
Abstract
Although the use of conductive polymers has invaded the electronics industry, the insolubility parameter presents a major problem against their direct application on the surfaces of materials. Attachment of cyclic compounds to polymer chains is recommended to increase the solubility of conductive polymers. The aim of this study is to synthesize a new type of intrinsic conductive polymer, soluble in common solvents. The technique is based on the copolymerization of thiophene with a synthesized monomer phenylazepane-2-one. The reaction was catalyzed by a solid catalyst, prepared by the acid treatment of natural clay, generating active sites responsible for the adhesion of the thiophene and benzene rings. Proton and carbon nuclear magnetic resonances (1H NMR/13C NMR), ultraviolet spectroscopy (UV–Visible), infrared spectroscopy (IR) and differential scanning calorimetry (DSC) were used to identify the material obtained poly [(phenylazepane-2-one)-co-(thiophene)] abbreviated poly (PAT). Thermogravimetric analysis (TGA) showed the thermal stability of poly (PAT) before 200 °C. The solubility of poly (PAT) has been tested and confirmed in various common solvents. The indirect bandgap was calculated at 1.12 eV using Tauc formula. Ac electrical conductivity and dielectric permittivity have been studied versus frequency and temperature, showing the semiconductor character of poly (PAT). The results obtained showed that this novel polymer can play a pioneering role to replace conventional insoluble conducting polymers.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The intrinsic electrical conductive polymers have become more and more attractive [1,2,3], in the face of the scarcity and high cost of the raw materials making up the inorganic semiconductors [4,5,6]. Their ease of implementation, lightness and low cost make them good candidates for technological applications [7, 8]. π-conjugated polymers consist of alternating single and double carbon–carbon bonds [9,10,11]. It is precisely this delocalized π-conjugated system which gives these polymers their particular electronic properties [12,13,14].
Thiophene based polymers are among the most studied and used polymers as π-conjugated materials. So work on thiophene and its derivatives is an important part of conductive polymer research [15]. Studies on these compounds as conductive polymers began in the early 1980s. In 1986, Elsenbaumer et al. [16], prepared polythiophene making it the first heterocyclic conjugate polymer able to be shaped. This step decisive for technological applications has been followed in 1988 by the use of polythiophene in the manufacture of the first real field-effect transistor based on organic materials [17]. Since then, polythiophene has been the subject of numerous publications and researches using this polymer in the field of optoelectronics [18, 19]. Polythiophene is part of the family of heterocyclic conductive polymers, which is to say that a different atom of the carbon atom is present in its cycle, it is a sulfur atom. Its chemical structure gives it interesting properties in terms of stability and resistance to heat. The major disadvantage of this polymer is its low solubility. To deal with this problem, alkyl chains have been introduced at the 3-position of the cycle. These chains provide a relatively high solubility, but this remains a solution to be avoided because of its unexpected and undesirable impact on the final polymer properties, which can come from crosslinking phenomena. The synthesis of copolymers has proven to be an effective means of compensating for certain defects in conductive polymers, by being able to modulate: chemical and thermal stability, optical properties and solubility, by varying the nature and composition of the monomers, resulting in a different polymer structure [20, 21].
Clays can serve as a support for a wide variety of chemical reagents. This is attributed to their ability to concentrate large amounts of reactive species between their aluminosilicate layers [22, 23]. Recently, for many chemical reactions, chemically modified clays have shown a great interest in its use as a catalyst support instead of homogeneous catalysts [24,25,26]. The main advantage lies in the ease of separating and recovering this type of catalyst from the reaction medium, it requires only simple filtration, as well as they are reusable and regenerable [27, 28].
The clay used in our study is Maghnite, obtained from the mountains of the Maghnia region (west of Algeria) from which its name was derived. It is characterized by a great cation exchange capacity [29]. Its aluminum/silicon portion is relatively higher when compared to other world clays [30]. The chemically activated Maghnite has shown a large catalytic capacity, which has enabled it to be used in a large number of polymerization reactions, published by the polymer chemistry laboratory of the University of Oran in Algeria [31,32,33].
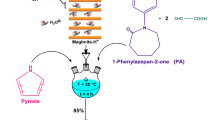
The originality of our work is to synthesize a novel conductive polymer, based on thiophene and a monomer synthesized from caprolactam and phenol (Fig. 1). The copolymerization reaction was catalyzed by a green ecological catalyst Maghnite-H+. Maghnite-H+ was prepared by an acid treatment of raw-Maghnite. The insertion of the initiating hydronium ions between the Maghnite layers was shown by X-ray powder diffraction (XRD) analysis. The chemical structure of the synthesized monomer and polymer was studied in detail by 1H NMR and 13C NMR spectroscopy. Operating conditions such as time, temperature and catalyst content have been well optimized in order to obtain a better yield, and at the same time linear molecular structures. The solubility of poly (PAT) has been tested in dimethylsulfoxide, tetrahydrofuran, N,N-dimethylformamide, sulfolane and water. The thermal stability of poly (PAT) has been confirmed at temperatures below 200 °C, these behaviors allow it to meet several industrial requirements. During the different stages synthesis of poly (PAT), we tried to follow a strategy which largely conforms to the principles of green chemistry.

The synthetic route of poly (PAT)
Experimental section
Materials
ε-Caprolactam, phenol, thiophene, acetic anhydride, dimethylsulfoxide, N,N-dimethylformamide, tetrahydrofuran, sulfolane, toluene, methanol and dichloromethane were purchased from Aldrich Chemical Co Algeria. Raw-Maghnite was procured from Algerian Society of Bentonite (BENTAL). XRD analysis of Maghnite and prepared Maghnite (Maghnite-H+) was recorded using Bruker X-Ray diffractometer from Germany at the rate of 2° min−1 in the 2θ range of 2.0–80.0°. Various methods of analysis were used to identify and characterize the polymer obtained such as FT-IR spectroscopy (Perkin-Elmer System, USA), UV–Vis spectroscopy (OPTIZEN 2120, Republic of Korea), 1H-NMR and 13C-NMR (300 MHz Bruker, Germany). The thermogravimetric analysis (ATG) was performed by Labsys Evo Setaram (France) with a heating rate of 10 °C/min. The DSC analysis was carried out on a Setaram 92 DSC apparatus (France) under a helium flow rate of 20 ml/min, the sample (4.8 mg) to be analyzed was preheated before registering the spectrum. The rate of heating was 10 °C/min. The ac electrical conductivity and the dielectric measurements of the poly (PAT) were determined by two-probe method at 1 V, in a frequency range from 20 Hz to 10 MHz, using a GW-Instek 82-LCR-Meter bridge with PC (Taiwan).
Catalyst preparation
Maghnite-H+ was prepared by treating the raw-Maghnite with a solution of sulfuric acid 0.25 M, at room temperature over a period of two days until saturation was achieved. The cation-exchanged Maghnite was: filtered, resuspended in deionized water continuously until no sulfate ions were detected in the filtrate by BaCl2, isolated by filtration, dried in an oven at 105 °C and then finely ground. The clay cation exchange capacity (CEC) was found to be 84 meq/100 g of dried Maghnite-H+ [12].
Monomer synthesis
Phenylazepane-2-one (PA) was prepared by the reaction of caprolactam with phenol. So in a three-neck flask, 5 g of caprolactam was solubilized in excess of toluene. After stabilizing the temperature at 70 °C, 4.25 g of phenol and 4.6 g of acetic anhydride were added to the flask. Then, 0.1 g of the previously prepared Maghnite-H+ was added to the reaction mixture. The mixture was left for 2 h with gentle stirring. At the end of the reaction, a light yellow precipitate in powder form was recovered by filtration, dried under vacuum and weighed. The yield of the reaction was about 86%.
Polymer synthesis
The copolymerization of thiophene and synthesized phenylazepane-2-one took place in solution under inert atmosphere (nitrogen), using Maghnite-H+ as catalyst. Maghnite-H+ was dried at 100 °C for one hour before being used. The role of Maghnite-H+ was dehydrogenation of thiophene (1 g) and phenylazepane-2-one (4.86 g) dissolved in dichloromethane. The reaction synthesis was carried out during 5 h at 25 °C. Finally, and after removing Maghnite-H+ from the mixture by filtration, methanol was added slowly to the mixture with gentle stirring to precipitate the polymer. The copolymer obtained was dried under vacuum at room temperature overnight. The yield of the reaction was about 91%. This previously described procedure was repeated several times at different temperatures, times and catalyst contents.
Results and discussion
XRD characterization of Maghnite-H+
The X-ray powder diffraction patterns of raw-Maghnite and Maghnite-H+ (Fig. 2) showed the presence of other crystalline phases in addition to montmorillonite, such as mica, calcite and quartz. Under the effect of the acid treatment of raw-Maghnite, the mica and calcite contents were clearly reduced [34]. this was deduced from the decrease in the intensity of the corresponding peaks located at 19.74°, 62.14° for mica, and 20.81°, 39.61° for calcite [10]. The removal of most of the quartz located at 20.81° and 26.54° [35] is probably due to the mechanical treatment of raw-Maghnite by the sedimentation process. The increase in basal spacing corresponding to the space of montmorillonite layers; from 10.5 Å in raw-Maghnite to 15.02 Å in Maghnite-H+ [10] was reflected by the shift of the montmorillonite peak towards the small values of θ (8.41° to 5.87°) [36]. This is explained by the replacement of interlamellar ions such as Mg2+ and Ca2+ by gigantic hydronium ions of the relatively large molecular radius [31]. In general, we note that acid treatment of Maghnite increased basal spacing of montmorillonite, but did not affect the original Maghnite structure, which was confirmed by the absence of the appearance or disappearance of peaks in XRD spectrum of Maghnite-H+.

X-ray powder diffraction of raw-Maghnite and Maghnite-H+ a structures, b montmorillonite peak and c XRD spectra.
Characterization
Figure 3 shows the optical absorption spectrum of dissolved poly (PAT) in DMSO. The color of the polymer solution was blackened red. The absorption spectrum makes appear three absorption bands located at 324, 347 and 418 nm assigned to; the C=C chromophore present in the aromatic benzene and thiophene rings, the nonbonding electronic level in nitrogen or sulfur atoms and the carbonyl groups of caprolactam, respectively. This curve obtained by UV–Visible spectroscopy makes it possible to calculate the absorption coefficient, which is a necessary parameter in order to then determine the optical bandgap of our synthesized material.

UV–visible spectrum of poly (PAT)
The 1H-NMR spectra of synthesized monomer and polymer were obtained to identify and confirm their chemical structure. Figure 4 shows the superposition of the 1H-NMR spectra of PA (a) and poly (PAT) (b). The resonances at 1.69, 2.54 and 3.33 ppm for the PA and 1.71, 2.53 and 3.36 ppm for the poly (PAT) are assigned to caprolactam ring protons. The peaks Hc (2H, d) at 6.66 ppm, Ha (1H, t) at 6.70 and Hb (2H, t) at 6.74 ppm for PA and Hc (2H, s) at 6.47 and Ha (1H, s) at 6.71 ppm for poly (PAT) correspond to phenyl ring protons. The disappearance of the peak Hb in the spectrum of the polymer and the conversion of Ha and Hc into singular gave the reason to conceive that the attachment of the thiophene ring on the phenyl ring was at the carbon atom in the meta position. The thiophene ring protons Hg (2H, d) appear at 6.89 ppm. The appearance of the Hg peak as a doublet peak shows that the attachment to the thiophene cycle was in atoms 1 and 4. The small peaks Hh at 7.12 ppm and Hi at 7.92 ppm are attributed to phenyl and thiophene ring protons, respectively, at the end of the polymer chain. The number average molecular weight (Mn) of poly (PAT) was calculated after integration, by comparing the signal areas corresponding to the protons of the thiophene ring in the repeating unit (Hg) to that of the terminal group (Hi), it is about 8900 g/mol. The molar ratio of thiophene and PA in the poly (PAT) chain is 31.71% and 68.29% respectively.

1H NMR spectra of PA (a) and poly (PAT) (b)
To complete the structural study of synthesized products, 1H NMR analysis was supported by 13C NMR analysis to identify and confirm the carbon–carbon bonds. Figure 5 shows the 13C NMR spectra of the monomer (curve above) and the polymer (curve below). The carbons of the phenyl ring are observed at 122.80, 128.86, 117.07 and 141.73 ppm for the monomer (C1′ C2′ C3 and C4) and at 122.80, 136.12, 117.10 and 141.66 ppm for the polymer (C′1, C′2, C′3 and C′4). The displacement of the C2 peak from 128.86 in the monomer to 136.12 in the polymer indicates the conversion of the secondary carbon to a tertiary carbon, due to the creation of a new bond with the thiophene carbon. The signals appeared at 172.45, 50.65, 34.56, 29.92, 24.20 and 29.21 ppm for the monomer (C5, C6, C7, C8, C9 and C10), and at 172.75, 50.29, 34.94, 29.93, 24.91 and 29.22 ppm for the polymer (C′5, C′6, C′7, C′8, C′9 and C′10) are assigned to carbon atoms belonging to the caprolactam ring. The attachment of the thiophene ring gave rise to the birth of two peaks C11 and C12 located, respectively, at 137.09 and 118.87 ppm. The two small peaks α and β saw at 128.88 and 122.14 ppm are, respectively, assigned to the carbons of thiophene and phenyl rings at the ends of the polymer chain. These results are to a large extent compatible with those obtained by 1H NMR analysis.

13C NMR spectra of PA a and poly (PAT) b
Figure 6 shows the IR absorption spectrum of poly (PAT) from which we will cite the main bands. The two mid-intensity bands at 2931 and 2854 cm−1 correspond to the C–H symmetric and asymmetric stretch, respectively. The band at 1729 cm−1 is C=O stretching vibration of the caprolactam ring. The band at 1600 cm−1 is assigned to C=C stretching vibration of the phenyl ring. The band at 1504 cm−1 is assigned to the C=C stretching vibration bond of the thiophene rings. The mid-intensity band at 1311 cm−1 corresponds to the valence vibration of the C–C bond. The band at 1248 cm−1 is assigned to the valence vibration of the C-H bond. The vibration band located at 805 cm−1 corresponds to the deformation of the C-S-C bond. The IR spectrum was in good agreement with the proposed structure.

IR spectrum of poly (PAT)
Thermal properties
Figure 7 shows the TGA, DTG and DSC thermograms of poly (PAT) obtained in a helium atmosphere, at a heating rate of 10 °C/min. This polymer has good thermal stability up to a temperature of around 200 °C, beyond this temperature; the polymer began to undergo thermal decomposition in a single main stage. This result showed a significant improvement in the degradation temperature compared to other synthesized polythiophene; 180 °C [37], polythiophene composites doped with copper; 170 °C [38] and similar to that of polythiophene-fullerenes diblock copolymer [39]. The DSC curve shows a small temperature variation at about 175 °C, which represents the glass transition temperature of the polymer, as well as an endothermic peak at about 259 °C due to the melting phenomenon. In the temperature range between 300 and 550 °C, several endothermic and exothermic effects are noticed in the DSC signal, due to the pyrolytic decomposition of the polymer.

TGA, DTG and DSC thermograms of poly (PAT)
Conductivity measurements
Figure 8a shows the evolution of ac electrical conductivity (σac), the real part of the permittivity (ɛ′), the imaginary part of the permittivity (ɛ′′) and the dielectric tangent lost tang (δ) at room temperature. The measurements were made in a frequency range between 20 Hz and 10 MHz. First of all, in Fig 8a, the ac electrical conductivity increased continuously with increasing frequency applied. This can be since, as the frequency increased, additional electrons are conducted across the interface due to the frequency-assisted electron hopping [40]. The study of the variation of dielectric permittivity constant as a function of the frequency makes it possible to fundamentally define the polarization capacity of the poly (PAT) during the application of the field. Its two real and imaginary parts decreased with increasing frequency; this is due to the dominance of the relaxation process. The values of ɛ′ and ɛ′′ became 7.98 and 0.81, respectively, at the maximum frequency. Tang (δ) is the ratio of ɛ′ to ɛ′′, it decreased with increasing frequency due to the dominance of the capacitive element against the ohmic component. The relaxation time has been calculated around 1953 µs. The ac electrical conductivity also measured as a function of temperature at different frequencies (Fig 8b). Remarkably, that the ac electrical conductivity of poly (PAT) increased with increasing temperature. This is attributed to the increased likelihood of electrons in the valence band getting enough energy to join the conduction band [41, 42]. This result confirmed that poly (PAT) exhibits semiconducting material behavior. The increase in ac electrical conductivity also with frequency is due to the existence of polymer defect sites, permitting the movement of charges. By way of comparison, Table 1 presents the values of electrical conductivity of polythiophene and of some of its derivatives obtained previously Fig. 9.

a Variation of σac, ɛ′, ɛ′′ and tang (δ) of poly (PAT) with frequency, b variation of σac of poly (PAT) with temperature at different frequencies

Determination of the indirect bandgap of poly (PAT) from a plot of (αhν)1/2 versus hν
Optical analysis
In order to determine the indirect bandgap of poly (PAF) film, the extrapolation of the plot of (αhv)1/2 versus hv was carried out, the results are shown in Fig. 9. The region of strong absorption was exploited to determine the indirect optical bandgap (Eg), using Tauc formula. The value of the indirect bandgap was obtained 1.12 eV, from the extrapolation of the line to the line base, where the value of the (αhv)1/2 is equal to zero. The result obtained allows us to conclude that poly (PAT) is a π-conjugated semiconducting at room temperature ( < 2 eV).

Solubility test results
Solubility test
The solubility of poly (PAT) in common organic solvents is one of the main objectives expected from the copolymerization of thiophene with PA. Therefore the solubility parameter was evaluated in dimethylsulfoxide, tetrahydrofuran, N,N-dimethylformamide, sulfolane and water. An amount of 1 g of poly (PAT) was introduced into 30 ml of solvent, the mixture was allowed to stir for 30 min at room temperature. The quantity of poly (PAT) which remained insoluble was recovered by filtration, dried and then weighed. The following expression made it possible to calculate the solubility index. The examination results are shown in Fig. 10.

Effect of the amount of Maghnite-H+, reaction time and temperature on the reaction yield of poly (PAT) synthesis
According to the values of the calculated solubility index, most of the poly (PAT) was found to be soluble in the solvents tested, relatively little in the water. Poly (PAT) has conjugated chains due to the degree of dehydrogenation, giving magenta solutions of high concentrations. The very good solubility of poly (PAT) despite their high degree of π-conjugation is due essentially to the large unitary groups (caprolactam/benzene), which are meta linked to the carbon atom of the thiophene ring, and also is largely due to the linear molecular structure of poly (PAT); absence of any kind of interchain bonds, that has been confirmed by the structural characterization.
Effect of the amount of Maghnite-H+, reaction time and temperature on the synthesis of poly (PAT)
In the first part, we followed the effect of time on the reaction synthesis of poly (PAT) for different amounts of Maghnite-H+. Figure 11a shows the effect of time on the polymerization yield, investigated by using various weight ratios Maghnite-H+/monomers. The polymerization was carried out in solution (CH2Cl2) and at a temperature of 25 °C. The polymerization yield increased over time, reaching 91% at 5 h by using 3% of Maghnite-H+. The reaction yield remained constant beyond 5 h. This can be explained by the total consumption of reagents. The remaining 9% is oligomers soluble in CH2Cl2. The continuous increase in reaction yield, depending on the amount of Maghnite-H+ used, is probably the result of the number of initiating active sites responsible for the induction of the polymerization, this number is proportional to the amount of catalyst used in the reaction.

Reaction mechanism of the synthesis of PAT using Maghnite-H+ as a catalyst
In the second party, the temperature effect on the polymerization reaction of thiophene with PA was followed. The polymerization was carried out for 5 h in the presence of 3 wt% of Maghnite-H+. The results obtained at different temperatures are shown in Fig. 11b. From these results, we conclude that the optimal temperature for the reaction is 25 °C (the yields reached a maximum). After this temperature, the reaction yield was reduced, indicating that the temperature 25 °C is the ceiling temperature, at which the rates of polymerization and depolymerization phenomena are equal. These results showed the double role in the opposite direction played by Maghnite-H+ as a cationic catalyst. Same results have been obtained during the synthesis of polyurethane using Maghnite-H+ as catalyst [30].
Figure 12 shows the proposed reaction mechanism of the copolymerization reaction of thiophene with PA, using Maghnite-H+ as a catalyst. The initiation step was based on the creation of active sites, by fixing the H+ ion of Maghnite-H+ on a thiophene ring sulfur atom, this led to the generation of a sulfonium ion. In the propagation step, the carbon atom adjacent to the sulfonium atom attaches to a phenyl ring carbon. This effect leads to the creation of the carbenium ion on another carbon atom of the phenyl ring, the latter becoming the new active site. This operation continues in an alternative way one by one, where there is each time a displacement of the active site, capable of capturing a free monomer. The process was propagated by chain-growth polymerization. The termination step is reached by adding water, thus deactivating Maghnite-H+. The proton of the carbenium ion which is located at the end of the chain returns to the Maghnite, achieving the ionic balance of the Maghnite-H+ and also allowing its reuse.
Conclusions
The effectiveness of the technique proposed in this study has been successfully demonstrated, in order to synthesize a novel soluble conductive polymer poly (PAT). It was based on the copolymerization of thiophene with PA. PA is a monomer synthesized from phenyl and caprolactam, the presence of its corresponding rings in the backbone gave the poly (PAT) its soluble appearance. The attachment between the phenyl and thiophene rings created alternating polymer chains of single and double bonds. The reaction catalysis method was heterogeneous and green, using Maghnite-H+ as activated clay. Maghnite-H+ was activated by an acidic treatment of raw-Maghnite. The reaction yield was 91%. The IR, 1H NMR and 13NMR analyzes identified in detail the chemical structure of poly (PAT), showing the linearity of the polymer chains. The attachment between the two monomers was in the meta position atom of the phenyl molecule, and in the 1 and 4 position atoms of the thiophene molecule. The study by TGA and DSC analyses has shown that poly (PAT) is thermally stable below the temperature of 200 °C. UV spectroscopy has shown that poly (PAT) has a narrow bandgap of 1.12 eV, which makes it a π-conjugated semiconducting material. Electrical properties were also evaluated; the electrical conductivity increased with increasing frequency, following polaron-hopping mechanism assisted by frequency. The relaxation time was about 1953 µs. In other words, when compared with many other common conductive polymers, the high intrinsic properties of poly (PAT) would be a significant advantage in view of realizing solar cells or field-effect transistors.
References
Derakhshankhah H, Mohammad-Rezaei R, Massoumi B, Abbasian M, Rezaei A, Samadian H, Jaymand M (2020) Conducting polymer-based electrically conductive adhesive materials: design, fabrication, properties, and applications. J Mater Sci Mater El 31:10947–10961. https://doi.org/10.1007/s10854-020-03712-0
Jaymand M (2013) Recent progress in chemical modification of polyaniline. Prog Polym Sci 38:1287–1306. https://doi.org/10.1016/j.progpolymsci.2013.05.015
Shirakawa H, Louis EJ, MacDiarmid AG, Chiang CK, Heeger AJ (1977) Synthesis of electrically conducting organic polymers: halogen derivatives of polyacetylene, (CH)x. J Chem Soc Chem Commun 16(578):580. https://doi.org/10.1039/c39770000578
Bouhadjar L, Chikh K, Kherroub DE (2016) Synthèse de matériau organique semi-conducteur. OMN.UNIV.EUROP, Paris
Patil YS, Salunkhe PH, Navale YH, Patil VB, Ubale VP, Ghanwat AA (2020) Tetraphenylthiophene–thiazole-based π-conjugated polyazomethines: synthesis, characterization and gas sensing application. Polym Bull 77:2205–2226. https://doi.org/10.1007/s00289-019-02856-2
Khelifa Baghdouche A, Guergouri M, Mosbah S, Bencharif M, Bencharif L (2020) Electrochemical synthesis and physicochemical properties of new conjugated polymers based on fluorine. J Electroanal Chem 857:113708. https://doi.org/10.1016/j.jelechem.2019.113708
Abou Elfadl A, Ismail AM, Mohammed MI (2020) Dielectric study and AC conduction mechanism of gamma irradiated nano-composite of polyvinyl alcohol matrix with Cd0.9Mn0.1S. J Mater Sci Mater Electron 3:8297–8307. https://doi.org/10.1007/s10854-020-03365-z
Yasa M, Goker S, Toppare L (2020) Selenophene-bearing low-band-gap conjugated polymers: tuning optoelectronic properties via fluorene and carbazole as donor moieties. Polym Bull 77:2443–2459. https://doi.org/10.1007/s00289-019-02872-2
Kadu RK, Thakur PB, Patil VR (2019) Photophysical properties of new fluorene-based conjugated polymers containing polyphenylene-substituted dendronized core. Polym Bull 76:595–613. https://doi.org/10.1007/s00289-018-2401-3
Kherroub DE, Bouhadjar L, Boukoussa B, Rahmouni A, Dahmani K, Belbachir M (2019) Highly conductive and soluble polymer synthesized by copolymerization of thiophene with para-methoxybenzaldehyde using clay catalyst. BCREC 14:413–420. https://doi.org/10.9767/bcrec.14.2.3793.413-420
Shahrivari S, Kowsari E, Shockravi A, Ehsani A (2019) Synthesis of different new copolyimides and influence of different molar ratios of diamines and dianhydride on pseudocapacitance performance of p-type conductive polymer. J Electroanal Chem 837:123–136. https://doi.org/10.1016/jjelechem.2019.02.024
Badaoui M, Bouhadjar L, Kherroub DE, Chikh K, Seghier A, Belarbi EH (2020) Green copolymerization of thiophene with para-cholorobenzaldehyde catalyzed by maghnite-H+. Polym Sci Ser B 62:256–263. https://doi.org/10.1134/s156009042003001x
Shabzendedar S, Modarresi-Alam AR, Noroozifar M, Kerman K (2020) Core-shell nanocomposite of superparamagnetic Fe3O4 nanoparticles with poly(m-aminobenzenesulfonic acid) for polymer solar cells. Org Electron 77:105462. https://doi.org/10.1016/j.orgel.2019.105462
Tongchao C, Xiuping J, Xiao H, Hongmei D, Yan Z, Jinsheng Z, Junhong Z (2019) Synthesis and electrochromic properties of cross-linked and soluble conjugated polymers based on 5, 8, 14, 17-tetrabromoquinoxaline[2′, 3’:9, 10]phenanthro[4,5 abc]phenazine as the multifunctionalized acceptor unit. Org Electron 73:43–54. https://doi.org/10.1016/j.orgel.2019.06.004
Jaymand M, Hatamzadeh M, Omidi Y (2014) Modification of polythiophene by the incorporation of processable polymeric chains: Recent progress in synthesis and applications. Prog Polym Sci 47:26–69. https://doi.org/10.1016/j.progpolymsci.2014.11.004
Elsenbaumer RL, Jen KY, Oboodi R (1986) Processible and environmentally stable conducting polymers. Synth Met 15:169–174. https://doi.org/10.1016/0379-6779(86)90020-2
Assadi A, Svensson C, Willander M, Inganäs O (1988) Field-effect mobility of poly(3-hexylthiophene). Appl Phys Lett 53:195–197. https://doi.org/10.1063/1.100171
Mahesh K, Karpagam S, Goubard FC (2018) Conductive and photoactive nature of conjugated polymer based on thiophene functionalized thiazole or benzothiadiazole. Express Polym Lett 12:238–255. https://doi.org/10.3144/expresspolymlett.2018.22
Singh R, Bajpai AK, Shrivastava AK (2020) CdSe nanorod-reinforced poly(thiophene) composites in designing energy storage devices: study of morphology and dielectric behavior. Polym Bull. https://doi.org/10.1007/s00289-020-03104-8
Wang GQ, Jiang M, Zhang Q, Wang R, Liang QD, Wang HH, Zhou GY (2019) Partially bio-based and tough polyesters, poly (ethylene 2,5-thiophenedicarboxylate-co-1,4-cyclohexanedimethylene 2,5-thiophenedicarboxylate)s. Express Polym Lett 13:938–947. https://doi.org/10.3144/expresspolymlett.2019.82
Temizkan K, Kaya İ (2019) Synthesis of soluble poly(azomethine)s containing thiophene and their fluorescence quantum yields. Polym Bull 77:3287–3303. https://doi.org/10.1007/s00289-019-02911-y
Youm JS, Ban HR, Chang JH, Kim JC (2019) Effects of the shape and surface treatment of clay on the process of uniaxially drawn low-density polyethylene/clay composites films. Macromol Res 28:356–364. https://doi.org/10.1007/s13233-020-8048-6
Kim HG, Kim EH, Kim SS (2018) The effects of clay platelets orientation achieved via a dry lamination process on the barrier properties of clay polymer nanocomposites. Macromol Res 26:454–458. https://doi.org/10.1007/s13233-018-6055-6
Mahmoudian M, Balkanloo PG, Nozad E (2017) A facile method for dye and heavy metal elimination by pH sensitive acid activated montmorillonite/polyethersulfone nanocomposite membrane. Chin J Polym Sci 36:49–57. https://doi.org/10.1007/s10118-018-2004-3
Gillani QF, Ahmad F, Mutalib MIA, Megat-Yusoff PSM, Ullah S (2018) Effects of halloysite nanotube reinforcement in expandable graphite based intumescent fire retardant coatings developed using hybrid epoxy binder system. Chin J Polym Sci 36:1286–1296. https://doi.org/10.1007/s10118-018-2148-1
Roufegari-Nejhad E, Sirousazar M, Abbasi-Chiyaneh V, Kheiri F (2019) Removal of methylene blue from aqueous solutions using poly(vinyl alcohol)/montmorillonite nanocomposite hydrogels: taguchi optimization. J Polym Environ 27:2239–2249. https://doi.org/10.1007/s10924-019-01514-y
Tekay E, Aydınoğlu D, Şen S (2019) Effective adsorption of Cr (VI) by high strength chitosan/montmorillonite composite hydrogels involving spirulina biomass/microalgae. J Polym Environ 27:1828–1842. https://doi.org/10.1007/s10924-019-01481-4
Souza JL, de Campos A, França D, Faez R (2019) PHB and montmorillonite clay composites as KNO3 and NPK support for a controlled release. J Polym Environ 27:2089–2097. https://doi.org/10.1007/s10924-019-01498-9
Kherroub DE, Belbachir M, Lamouri S (2014) Study and optimization of the polymerization parameter of furfuryl alcohol by algerian modified clay. Arab J Sci Eng 40:143–150. https://doi.org/10.1007/s13369-014-1512-x
Boulaouche T, Kherroub DE, Khimeche K, Belbachir M (2019) Green strategy for the synthesis of polyurethane by a heterogeneous catalyst based on activated clay. Res Chem Intermed 45:3585–3600. https://doi.org/10.1007/s11164-019-03810-7
Derkaoui S, Kherroub DE, Belbachir M (2019) Green synthesis, anionic polymerization of 1,4-bis(methacryloyl)piperazine using Algerian clay as catalyst. Green Process Synth 8:611–621. https://doi.org/10.1515/gps-2019-0031
Kherroub DE, Belbachir M, Lamouri S, Chikh K (2018) Acid-activated bentonite (Maghnite-H+) as a novel catalyst for the polymerization of decamethylcyclopentasiloxane. Bull Mater Sci. https://doi.org/10.1007/s12034-018-1563-9
Kherroub DE, Belbachir M, Lamouri S (2015) Synthesis of poly(furfuryl alcohol)/montmorillonite nanocomposites by direct in-situ polymerization. Bull Mater Sci 38:57–63. https://doi.org/10.1007/s12034-014-0818-3
Kherroub DE, Belbachir M, Lamouri S, Chikh K (2017) Cationic ring opening polymerization of octamethylcyclotetrasiloxane using a cost-effective solid acid catalyst (Maghnite-H+). Iran J Sci Technol Trans Sci 43:75–83. https://doi.org/10.1007/s40995-017-0269-y
Kherroub DE, Belbachir M, Lamouri S (2017) Synthesis and characterization of polyvinylmethylsiloxanes by cationic polymerization using a solid green catalyst. e-Polymers 17:439–448. https://doi.org/10.1515/epoly-2017-0039
Kherroub DE, Khodja M, Belbachir M, Lamouri S, Bouhadjar L, Boucherdoud A (2018) Maghnite-H+ as inorganic acidic catalyst in ring opening polymerization of dodecamethylcyclohexasiloxane. Silic 11:1165–1173. https://doi.org/10.1007/s12633-018-9769-4
khezri T, Sharif M, Pourabas B, (2016) Polythiophene–graphene oxide doped epoxy resin nanocomposites with enhanced electrical, mechanical and thermal properties. RSC Adv 6:93680–93693. https://doi.org/10.1039/c6ra16701b
Erdönmez S, Ozkazanc E (2013) Power-law conductivity in polythiophene/copper(II) acetylacetonate composites. Polym Int 63:31–36. https://doi.org/10.1002/pi.4536
Lee JU, Cirpan A, Emrick T, Russell TP, Jo WH (2009) Synthesis and photophysical property of well-defined donor–acceptor diblock copolymer based on regioregular poly(3-hexylthiophene) and fullerene. J Mater Chem 19:1483. https://doi.org/10.1039/b813368a
Tiwari DC, Sen V, Sharma R (2012) Temperature dependent studies of electric and dielectric properties of polythiophene based nano composite. Indian J Pure Appl Phy 50:49–56
Kelkar D, Chourasia A (2013) Electrical and magnetic conduction properties of polythiophene doped with FeCl3. Macromol. Symp 327:45–53. https://doi.org/10.1002/masy.201350505
El-Shekeil A, Abu-Bakr AO (2011) DC electrical conductivity of the direct electrochemically synthesized polythiophene metal complexes. J Macromol Sci A 48:233–240. https://doi.org/10.1080/10601325.2011.544943
Kumara Swamy N, Sandeep S, Santhosh AS (2017) Conductive polymers and their nanohybrid transducers for electrochemical biosensors applications: a brief review. Indian J Adv Chem Sci S2:6–9. https://doi.org/10.22607/IJACS.2017.S02002
Zhang J, Song G, Qiu L, Feng Y, Chen J, Yan J, Liu L, Huang X, Cui Y, Sun S, Xu W, Zhu D (2018) Highly conducting polythiophene thin films with less ordered microstructure displaying excellent thermoelectric performance. Macromol Rapid Comm 39:1800283. https://doi.org/10.1002/marc.201800283
Konstantinov II (1986) Ein molekulares strukturmodell flexibelkettiger polymere mit mesogenen seitengruppen. Acta Poly 37:255–263. https://doi.org/10.1002/actp.1986.010370501
Cao G, Cui H, Wang L, Wang T, Tian Y (2020) Highly conductive and highly dispersed polythiophene nanoparticles for fabricating high-performance conductive adhesives. ACS Appl Electron Mater. https://doi.org/10.1021/acsaelm.0c00457
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Boucherdoud, A., Kherroub, D.E. Physicochemical, optical and electrical investigation on poly [(phenylene-2-one)-co-(thiophene)] novel soluble conductive polymer as-synthesized through heterogeneous catalysis route. Polym. Bull. 78, 6367–6385 (2021). https://doi.org/10.1007/s00289-020-03419-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-020-03419-6