces of glass, mica and platinum. J Am Chem Soc 40:1361–1403
Temkin MI (1940) Kinetics of ammonia synthesis on promoted iron catalysts. Acta physiochim URSS 12:327–356
Gulicovski JJ, Čerović LS, Milonjić SK (2008) Point of zero charge and isoelectric point of alumina. Mater Manuf Processes 23:615–619
Bakatula EN, Richard D, Neculita CM, Zagury GJ (2018) Determination of point of zero charge of natural organic materials. Environ Sci Pollut Res 25:7823–7833
Agassin STR, Dognini J, Paulino AT (2023) Raw rice husk biochar as a potential valuable industrial byproduct for the removal of rhodamine B from water. Water 15:3849
Iconaru SL, Motelica-Heino M, Predoi D (2013) Study on europium-doped hydroxyapatite nanoparticles by fourier transform infrared spectroscopy and their antimicrobial properties. J Spectrosc 2013:1–9
Dennis G, Harrison W, Agnes K, Erastus G (2016) Effect of biological control antagonists adsorbed on chitosan immobilized silica nanocomposite on Ralstonia solanacearum and growth of tomato seedlings. Advances in Research 6(3):1–23
Sabaa MW, Elzanaty AM, Abdel-Gawad OF, Arafa EG (2018) Synthesis, characterization and antimicrobial activity of Schiff bases modified chitosan-graft-poly (acrylonitrile). Int J Biol Macromol 109:1280–1291
Vokhidova NR, Mamasoliyev UM, Yugay SM, Rashidova SS (2023) Synthesis and study of the structure of N-succinyl chitosan Bombyx mori and their biological applications. Polym Bull 80(12):12907–12921
Gieroba B, Kalisz G, Krysa M, Khalavka M, Przekora A (2023) Application of vibrational spectroscopic techniques in the study of the natural polysaccharides and their cross-linking process. Int J Mol Sci 24:2630
Wu LM, Tong DS, Zhao LZ, Yu WH, Zhou CH, Wang H (2014) Fourier transform infrared spectroscopy analysis for hydrothermal transformation of microcrystalline cellulose on montmorillonite. Appl Clay Sci 95:74–82
Fathy M, Abdel Moghny T, Mousa MA, El-Bellihi AHA, Awadallah AE (2016) Absorption of calcium ions on oxidized graphene sheets and study its dynamic behavior by kinetic and isothermal models. Appl Nanosci 6:1105–1117
Zhang Y, Kuga S, Wu M, Huang Y (2019) Cellulose nanosheets formed by mild additive-free ball milling. Cellulose 26:3143–3153
Gieroba B, Sroka-Bartnicka A, Kazimierczak P, Kalisz G, Lewalska-Graczyk A, Vivcharenko V, Nowakowski R, Pieta IS, Przekora A (2022) Surface chemical and morphological analysis of chitosan/1,3-β-d-glucan polysaccharide films cross-linked at 90 °C. Int J Mol Sci 23:5953
Predoi SA, Ciobanu SC, Chifiriuc CM, Iconaru SL, Predoi D, Negrila CC, Marinas IC, Raaen S, Rokosz K, Motelica-Heino M (2024) Sodium bicarbonate-hydroxyapatite used for removal of lead ions from aqueous solution. Ceram Int 50(1):1742–1755
Aissa A, Agougui H, Debbabi M (2011) Surface modification of calcium fluoro andhydroxyapatite by 1-octylphosphonic dichloride. Appl Surf Sci 257:9002–9007
Dara PK, Mahadevan R, Digita PA, Visnuvinayagam S, Kumar LRG, Suseela Mathew CN, Ravishankar RA (2020) Synthesis and biochemical characterization of silver nanoparticles grafted chitosan (Chi-Ag-NPs): In vitro studies on antioxidant and antibacterial applications. SN Appl Sci. https://doi.org/10.1007/s42452-020-2261-y
Burkhanova ND, Yugai SM, Pulatova KP, Nikononvich GV, Milusheva RY, Voropaeva NL, Rashidova SS (2000) Structural investigations of chitin and its deacetylation products. Chem Nat Compd 36:352–355
Mora-Herrera D, Pal M (2022) A comprehensive study of X-ray peak broadening and optical spectrum of Cu2ZnSnS4 nanocrystals for the determination of microstructural and optical parameters. Appl Phys A 128:1008
Besleaga C, Nan B, Popa A-C, Balescu LM, Nedelcu L, Neto AS, Pasuk I, Leonat L, Popescu-Pelin G, Ferreira JMF, Stan GE (2022) Sr and Mg doped Bi-phasic calcium phosphate macroporous bone graft substitutes fabricated by robocasting: a structural and cytocompatibility assessment. J. Funct. Biomater 13(3):123
Yan L, Wang L, Gao S, Liu C, Zhang MA, Zheng L (2020) Celery cellulose hydrogel as carriers for controlled release of short-chain fatty acid by ultrasound. Food Chem 309:125717
Jia Z, Yang C, Zhao F, Chao X, Li Y, Xing H (2020) One-Step Reinforcement and deacidification of paper documents: application of Lewis base—chitosan nanoparticle coatings and analytical characterization. Coatings 10(12):1226
Ţălu, Ș. (2015) Micro and nanoscale characterization of three dimensional surfaces. Basics and applications, Napoca Star Publishing House, Cluj-Napoca, Romania
Talu S, Abdolghaderi S, Pinto EP, Matos RS, Salerno M (2020) Advanced fractal analysis of nanoscale topography of Ag/DLC composite synthesized by RF-PECVD. Surf Eng 36(7):713–719
Ţălu Ş, Stach S, Zaharieva J, Milanova M, Todorovsky D, Giovanzana S (2014) Surface roughness characterization of poly(methylmethacrylate) films with immobilized Eu (III) β-Diketonates by fractal analysis. Int J Polym Anal Charact 19(5):404–421
Elenkova D, Zaharieva J, Getsova M, Manolov I, Milanova M, Stach S, Ţălu Ş (2015) Morphology and optical properties of SiO2-Based composite thin films with immobilized Terbium(III) complex with a biscoumarin derivative. Int J Polym Anal Charact 20(1):42–56
Ţălu Ş, Bramowicz M, Kulesza S, Ghaderi A, Dalouji V, Solaymani S, Khalaj Z (2016) Microstructure and micromorphology of Cu/Co nanoparticles: surface texture analysis. Electron Mater Lett 12(5):580–588
Ţălu Ş, Patra N, Salerno M (2015) Micromorphological characterization of polymer-oxide nanocomposite thin films by atomic force microscopy and fractal geometry analysis. Prog Org Coat 89:50–56
Solaymani S, Slawomir S, Ţălu Ş, Bramowicz M, Nezafat NB, Dalouji V, Rezaee S, Karami H, Malekzadeh M, Dorbidi ES (2018) The effect of different laser irradiation on rugometric and microtopographic features in zirconia ceramics: study of surface statistical metrics. J Alloy Compd 765:180–185
Das A, Jaiswal J, Borah CK, Ruti I, Matos RS, Pinto EP, Yadav RP, Ţălu Ş, Kumar S (2023) Correlating the nonlinear roughening and optical properties of anatase thin films - a fractal geometric approach. Adv Theory Simulat 2300238:1–12
Gucek A, Sener S, Bilgen S, Mazmanci A (2005) Adsorption and kinetic studies of cationic and anionic dyes on pyrophyllite from aqueous solutions. J Coll Interf Sci 286:53–60
Abdulhameed AS, Jawad AH, Mohammad AT (2019) Synthesis of chitosan–ethylene glycol diglycidyl ether/TiO2 nanoparticles for adsorption of reactive orange 16 dye using a response surface methodology approach. Bioresour Technol 2019(293):122071
Acknowledgements
The author extends the appreciation to the Deanship of Postgraduate Studies and Scientific Research at Majmaah University for funding this research work through the project number (R-2024-1289). The authors thank the National Institute of Materials Physics-Romania and the University of Gafsa (Tunisia).
Author information
Authors and Affiliations
Contributions
Yassine Benali and Nabil Mabrouki contributed to Conceptualization, Methodology, Software, Data curation, and Writing—original draft. Hassen Agougui and Mahjoub Jabli contributed to Methodology, Software, Data curation, and Writing—original draft. Simona Liliana Iconaru, Hatem Majdoub, Daniela Predoi, Steluța Ciobanu, Ştefan Ţălu and Khaled Boughzala contributed to Software, Data curation, Visualization, and Investigation.
Corresponding authors
Ethics declarations
Conflict of interest
The author declare that no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Benali, Y., Mabrouki, N., Agougui, H. et al. A new porous composite hydroxyapatite/chitosan/microcrystalline-cellulose: synthesis, characterization and application to the adsorption of Eriochrome Black T. Polym. Bull. (2024). https://doi.org/10.1007/s00289-024-05496-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00289-024-05496-3