
Abstract
In this study, a low-cost thermoset such as unsaturated polyester resin (UPR) was used for the preparation of lightweight and thermal insulation polymer composite using rice husk-derived silica aerogel (SA) as filler. For the first time, hydrophilic and hydrophobic silica aerogel (SA) of similar physical properties were added to the UPR to study the effects of SA surface polarity on the mechanical tensile and thermal properties of the composites. The composites with 40% and 60% of SA filler by volume were prepared via direct mixing and cured at room temperature using methyl ethyl ketone peroxide. The UPR composites were characterized and compared using density measurement, hot-disc thermal conductivity analyzer, universal testing machine, Fourier transform infrared spectroscopy, scanning electron microscopy and thermogravimetric analysis. The results of this study indicate that the filler–matrix interaction appears to be dependent on the type of SA (hydrophobic or hydrophilic), due to noticeable differences in the data values. UPR composites containing hydrophilic SA exhibit lower density and thermal conductivity due to a higher volume of preserved SA pores. Both hydrophobic and hydrophilic SA could increase the tensile stiffness, but composite with hydrophilic SA exhibit higher fracture strain, indicating higher toughness. On the other hand, composites with hydrophobic SA produced stronger hydrogen bonding interaction which increases resin viscosity and led to rougher surface morphology. However, the addition of SA, regardless of surface polarity and volume concentration had little or no effect on thermal stability except that the composite with hydrophobic SA gives a slightly higher char yield.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Silica (SiO2) is one of the most common ceramic fillers used in polymer resins to improve properties and/or reducing the cost. Natural mineral silica such as quartz and cristobalite typically exists in crystalline form while colloidal silica, pyrogenic (fumed) silica and precipitated silica are the three main classes of amorphous silica. Due to their high specific surface area, commercial availability and competitive cost, the effects of amorphous-nano silica in thermosetting compositions such as epoxies and unsaturated polyester resins (UPRs) have been intensively investigated [1]. From the available literature, improvement in the flexural strength, impact strength, and surface hardness of the composite is commonly observed by incorporating silica nanoparticles into epoxies and UPR [2,3,4,5,6,7,8]. More interestingly, several studies also demonstrate the potential of the nano-silica as a flame retardant synergist to increase polymer flame retardancy and thermal stability [8,9,10,11].
Recently, silica aerogel (SA) is a relatively new form of amorphous silica used as additives in various types of polymers. Unlike most other forms of amorphous silica, SA exhibits many interesting properties due to its three-dimensional mesoporous structure. Due to the presence of high pore volume, SA has a large internal surface area (500–1000 m2/g), high porosity (85–95%), extremely low density (0.03–0.1 g/cm3) and ultrafine pore size (5–20 nm) which results in a very low thermal conductivity (0.01–0.05 W/mK at 25 °C) [12][12]. Previously, numerous investigations have been carried out to study the effects of SA fillers in polymer resins as systematically reviewed in a review article by Salimian et al. [14]. However, SA is a lot more expensive when compared to other forms of silica due to expensive raw materials based on silicon alkoxides which dominate the overall cost [15]. To reduce cost, researchers have begun to shift their attention to natural silica sources, such as agricultural waste. Among the many types of agricultural wastes, rice husk is the optimal choice for this objective because it is very rich in silica. Nano-sized amorphous silica can be extracted via acid leaching followed by the combustion of the rice husk at 700–850 °C. This active form of silica source was found suitable as starting material for SA. The potentials of rice husk-derived silica as reinforcement in epoxies and UPRs have been investigated recently [6, 7, 10, 16, 17].
Fundamentally, SA is relatively polar (hydrophilic) since the surface is mostly covered with silanol (Si–OH) functional groups but chemical treatment can make them hydrophobic. The hydrophilic SA is vulnerable to moisture from the environment and the wetting of the nanopores will lead to the collapse of its brittle-porous structure due to capillary forces [18]. This has resulted in a higher demand for hydrophobic SA which is attributed to its longer shelf life extension compared with the hydrophilic SA [19]. Among polymer resins, the use of SA in unsaturated polyester resin (UPR) is advantageous because the resin can be cured without applying pressure; hence, it is possible to preserve the SA pores in the composite [20]. Nevertheless, studies related to the use of SA in UPR are scarce, presumably due to the expensive cost of the commercial SA and high filler loading requirement for UPR which resulted in its use being limited to certain polymers only. Filler’s surface polarity is one of the important factors to ensure good filler–matrix interaction, for example, through the formation of hydrogen bonds [21]. Since UPR comprises polar polyester segments (ketones, esters and alcohols) in the main chain and nonpolar polystyrene segments in the crosslinks [22], it is important to experimentally clarify the effect of hydrophilic and hydrophobic SA on the composite as the difference in surface functionality could lead to certain intermolecular interactions with the polymer structure which consequently affect the extrinsic properties of the composite.
Most recent aerogel scholarships, however, rather focus on common factors such as filler’s amount, particle sizes or curing techniques, while the effect of SA surface polarity on the properties of the polymer composites tends to be overlooked. As the development of SA–polymer composites are still largely in their infancy stage, researchers often select the SA based on convenience (i.e., using commercial SA) and only reconsider when there is a failure or an unexpected result. To the end, no study can be found in the literature which investigates the effects of hydrophilic and hydrophobic SA on the properties of the composite. Understanding this topic is important as surface polarity commonly affects the wettability and dispersibility of the filler in the polymer matrix. Both the dispersion and wettability of the filler on the resin are key factors that can affect the final properties of the materials and need to be assessed [23].
For the first time, this paper provides a comparative investigation of the effects of hydrophobic and hydrophilic SA on the final properties of the UPR composites. Both types of SA were proportionally mixed with UPR and the mechanical properties, morphological and chemical interaction of the composites were studied using uniaxial tensile test, scanning electron microscopy and Fourier transform infrared spectroscopy. The thermal properties of the composites were also evaluated using a thermal conductivity test and thermogravimetric analysis. The comparative results among several tests show that the composites filled with hydrophilic SA exhibit lower density, lower thermal conductivity, higher specific modulus and better toughness as compared to the composites with hydrophobic SA.
Experimental details
Materials
Unsaturated polyester resins (UPR)
Low viscosity (300–500 cP at 25 °C) orthophthalic type UPR (poly-maleic anhydride-phthalic anhydride-1,2-propanediol) containing 35–40 wt% of styrene and methyl ethyl ketone peroxide (MEKP) hardener was purchased from Reversol, Revertex Pvt. Ltd. Co., Malaysia. This cobalt accelerated resin tends to be pale pink color and fairly translucent after curing with the bulk density of around 1.12–1.15 g/cm3 when properly cured. Figure 1 shows the physical appearance of the UPR used in this study and its chemical structure.

UPR resin used in this study a uncured resin b cured resin c chemical structure of cross-linked UPR
Preparation and characterization of hydrophilic and hydrophobic silica aerogels (SA)
Figure 2a shows the preparation of the SA from rice husk in which the detail of the process was described in our earlier publication [16]. From the process, the SA particles having hydrophilic surfaces as depicted in Fig. 2b, c were first obtained before being modified. To obtain the hydrophobic SA, the as-synthesized hydrophilic SA was further modified with 5% TMCS (Trimethylchlorosilane, Cl-Si(CH3)3, 98% Tokyo Chemical Industry Co., Ltd) in n-hexane solution (RCI Labscan Ltd) for 1 h and subsequently dried at 80 °C under ambient pressure. Both types of SA exhibit the following properties; bulk density of 0.07–0.08 g/cm3, the porosity was greater than 85%, the pore diameter was less than 20 nm and surface area between 600 and 800 m2/g. Henceforth, the hydrophilic and hydrophobic SA will also be denoted as SiO2(OH) and SiO2(CH3), respectively.

a Process flow of SA synthesis from rice husk b Photograph of SA particles c SEM image of the SA particles at 20,000 × magnification, revealing pore structure
Figure 3 shows the FTIR spectra of the SiO2(OH) and SiO2(CH3). The spectra were measured in transmission mode and the peak intensity corresponds to the relative population of the functional groups on the molecular structure. The broad and intense bands for both types of SA, centered at 1100 cm−1 and 495 cm−1, are ascribed to the asymmetric and symmetric stretching vibration of siloxane (Si–O–Si) bonds [24]. More intense siloxane functional groups (1100 cm−1 and 495 cm−1) can be observed for the SiO2(CH3) as the attached hydrophobic (Si–CH3) surface groups change the porous structure and the surface area of the SA because of polymerization. The structural change on the surface of the SiO2(OH) reacted with a small concentration of TMCS is shown as follows:

Comparison of FTIR spectra for hydrophilic and hydrophobic SA
The non-polar groups as represented by the Si–C bonds (890 cm−1) and the absorption peaks at around 2980 cm−1 corresponding to C–H bonds are quite obvious for the SiO2(CH3), confirming the Si–CH3 formation during the TMCS modification [24]. Hydrophobization of the SiO2(CH3) is also evidenced by the decrease of the O–H absorption peaks at 3400 cm−1 and 1630 cm−1 as compared to the unmodified SiO2(OH). The hydrophobicity of the SiO2(OH) and SiO2(CH3) was further demonstrated by the water droplet–contact angle, θ, as depicted in Fig. 4. Good water repellency can be observed for the SiO2(CH3) based on the contact angle (higher than 90°), indicating a higher population of the Si–CH3 groups on the silica surface.

Profiles of water droplets on surfaces coated with a hydrophilic and b hydrophobic SA
Preparation and characterization of UPR/SA composites
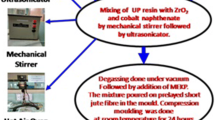
Based on the volume ratio, UPR mixtures containing 40% and 60% of SA particles were prepared as described in Table 1. The SA particles were gradually added into UPR under vigorous stirring using an overhead stirrer (HS 30E, Daihan Scientific, Korea). Then an appropriate amount of MEKP was added to initiate cross-linking and the mixtures were stirred well until their physical color changes from light pink to pale yellow. The UPR mixtures were degassed under vacuum (5–10 mmHg) for about 2 h to remove trapped air and subsequently poured into waxed molds of desired dimensions. The mixtures were left in room condition for 48 h to allow complete curing into UPR composites. For comparison, a neat UPR sample was also prepared using the same procedure.
Characterization of composites
Densities and void contents
The measured densities (Mρ) of composites were calculated by dividing the measured weight by measured volume for each composite, expressed as grams per cubic centimeter (g/cm3). The theoretical densities (Tρ) of the composites were calculated through the volume rule of mixture, by adding up the density of the SA filler and the UPR matrix respective to the volume fraction of each component as shown in Eq. (2):
where ρc, ρSA and ρupr represent the density of the composite, SA filler and the UPR matrix, respectively. By comparing the measured density to the theoretical density, the void content or porosity (Φ) was calculated based on ASTM D2734 (Void Content of Reinforced Plastics) using the equation below:
where Tρ and Mρ represent the theoretical density and measured density of the composite. However, since the SA particles themselves are already porous, the above equation of (3) was modified into Eq. (4) as the actual porosity of the filled composite is determined based on the ratio between the measured density of the filled composite and the measured density of the cured unfilled resin (neat UPR).
Thermal conductivity
The effect of porosity (void content) on the thermal conductivity of the composites was measured at room temperature using a hot disc thermal constant analyzer (Hot disc TPS 2500 s, Hot Disc AB, Sweden). Specimens in the form of 11 mm-diameter and 3 mm thick disks were prepared from the composites. The hot disk sensor (model 4922) is sandwiched between two identical disks and the measurements were carried out based on the transient plane source (TPS) method (ISO DIS 22007–2). The average thermal conductivity of each specimen was calculated from three measurements.
Tensile test and morphology of the fractured surface
The tensile properties were determined using a universal testing machine (Instron 4467, Instron Corp., Canton (MA), USA) at a crosshead speed of 1.5 mm min−1 until failure. The samples were prepared according to the standard dumbbell-shaped specimens as per ASTM D638 Type IV. The load-displacement data were plotted to evaluate the tensile properties. The tensile-fracture surfaces were examined using scanning electron microscopy (VPSEM-SU3500, Hitachi High-Technologies Co., Ltd. Japan).
Chemical bonds
The surface chemistry of the SA fillers and the UPR composites were determined using attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR, Spectrum 100, PerkinElmer Inc, USA). The infrared radiation wavenumbers used in this study was set as a mid-infra-red region (600–4000 cm−1) with a resolution of 5 cm−1.
Thermogravimetric analysis
Thermal behaviors of the UPR composites were evaluated using thermogravimetric analysis (TGA 2, Mettler Toledo, U.S) on 10 mg specimen at a heating rate of 10 °C/min under nitrogen atmospheres; with a gas flow rate of 20 ml/min. The weight loss was recorded up to 600 °C.
Results and discussions
Relationship between density, void content and thermal conductivity
Table 2 shows the comparison between the theoretical (Tρ) and the measured densities (Mρ) of the composites prepared in this study. The void contents (Φ) measures the porosity of the composites due to the addition of porous SA as fillers. A negligible difference between the Tρ and Mρ for neat UPR suggests the good quality of the cured resin, with very minimal porosity. For composites filled with SiO2(OH) and SiO2(CH3) particles, the void content or porosity of each respective composite was majorly contributed by the volume of the preserved SA pores. From the result, it is clear that the measured densities for all SA-filled composites are marginally lower than the neat UPR between 2.5 and 9%, indicating that the composites were relatively porous. The distinction in the Mρ and Φ% between the composites containing SiO2(OH) and SiO2(CH3) can be observed. The composites filled with SiO2(OH) showed lower density and therefore higher porosity, as compared to other composites with SiO2(CH3). Higher porosity means lower capillary absorption of the liquid UPR into the SA pores during the resin-filler mixing process.
Further, the relationship between the composite’s porosity and their thermal conductivity is elucidated in Fig. 5. In general, our finding is in good agreement with other studies that have reported a decrease in thermal conductivity by the addition of SA into polymer matrices [25,26,27,28,29,30]. Empirically, reductions in thermal conductivity have been found for UPR composites with increasing levels of porosity. As expected, the composites containing SiO2(OH) exhibit lower thermal conductivity and better thermal insulation, due to the higher volume of preserved SA pores in the composites. The 60SiO2(OH) has the lowest thermal conductivity (0.345 ± 0.005 W/mK), which is 24.5% lower than the neat UPR. Since porosity was mostly contributed by the nano-sized pores of SA in the composite, it is clear that the preservation of SA pores from resin intrusion is important to improve the thermal insulation of the composite as highlighted in previous works [25,26,27,28,29,30]. The nano-sized pores of SA are not only effectively limit the convection but also the conduction heat transfer [28].

Relation between thermal conductivity and porosity for neat UPR and UPR composites
Tensile testing
The load (N) versus extension graph (mm) as shown in Fig. 6 are the average values obtained from three repetitions. Table 3 shows the average values of the maximum tensile stress, strain and elastic modulus were calculated using the in-built software (Bluehill 3, Instron). In general, the load extension curves for all samples were steadily increased up to their maximum breaking load before there was a sudden drop, indicating a brittle failure. It is worth noting that all samples revealed a visco-elastic behavior in which the linear elastic region of the sample is difficult to determine. Therefore, the secant modulus of elasticity is applied to calculate the elastic modulus. The tensile data for neat UPR were fairly consistent after three repetitions with a smaller standard deviation, indicating a structural uniformity but the standard deviation for composites with SA was increased around 10–12%.

Tensile load versus extension curves of neat UPR and UPR composites
The neat UPR was found to be less elastic than the SA-filled composites as indicated by higher elongation at the break due to plastic deformation. The addition of SA fillers in UPR has significantly increased the stiffness of the matrix as evidenced by the increase in the elastic modulus with the SA loading. The improvement in the mechanical properties of polymer matrix including the modulus, strength, elasticity, and toughness by the addition of SA was also reported in many works as summarized by Salimian et.al [14]. The distribution of the SA particles has a prominent effect on the strength of the polymer via mechanical interlocking which restricts the mobility of the surrounding matrix chains and limits the plastic deformation. Moreover, the three-dimensional network of nanopores of isotropic silica particles can be partially aligned in the direction of stress, leading to further enhance the tensile properties [14].
When the SA loading was increased from 40 to 60 vol%, the composites filled with SiO2(OH) showed a higher increment of elastic modulus as compared to the composites filled with SiO2(CH3). It is interesting to found that the composites with SiO2(OH) tend to have higher fracture strain, indicating better toughness. However, composites with SiO2(CH3) were relatively stiffer as they failed at smaller strain (i.e., higher elastic modulus). This finding suggests that the addition of SiO2(OH) and SiO2(CH3) could increase the toughness and stiffness of the UPR, respectively. Similar result was also reported by [31] after incorporating hydrophobic and hydrophilic minerals into UPR. The highest elastic modulus of 263 MPa was recorded for the 60SiO2(CH3) composite while the 60SiO2(OH) composite shows the highest specific modulus of 238 MPa, due to its lower density.
Microstructure of fractured surfaces
Figure 7a is an example photo image of the failed tensile specimen depicting the brittle fracture mode of the UPR. The SEM micrograph of the fracture surface of neat UPR as shown in Fig. 7b reveals a smooth, glassy and homogeneous microstructure. Meanwhile, a similar cleavage fracture with river-like patterns was observed for 40SiO2(OH) and 60SiO2(OH) composites in Fig. 7c, d. This type of fracture surface usually involves high energy absorption, which could explain the higher values of the tensile strain for the SiO2(OH) composites [32]. Besides, no macroscopic agglomeration of SA particles or macro-voids can be seen on the morphologies, indicating good wettability of the SiO2(OH) particles with the UPR matrix. Meanwhile, Fig. 7e, f depicts rougher surface textures for the 40SiO2(CH3) and 60SiO2(CH3) composites in which the area is mostly occupied with irregular aggregates. These aggregates were formed as a result of poor filler dispersibility due to the increase of the resin viscosity during the mixing process [33]. Although some voids as presented in the 60SiO2(CH3) composite are because of the pulled-out aggregates, most of the aggregate structures, however, are remain intact after the tensile pull-off, thus implying a good mechanical interlocking between the SiO2(CH3) and the UPR matrix [33].

a Example of fractured tensile specimen. SEM images of fractured surfaces of b neat UPR c 40SiO2(OH) d 60SiO2(OH) e 60SiO2(CH3) f 60SiO2(CH3)
FTIR studies
The FTIR spectra as shown in Fig. 8 reveal the functional groups for neat UPR, 60SiO2(OH) and 60SiO2(CH3) composites after complete curing. In general, the peaks generated for all specimens are characteristic of orthophthalic UPR as further described in Table 4. Apparently, none of the SA characteristic peaks can be seen on the spectra, likely due to a relatively lower amount of SA particles on the sample’s surface for IR absorption. It is worth noting that the addition of SA fillers did not cause any change in the molecular structure of the UPR as the peaks and the corresponding wavelengths are quite identical for all specimens. Nevertheless, noticeable changes for some IR bands could still be observed between the specimens, indicating a variation in the concentration or path length for certain functional groups due to molecular interaction between SA and UPR [34]. Notable differences can be seen in the intensities of the saturated carbonyl (C=O) group near 1720 cm−1 (j) and the hydroxyl (O–H) group near 3450 cm−1 (l) due to intermolecular reaction between the UPR and the SA through the hydrogen bonding [34, 34].

FTIR spectra of neat UPR, 60SiO2(OH) and 60SiO2(CH3)
The correlation between the intensity changes and the type of SA used is shown in Fig. 9. By using the C–O–C peak near 1070 cm−1 as a reference, it is found that the peak intensity for the C=O groups near 1720 cm−1 was increased by the addition of SiO2(OH), but decreased with SiO2(CH3). The result suggests that covalent bonding has taken place through an esterification reaction between the silanols –OH from the hydrophilic SA and UPR ester (–COO–) group, forming the carboxyl (COOH) groups [36]. On the other hand, the addition of SiO2(CH3) led to an increase of the hydroxyl (–OH) peak intensity near 3450 cm−1, due to cooperative hydrogen bonding the –CH3 groups of the SiO2(CH3) with the available sites on UPR chains such as carbonyl and ester groups [34, 34]. Based on the relative OH: C–O ratio, it is clear that the composite with SiO2(CH3) contains more hydrogen bonding than the composite with SiO2(OH). The increases of the intermolecular forces between the UPR and the SA through the hydrogen bonding could increase the viscosity of the resin which affects the morphology of the composite and determines the tensile behavior [38, 38].

Calculated C=O:C–O and O–H:C–O ratios from FTIR spectra of neat UPR, 60SiO2(OH) and 60SiO2(CH3)
Thermogravimetric analysis
The TGA curves of neat UPR and UPR composites are presented in Fig. 10. For further details, the weight losses at a specific temperature for each sample are recorded in Table 5. In general, the TGA curves of neat UPR and UPR composites are relatively similar, corresponding to three degradation stages of UPR as commonly observed [40]. The first degradation stage occurs below 200 °C. Around 5 wt% of weight losses were recorded from all samples which were associated with the slow evaporation of the adsorbed moisture and volatilization of uncured substances on the surface [40]. The second degradation stage is represented by major weight loss of the UPR matrix above 300 °C up to 500 °C. More than 90 wt% losses were recorded during this stage which is ascribed to the primary chain scission of the cross-linked styrene and the main polyester chain, leaving a small amount of carbon soot as residual char [40]. The final degradation stage as observed until 600 °C shows gradual weight loss due to the decomposition of residual char. The final residual weight of the degraded composite can be used as a measure of the filler content since the UPR is completely decomposed into volatiles at 600° C.

TGA weight versus temperature curves for neat UPR and UPR composites
The degradation behavior of the UPR matrix was slightly affected by the addition of SA, whether hydrophilic or hydrophobic until the temperature reaches 400 °C. Above 400 °C, a noticeable variation in the rate of weight loss can be observed between 60SiO2(OH) and 60SiO2(CH3) composites. Comparing these two, the 60SiO2(CH3) composite showed a lower weight loss at 400 °C which finally yield a higher amount of residue. The higher char yield of the 60SiO2(CH3) indicates that the presence of SiO2(CH3) could increase char formation due to the carbonization of the –Si(CH3)3 surface groups [41].
Conclusions
In the present study, rice husk-derived SA with similar surface area and particle size but different surface polarity (hydrophilic and hydrophobic) were mixed with UPR into polymer composites. The effect of the SA surface polarity on the UPR composites was investigated using tensile test, thermal conductivity, TGA, FTIR and SEM. The noticeable differences in the data values obtained from each experiments indicate the dependency of the filler–matrix interaction on the SA surface polarity. In general, composites containing hydrophilic SA tends to be more porous, due to the higher volume of preserved SA pores in the composite. The lowest density (i.e., highest porosity) and lowest thermal conductivity were found to be 1.05 g/cm3 and 0.345 W/mK for the composite filled with 60 vol% of hydrophilic SA (60SiO2(OH)). The composites containing hydrophilic SA also showed higher values of fracture strain during tensile tests. On the other hand, the addition of hydrophobic SA results in a rougher composite’s morphology with irregular and randomly distributed aggregates, indicating that interaction between UPR and hydrophobic SA could induce a higher resin viscosity. An increase in resin viscosity was evidenced by the changes in FTIR peak intensities corresponding to C=O and –OH functional groups. A higher number of hydrogen bonding between the hydrophobic SA and UPR decreases the dispersibility of the hydrophobic SA in UPR and consequently affects the tensile behavior. In TGA studies, the thermal stability of the UPR composites was less affected by the SA surface polarity, regardless of filler loading, except that the composite with 60vol% of hydrophobic SA (60SiO2(CH3)) yield slightly higher residue at the end of the decomposition.
Declaration
On behalf of all Co-Authors, the corresponding author states that there is no conflict of interest. The corresponding author shall bear full responsibility for the submission and this research has not been submitted for publication elsewhere. We attest to the fact that all authors listed on the title page have contributed significantly to the work, have read the manuscript, attest to the validity and legitimacy of the data and its interpretation, and agree to its submission to the journal of Polymer Bulletin.
References
Rothon R (2017) Particulate fillers in thermoset plastics. In: Rothon R (ed) Fillers for polymer applications. Springer International Publishing
Rusmirović JD, Radoman T, Džunuzović ES, Džunuzović JV, Markovski J, Spasojević P, Marinković AD (2017) Effect of the modified silica nanofiller on the mechanical properties of unsaturated polyester resins based on recycled polyethylene terephthalate. Polym Compos 38(3):538–554. https://doi.org/10.1002/pc.23613
Marinković AD, Radoman T, Džunuzović ES, Džunuzović JV, Spasojević P, Isailović B, Bugarski B (2013) Mechanical properties of composites based on unsaturated polyester resins obtained by chemical recycling of poly(ethylene terephthalate). Hem Ind 67(6):913–922. https://doi.org/10.2298/HEMIND130930077M
Morote-Martínez V, Pascual-Sánchez V, Martín-Martínez JM (2008) Improvement in mechanical and structural integrity of natural stone by applying unsaturated polyester resin-nanosilica hybrid thin coating. Eur Polym J 44(10):3146–3155. https://doi.org/10.1016/j.eurpolymj.2008.07.027
Rajaee P, Ashenai Ghasemi F, Fasihi M, Saberian M (2019) Effect of styrene-butadiene rubber and fumed silica nano-filler on the microstructure and mechanical properties of glass fiber reinforced unsaturated polyester resin. Compos B Eng 173:106803. https://doi.org/10.1016/j.compositesb.2019.05.014
Pham TD, Vu CM, Choi HJ (2017) Enhanced fracture toughness and mechanical properties of epoxy resin with rice husk-based nano-silica. Polym Sci Ser A 59(3):437–444. https://doi.org/10.1134/S0965545X17030026
Bach Q-V, Vu CM, Vu HT, Nguyen DD (2019) Enhancing mode I and II interlaminar fracture toughness of carbon fiber-filled epoxy-based composites using both rice husk silica and silk fibroin electrospun nanofibers. High Perform Polym 31(9–10):1195–1203. https://doi.org/10.1177/0954008319840404
Bach Q-V, Vu CM, Nguyen DD, Kien BX (2020) Impacts of phosphorous-linked epoxidized vegetable oil on mechanical behaviors and flammability properties of silica reinforced epoxy composite. Thermochim Acta 691:178722. https://doi.org/10.1016/j.tca.2020.178722
Vu CM, Bach Q-V (2020) Effects of DOPO-grafted epoxidized soybean oil on fracture toughness and flame retardant of epoxy resin/rice husk silica hybrid. Macromol Res 28(9):826–834. https://doi.org/10.1007/s13233-020-8102-4
Vu CM, Nguyen V-H, Bach Q-V (2020) Phosphorous-jointed epoxidized soybean oil and rice husk-based silica as the novel additives for improvement mechanical and flame retardant of epoxy resin. J Fire Sci 38(1):3–27. https://doi.org/10.1177/0734904119900990
Halim ZAA, Yajid MAM, Nurhadi FA, Ahmad N, Hamdan H (2020) Effect of silica aerogel—Aluminium trihydroxide hybrid filler on the physio-mechanical and thermal decomposition behaviour of unsaturated polyester resin composite. Polym Degrad Stab 182:109377. https://doi.org/10.1016/j.polymdegradstab.2020.109377
Ebert HP (2011) Thermal properties of aerogels. In: Aegerter M, Leventis N, Koebel M (eds) Aerogels handbook. Advances in sol-gel derived materials and technologies. Springer, New York
Koch, M. L. (1986) Silica aerogel and space astrophysics, In: Aerogels, Springer Proceeding in Physics. Springer, Berlin.
Salimian S, Zadhoush A, Naeimirad M, Kotek R, Ramakrishna S (2017) A review on aerogel: 3D nanoporous structured fillers in polymer-based nanocomposites. Polym Compos 39(10):3383–3408. https://doi.org/10.1002/pc.24412
Stojanovic A, Paz Comesaña S, Rentsch D, Koebel MM, Malfait WJ (2019) Ambient pressure drying of silica aerogels after hydrophobization with mono-, di- and tri-functional silanes and mixtures thereof. Microporous Mesoporous Mater 284:289–295. https://doi.org/10.1016/j.micromeso.2019.04.038
Halim, Zulhelmi Alif Abd, Yajid, M. A. M., Idris, M. H., & Hamdan, H. (2018). Effects of rice husk derived amorphous silica on the thermal-mechanical properties of unsaturated polyester composites. Journal of Macromolecular Science, Part B. https://www.tandfonline.com/doi/abs/https://doi.org/10.1080/00222348.2018.1476440
Abdul Halim ZA, Yajid MAM, Idris MH, Hamdan H (2017) Effects of silica aerogel particle sizes on the thermal–mechanical properties of silica aerogel—Unsaturated polyester composites. Plastics Rubber Compos 46(4):184–192. https://doi.org/10.1080/14658011.2017.1306913
Christy AA (2014) The nature of silanol groups on the surfaces of silica, modified silica and some silica based materials. Adv Mater Res 998–999:3–10
Nah H, Parale VG, Lee K et al (2018) Silylation of sodium silicate-based silica aerogel using trimethylethoxysilane as alternative surface modification agent. J Sol-Gel Sci Technol 87:319–330. https://doi.org/10.1007/s10971-018-4729-4
Vu CM, Nguyen DD, Sinh LH, Choi HJ, Pham TD (2018) Micro-fibril cellulose as a filler for glass fiber reinforced unsaturated polyester composites: fabrication and mechanical characteristics. Macromol Res 26(1):54–60. https://doi.org/10.1007/s13233-018-6006-3
Jean B, Emmanuel C (2013) Chapter 8—Reinforcement of elastomers by particulate fillers. In: James EM, Burak E, Michael R (eds) The science and technology of rubber, 4th edn. Academic Press, New York and London
de Menezes EA, Friedrich L, Colpo A, Amico SC (2019) Micromechanics of short-fiber and particulate composites. In: Sabu T, Mahesh H, Cintil JC (eds) Unsaturated polyester resins: fundamentals, design, fabrication, and applications. Elsevier
Vu CM, Sinh LH, Nguyen DD, Thi HV, Choi HJ (2018) Simultaneous improvement of the fracture toughness and mechanical characteristics of amine-functionalized nano/micro glass fibril-reinforced epoxy resin. Polym Test 71:200–208. https://doi.org/10.1016/j.polymertesting.2018.09.005
AlOweini R, El Rassy H (2009) Synthesis and characterization by FTIR spectroscopy of silica aerogels prepared using several Si(OR)4 and R′′Si(OR′)3 precursors. J Mol Struct 919:140–145. https://doi.org/10.1016/j.molstruc.2008.08.025
Chang K-J, Wang Y-Z, Peng K-C, Tsai H-S, Chen J-R, Huang C-T, Ho K-S, Lien W-F (2014) Preparation of silica aerogel/polyurethane composites for the application of thermal insulation. J Polym Res 21(1):1–9. https://doi.org/10.1007/s10965-013-0338-7
Kim SY, Noh YJ, Lim J, You N-H (2013) Silica aerogel/polyimide composites with preserved aerogel pores using multi-step curing. Macromol Res 22(1):108–111. https://doi.org/10.1007/s13233-014-2006-0
Ge D, Yang L, Li Y, Zhao J (2009) Hydrophobic and thermal insulation properties of silica aerogel/epoxy composite. J Non-Cryst Solids 355(52–54):2610–2615. https://doi.org/10.1016/j.jnoncrysol.2009.09.017
Motahari S, Motlagh GH, Moharramzadeh A (2015) Thermal and flammability properties of polypropylene/silica aerogel composites. J Macromol Sci Part B 54(9):1081–1091. https://doi.org/10.1080/00222348.2015.1078619
Kim HM, Noh YJ, Yu J, Kim SY, Youn JR (2015) Silica aerogel/polyvinyl alcohol (PVA) insulation composites with preserved aerogel pores using interfaces between the superhydrophobic aerogel and hydrophilic PVA solution. Compos A Appl Sci Manuf 75:39–45. https://doi.org/10.1016/j.compositesa.2015.04.014
Halim ZAA, Yajid MAM, Idris MH, Hamdan H (2018) Dispersion of polymeric-coated–silica aerogel particles in unsaturated polyester composites: effects on thermal-mechanical properties. J Dispersion Sci Technol 39(8):1093–1101. https://doi.org/10.1080/01932691.2017.1382375
Shamsuri AA, Sumadin ZA (2018) Influence of hydrophobic and hydrophilic mineral fillers on processing, tensile and impact properties of LDPE/KCF biocomposites. Compos Commun 9:65–69. https://doi.org/10.1016/j.coco.2018.06.003
Jing W, Qingsong Y, Zhiyuan Y (2018) Effect of loading chemically and mechanically pre-treated fumed silica as filler on an etch & rinse model dental adhesive. J Adhes Sci Technol 32(5):527–541. https://doi.org/10.1080/01694243.2017.1370167
Gorelov BM, Gorb AM, Polovina OI, Wacke S et al (2018) Filler’s impact on structure and physical properties in polyester resin–oxide nanocomposites. Adsorpt Sci Technol 36(1–2):549–570. https://doi.org/10.1177/0263617417706797
Yan Z, Xiaoyun Y, Demin J (2007) Cure behaviour of unsaturated polyester/modified montmorillonitenanocomposites. Polym Int 56(2):267–274. https://doi.org/10.1002/pi.2155
Reddy KO, Shukla M, Maheswari CU, Rajulu AV (2012) Evaluation of mechanical behavior of chemically modified Borassus fruit short fiber/unsaturated polyester composites. J Compos Mater. https://doi.org/10.1177/0021998312454032
Yang P, Wang X, Fan H, Gu Y (2013) Effect of hydrogen bonds on the modulus of bulk polybenzoxazines in the glassy state. Phys Chem Chem Phys 15(37):333–338. https://doi.org/10.1039/c3cp51001h
Hunt PA (2007) Why does a reduction in hydrogen bonding lead to an increase in viscosity for the 1-butyl-2,3-dimethyl-imidazolium-based ionic liquids? J Phys Chem B 111(18):4844–4853. https://doi.org/10.1021/jp067182p
Fang H, Ni K, Wu J, Li J, Huang L, Reible D (2019) The effects of hydrogen bonding on the shear viscosity of liquid water. Int J Sedim Res 34(1):8–13. https://doi.org/10.1016/j.ijsrc.2018.10.008
Ewa KW (2018) Studies on the mechanism of thermal decomposition of unsaturated polyester resins with reduced flammability. Polym Polym Compos 12(2):127–134. https://doi.org/10.1177/096739110401200204
Zhang G, Yu Y, Zhang Y, Chen Z, Chen T, Jiang J (2020) Preparation of microencapsulated aluminum hypophosphite and its flame retardancy of the unsaturated polyester resin composites. Polym Bull. https://doi.org/10.1007/s00289-020-03377-z
Zhi L, Xudong C, Long S, Song H, Lunlun G et al (2016) Flammability and oxidation kinetics of hydrophobic silica aerogels. J Hazard Mater 320:350–358. https://doi.org/10.1016/j.jhazmat.2016.07.054
Acknowledgements
The author would like to thank the Research Management Centre (RMC) of Universiti Teknologi Malaysia for funding this project through Post-Doctoral Fellowship Scheme for the project: Design and Development of Eco-Friendly Silica-Alumina Hybrid Flame Retardant for Commodity Plastics.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Abdul Halim, ., Awang, N., Yajid, M.A.M. et al. A comparison between the effects of hydrophobic and hydrophilic silica aerogel fillers on tensile and thermal properties of unsaturated polyester composites. Polym. Bull. 79, 6173–6191 (2022). https://doi.org/10.1007/s00289-021-03798-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-021-03798-4