
Abstract
The aim of this work was to synthesize a novel hydrophilic structure based on polystyrene (PS) cross-linked by using N,N′-(hexane-1,6-diyl)diacrylamide (HDBPA). FT-IR was carried out for chemical structure characterization of HDBPA and cross-linked samples. In addition to FT-IR, 1H NMR was also applied on HDBPA for more investigations. The results showed that HDBPA was synthetized through a condensation reaction, and consequently, it could successfully cross-link PS structure. Moreover, the determination of nitrogen content in HDBPA within PS network was investigated by means of elemental analysis. Afterward, the reactivity ratios between HDBPA and styrene were calculated via Fineman–Ross equation in which the obtained values were 0.66 and 0.23, respectively. On the other hand, by adding the HDBPA to PS the water contact angle was decreased from 73.6° to 57.4°, thereby proving the indispensable role of the cross-linking agent on hydrophilicity of the samples. Finally, the thermal behavior of the samples was studied using DSC and TGA analyses. According to DSC thermograms, there was a shift in Tg up to 30 °C for the samples with cross-linking densities ranging from 2.5 to 20 mol.%. In regard to the thermal stability of the cross-linked samples, the TGA curves revealed a reduction in this property owing to the presence of amide groups in HDBPA through the PS structure.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The hydrophobic property of widely well-known polymers like polystyrene (PS) has yet remained a big challenge which leads to restrictions for many applications. Those polymers containing at least a hydrophilic head are being received a great deal of attention to employ in many new fields such as electrochemistry [1], pharmacy [2], microprinting [3] and templating agent [4]. To increase the hydrophilic characteristics of a polymer, different strategies such as its blending with hydrophilic micro-/nanoparticles [5, 6] and also surface modification [7] have been studied.
PS is a general-purpose and brittle synthetic polymer with an aromatic side group which has been extensively used in several industries. Hydrophilic-modified PS has many applications in membrane fabrication [8, 9], packaging materials [10] and cell culture dishes [11]. Two main strategies for increasing the PS hydrophilicity are plasma treatment [12] and copolymerization [9, 13]. The conventional method to modify the structure of PS for cell culture application is to cover the surface of PS-based dishes with a hydrophilic polymer. This solution cannot be an efficient method because of cell death and impairment of cellular test results [14].
Acrylic monomers are used to add some important properties to styrene including high polarity, hydrophilicity and stability in aqueous solutions. In recent years, copolymers produced by hydrophilic blocks of acrylic acid and hydrophobic chains of PS have received much attention [15,16,17].
This work aims at synthesizing a hydrophilic-modified and cross-linked PS using N,N′-(hexane-1,6-diyl)diacrylamide (HDBPA) as a cross-linker. The HDBPA was also prepared by a condensation reaction between acrylic acid and hexamethylene diisocyanate. The properties of the cross-linked PS are changed by altering the HDBPA ratio. The aim of the present study is to offer a method to synthetize hydrophilic HDBPA and a comprehensive study on structural and thermal behavior of PS with different amounts of HDBPA as a cross-linker.
Experimental
Materials
Styrene (99%, Merck, Germany) was stored in a refrigerator (− 10 °C) after being purified by distillation over CaH2 under reduced pressure and stored under nitrogen atmosphere. Hydroquinone (analytical grade, Merck, Germany) was used as the inhibitor of the polymerization. The hexamethylene diisocyanate (HDI), acrylic acid, azobisisobutyronitrile (AIBN) and dimethylformamide (DMF) (Merck, Germany) were used as received.
Synthesis of N,N′-(hexane-1,6-diyl)diacrylamide (HDBPA)
HDBPA was synthesized on the basis of a condensation reaction. Acrylic acid (2.46 mL) was added to a glass vial reactor (50 mL) and mixed with HDI (2.87 mL) and DMF (30 mL) at ice bath. (The overall molar ratio of acrylic acid to HDI was kept at 2:1.) Then, the reaction was done at room temperature for 24 h using a magnetic heater–stirrer under partial vacuum without a reflux condenser.
Synthesis of HDBPA cross-linked PS
Cross-linking reaction was conducted in DMF medium using styrene and AIBN (initiator) by general solution polymerization. The polymerization of styrene was carried out in sealed glass bottles as reactor. In brief, to produce samples containing 2.5 mol.% HDBPA cross-linker, 13.1 mol (1.5 mL) styrene was added to 0.011 mol (2.5 mL) of HDBPA in DMF solution (3.1 × 10−2 wt.%). Then, DMF was added until the reaction medium reached to the total volume of 15 mL and AIBN (3 mg) was used as the polymerization initiator. The prepared medium was vacuumed after adding to the reactor and then deoxygenated by bubbling nitrogen gas with 99.999% purity at room temperature for 5 min. Reaction was performed by stirring at 70 °C for 24 h. To remove the unreacted substances, reaction products were washed three times and the synthetized gel was dried in vacuum oven at 40 °C for 48 h. The same reaction was repeated, for samples containing 10 and 20 mol.% HDBPA except for altering the amount of HDBPA-to-styrene molar ratios. The same reaction was carried out for samples containing 10 and 20 mol.% HDBPA except for altering the amount of HDBPA-to-styrene molar ratios. P1, P2 and P3 represent samples with 2.5, 10 and 20 mol.% cross-linker, respectively.
Measurements
FT-IR, 1H NMR and elemental analysis studies
For structural composition analysis, FT-IR spectra of samples were recorded using KBr method from 4000 to 400 cm−1 at a spectral resolution of 4 cm−1 with a Thermo Nicolet 670 spectrometer. To understand the HDBPA and styrene content, elemental analysis was performed by CHNS elemental analyzer (Flash EA 1112 series, Thermo Finnigan). Liquid proton nuclear magnetic resonance (1H NMR) spectrometer (300 MHz, Bruker Co.) was also conducted on HDBPA to determine the molecular structure.
To determine monomer conversion, gravimetric analysis was used. During the polymerization, the samples (1 mL) were periodically taken from the reactor. In order to stop polymerization reaction in samples, hydroquinone was used. Monomer conversion was calculated using Eq. (1):
where \(M_{0}\), \(M_{1} , M_{2}\) and \(M_{T}\) are mass of blank aluminum foil, aluminum foil containing the reaction solution and dried aluminum foil containing dried sample and the total mass of reactor content, respectively. The results are shown in Fig. 1. Total monomer conversion was determined via gravimetry with amounts of 40.7%, 53.6% and 65.4% for P1, P2 and P3, respectively.

Styrene conversion plot as a function of time
Water contact angle
Contact angle measurements were taken using a contact angle measuring system (G40, Krüss GmbH, Hamburg, Germany) at room temperature. The common sessile drop method was used to determine contact angle values [18].
DSC and TGA analyses
Thermal properties of the samples were evaluated by differential scanning calorimetry (DSC, PerkinElmer Diamond DSC) and thermogravimetric analyses (TGA, Pyris 1) in nitrogen atmosphere and heating rate of 10 °C/min.
Results and discussion
As reported in the literature [19], isocyanate-containing structures (R–N = C=O), where R is an alkyl or aryl groups, are able to react with active hydrogen atoms in amine, water, alcohol, carboxylic acid, urethanes and urea at room temperature. In this study, two isocyanate groups of HDI reacted with carboxylic acid functional groups of acrylic acid. The mechanism is shown in Fig. 2. In the first step, isocyanate groups of HDI reacted with active hydrogen in acrylic acid and resulted in an anhydride structure (highlighted in Fig. 2). In the next step, the intermediate compound converted to the final product containing two amide groups (HDBPA). The by-product of the reaction was CO2 gas which released. Figure 3 illustrates the mechanism of cross-linking reaction.

Schematic illustration of HDBPA synthesis reaction

Schematic illustration of cross-linking reaction
Characterization of chemical structure of HDBPA and cross-linked PS
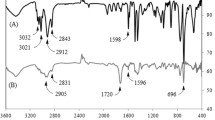
As shown by FT-IR spectroscopy (Fig. 4a), HDBPA characteristic peaks were as follows: absorbance peaks at 2860 and 2930 cm−1 attributed to –CH2 stretching vibrations [20] and 3415 cm−1 ascribed to amide groups of HDBPA formed during the condensation reaction [21]. Finally, 1666, 1386 and 1715 cm−1 are assigned to the amide carbonyl group stretching [22], N–C bond in amide group [23] and –C = O bond in unreacted acrylic acid monomer, respectively.

a FT-IR spectrum of HDBPA and cross-linked PS (P2: 10% mol.), b 1H NMR spectra of HDBPA
The absence of isocyanate group absorbance peak at 2273 cm−1 indicates that both isocyanate groups present in HDI have participated in the reaction. The cross-linked samples were also investigated using FT-IR technique, and as it was expected, the structure remained intact except for the C = C bond in benzene group of PS by which the absorbance peak at 1441 cm−1 was observed [24].
To investigate the molecular structure of the synthetized cross-linker, 1H NMR was also performed. The 1H NMR spectrum and chemical shifts attributed to each proton are illustrated in Fig. 4b [25,26,27] which is in accordance with chemical structure of HDBPA. The chemical shift at 2.5 ppm is known to be related to solvent (DMSO) [26]. According to the spectrum, the 1H NMR peaks split into a triplet at 1.4, 3.1 and 5.5 ppm a doublet at 6.2 and 8.0 ppm. The area under each peak related to the carbons in the structure of HDBPA was also calculated and is depicted in Fig. 4b.
To examine the successful copolymerization of styrene and HDBPA cross-linker, the elemental analysis was performed. The copolymerization reaction product was washed for several times to remove solvent and unreacted substances. Then, HDBPA content in each sample was calculated using nitrogen percentage data obtained by elemental analysis. The results are represented in Table 1. Accordingly, mol.% of cross-linker was increased from 7.7 in P1 to 31.4 in P3. Reactivity ratio of monomers was calculated using Fineman–Ross equation (FR, Eq. 2) [28].
where F is the monomer ratio in feed (\(F = \left[ {{\text{HDBPA}}]/} \right[{\text{Styrene}}]\)) and ƒ is the molar fraction of comonomer units in molecular structure (\(f = m_{1} /m_{2}\), HDBPA (\(m_{1}\)) and styrene (\(m_{2}\))).
In this method, copolymerization constants are evaluated by plotting \((F^{2} /f)\) versus \([F\left( {f - 1} \right)/f]\) (Fig. 5). Considering HDBPA as \(M_{1}\) and styrene monomer as \(M_{2}\), the reactivity ratios are \(r_{1} = 0.66\) and \(r_{2} = 0.23\). It should be noted that the obtained values for \(r_{1}\) and \(r_{2}\) declare a rapid gelation. However, low monomer loading in reaction mixture facilitates the product processing [28]. According to the results, the higher amount of HDBPA in molecular structure compared to feed was attributed to higher reactivity ratio of HDBPA.

FR method to determine reactivity ratio (slope is r1 and intercept is r2)
Wettability of PS in the presence and absence of HDBPA
The water contact angle between a surface and water is governed by its surface energy. It is also known that surface energy and contact angle have an inverse relation, i.e., for a high wetting surface, the surface energy is stronger than the surface tension of the liquid [29]. Hence, the hydrophilic nature of HDBPA (cross-linker) contributed to an increase in surface energy and hydrophilicity of modified PS which led to a decrease in contact angle (Fig. 6).

Contact angle variation as a function of cross-linking density of samples
Water absorbing capability of cross-linked materials is also an indicator of its hydrophilic nature [30,31,32]. By increasing the HDBPA content in structure, the structure becomes more hydrophilic with expected higher water uptake. However, at the higher cross-linker content, the water uptake efficiency reduces due to more extensive 3-D network structure. Hence, it is predicted that the overall water uptake is governed via these two factors.
Thermal characterization of cross-linked PS
DSC
The glass transition temperatures (Tg) were determined using DSC. The results were 87, 102 and 117 °C for P1, P2 and P3, respectively (Fig. 7). To remove the thermal history, the prepared samples were first heated from ambient temperature to 400 °C at the heating rate of 10 °C/min and then cooled again to ambient temperature. By plotting obtained Tg versus degree of cross-linking, Tg of the pure PS could be calculated via data extrapolation. The PS Tg was estimated about 84 °C which is in complete agreement with the results obtained by Kanig et al. [33]. Yamasaki et al. [34] also studied Tg for different structures. They concluded that a Tg about 84 °C is an expected result for PS-based structures. The observed increment in glass transition temperature is expected due to a decrease in the degree of chains freedom imposed by cross-linking.

DSC plot of P1, P2 and P3
TGA
To investigate the effects of cross-linking on thermal stability of the final structure, TGA analysis was performed. As shown in Fig. 8, thermogram showed a weight loss at 100–260 °C which might be a result of residual moisture and solvent entrapped in cross-linked structure.

a Derivation of TGA plot. b TGA plot of P1, P2 and P3
The second step for P2 and P3 was observed at 260–350 °C which was due to the backbone fragmentation. It should be noted that the second weight loss was not observed in P1 at temperature below 400 °C. The higher thermal stability in P1 is consistent with the reports of other researchers [35,36,37]. The benzene ring in PS is mostly the reason for thermal stability of this commercial polymer. Delman et al. [35] conducted a comprehensive study on the effects of different functional groups on the thermal stability of compounds containing benzene groups. They concluded that the substitution of a benzene ring with an N–H bond decreases the thermal stability of structure. Similar results were obtained by Wu and Uhl et al. [38, 39]
Conclusion
Hydrophilic modification of PS was carried out by simultaneously polymerization and cross-linking of PS with synthetized HDBPA cross-linker. FT-IR and 1H NMR peaks revealed successful synthesis of HDBPA. PS hydrophilic modification was also verified based on FT-IR data. HDBPA content was calculated based on the nitrogen percentage data measured from elemental analysis. Reactivity ratio values were calculated by plotting FR equation parameters (\(r_{\text{HDBPA}} = 0.66\) and \(r_{{{\text{St}} .}} = 0.23\)). The results showed a very good fitting with R2 of 0.99. The improvement in hydrophilic properties was confirmed by contact angle measurements. To investigate the thermal properties, DSC and TGA analyses were also applied on the cross-linked samples. As expected, in DSC, the Tg of samples was increased by raising in cross-link density. Also, in TGA a decrease in thermal stability of samples was occurred as a result of the substitution of a benzene ring with amide groups.
References
Wang T, Zhu H, Xue H (2016) Reversible pH stimulus-response material based on amphiphilic block polymer self-assembly and its electrochemical application. Materials 9(6):478
Sarmento B, Campana-Filho SP, Azevedo C, Almeida A, Silva D (2018) Synthesis and applications of amphiphilic chitosan derivatives for drug delivery applications. In: Neves AR, Reis S (eds) Nanoparticles in life sciences and biomedicine. Pan Stanford, New York, pp 45–73
Oh JY, Kim S, Baik HK, Jeong U (2016) Conducting polymer dough for deformable electronics. Adv Mater 28(22):4455–4461
Zeynep EY, Antoine D, Brice C, Frank B, Christine J (2015) Double hydrophilic polyphosphoester containing copolymers as efficient templating agents for calcium carbonate microparticles. J Mater Chem B 3(36):7227–7236
Gao H, Jones M-C, Chen J, Liang Y, Prud’homme RE, Leroux J-C (2008) Hydrophilic nanoreservoirs embedded into polymeric micro/nanoparticles: an approach to compatibilize polar molecules with hydrophobic matrixes. Chem Mater 20(13):4191–4193
Ansary RH, Awang MB, Rahman MM (2014) Biodegradable poly (D, L-lactic-co-glycolic acid)-based micro/nanoparticles for sustained release of protein drugs-a review. Trop J Pharm Res 13(7):1179–1190
Surmenev R, Chernozem R, Syromotina D, Oehr C, Baumbach T, Krause B, Boyandin A, Dvoinina L, Volova T, Surmeneva M (2019) Low-temperature argon and ammonia plasma treatment of poly-3-hydroxybutyrate films: surface topography and chemistry changes affect fibroblast cells in vitro. Eur Polym J 112:137–145
Garcia-Ivars J, Wang-Xu X, Iborra-Clar M-I (2017) Application of post-consumer recycled high-impact polystyrene in the preparation of phase-inversion membranes for low-pressure membrane processes. Sep Purif Technol 175:340–351
Ma Y, Dai J, Wu L, Fang G, Guo Z (2017) Enhanced anti-ultraviolet, anti-fouling and anti-bacterial polyelectrolyte membrane of polystyrene grafted with trimethyl quaternary ammonium salt modified lignin. Polymer 114:113–121
Hannon JC, Kerry JP, Cruz-Romero M, Azlin-Hasim S, Morris M, Cummins E (2017) Kinetic desorption models for the release of nanosilver from an experimental nanosilver coating on polystyrene food packaging. Innov Food Sci Emerg Technol 44:149–158
Okano T, Yamada N, Sakai H, Sakurai Y (1993) A novel recovery system for cultured cells using plasma-treated polystyrene dishes grafted with poly (N-isopropylacrylamide). J Biomed Mater Res 27(10):1243–1251
Hwang S-J, Tseng M-C, Shu J-R, Yu HH (2008) Surface modification of cyclic olefin copolymer substrate by oxygen plasma treatment. Surf Coat Technol 202(15):3669–3674
Qu J-B, Liu Y, Liu J-Y, Huan G-S, Wei S-N, Li S-H, Liu J-G (2018) One-pot synthesis of bimodal gigaporous polystyrene microspheres with hydrophilic surfaces. Macromolecules 51(11):4085–4093
Fekete N, Béland AV, Campbell K, Clark SL, Hoesli CA (2018) Bags versus flasks: a comparison of cell culture systems for the production of dendritic cell–based immunotherapies. Transfusion 58(7):1800–1813
Borisova O, Billon L, Zaremski M, Grassl B, Bakaeva Z, Lapp A, Stepanek P, Borisov O (2012) Synthesis and pH-and salinity-controlled self-assembly of novel amphiphilic block-gradient copolymers of styrene and acrylic acid. Soft Matter 8(29):7649–7659
Oikonomou EK, Bethani A, Bokias G, Kallitsis JK (2011) Poly (sodium styrene sulfonate)-b-poly (methyl methacrylate) diblock copolymers through direct atom transfer radical polymerization: influence of hydrophilic–hydrophobic balance on self-organization in aqueous solution. Eur Polym J 47(4):752–761
Luo T, Lin S, Xie R, Ju X-J, Liu Z, Wang W, Mou C-L, Zhao C, Chen Q, Chu L-Y (2014) pH-responsive poly (ether sulfone) composite membranes blended with amphiphilic polystyrene-block-poly (acrylic acid) copolymers. J Membr Sci 450:162–173
Richard M-N, Bell M, Srinivasan R, Borhan A, Nagarajan R (2019) An approximate analytical approach to estimate the diffusivity of toxic chemicals in polymer barrier materials from the time evolution of sessile drop profiles. Polym Bull 76:339–364
Sharmin E, Zafar F (eds) (2012) Polyurethane. In: Polyurethane an introduction. InTechOpen, London, pp 3–16
Modarresi-Saryazdi SM, Haddadi-Asl V, Salami-Kalajahi M (2018) N, N’-methylenebis (acrylamide)-crosslinked poly (acrylic acid) particles as doxorubicin carriers: a comparison between release behavior of physically loaded drug and conjugated drug via acid-labile hydrazone linkage. J Biomed Mater Res Part A 106(2):342–348
Kong J, Yu S (2007) Fourier transform infrared spectroscopic analysis of protein secondary structures. Acta Biochim Biophys Sin 39(8):549–559
Mu B, Shen R, Liu P (2009) Crosslinked polymeric nanocapsules from polymer brushes grafted silica nanoparticles via surface-initiated atom transfer radical polymerization. Colloids Surf B 74(2):511–515
Ma Y, Jiang Y, Tan H, Zhang Y, Gu J (2017) A rapid and efficient route to preparation of isocyanate microcapsules. Polymers 9(7):274
Yang H, Yan R, Chen H, Lee DH, Zheng C (2007) Characteristics of hemicellulose, cellulose and lignin pyrolysis. Fuel 86(12–13):1781–1788
Abraham RJ, Griffiths L, Perez M (2013) 1H NMR spectra. Part 30: 1H chemical shifts in amides and the magnetic anisotropy, electric field and steric effects of the amide group. Magn Reson Chem 51(3):143–155
Nikdel M, Salami-Kalajahi M, Hosseini MS (2014) Synthesis of poly (2-hydroxyethyl methacrylate-co-acrylic acid)-grafted graphene oxide nanosheets via reversible addition–fragmentation chain transfer polymerization. RSC Adv 4(32):16743–16750
Yu L, Xu K, Ge L, Wan W, Darabi A, Xing M, Zhong W (2016) Cytocompatible, Photoreversible, and Self-Healing Hydrogels for Regulating Bone Marrow Stromal Cell Differentiation. Macromol Biosci 16(9):1381–1390
Fineman M, Ross SD (1950) Linear method for determining monomer reactivity ratios in copolymerization. J Polym Sci 5(2):259–262
Strobel M, Lyons C (2011) An essay on contact angle measurements. Plasma Process Polym 8(1):8–13
Pal P, Singh S-K, Mishra S, Pandey J-P, Sen G (2019) Gum ghatti based hydrogel: microwave synthesis, characterization, 5-Fluorouracil encapsulation and ‘in vitro’drug release evaluation. Carbohydr Polym 222:114979
Pal P, Pandey J, Sen G (2018) Sesbania gum based hydrogel as platform for sustained drug delivery: an ‘in vitro’study of 5-Fu release. Int J Biol Macromol 113:1116–1124
Pal P, Pandey J-P, Sen G (2017) Modified PVP based hydrogel: synthesis, characterization and application in selective abstraction of metal ions from water. Mater Chem Phys 194:261–273
Ueberreiter K, Kanig G (1950) Second-order transitions and mesh distribution functions of cross-linked polystyrenes. J Chem Phys 18(4):399–406
Yamasaki R, Endo T (2013) Synthesis and polymerization of styrene monomer carrying isothiocyanate moiety and its copolymerization with HEMA based on chemo-selectivity to nucleophiles. J Polym Sci Part A Polym Chem 51(24):5215–5220
Delman A, Stein A, Simms B (1967) Synthesis and thermal stability of structurally related aromatic Schiff bases and acid amides. J Macromol Sci Chem 1(1):147–178
Wu HS, Chuang MH, Hwang JW (1999) Kinetics and thermal analysis of copolymerization of m-isopropenyl–α, α′-dimethylbenzyl isocyanate with styrene. J Appl Polym Sci 73(13):2763–2770
Uhl FM, Levchik GF, Levchik SV, Dick C, Liggat JJ, Snape C, Wilkie CA (2001) The thermal stability of cross-linked polymers: methyl methacrylate with divinylbenzene and styrene with dimethacrylates. Polym Degrad Stab 71(2):317–325
Hsieh W-h, Cheng W-t, Chen L-c, Lin S-y (2018) Non-isothermal dehydration kinetic study of aspartame hemihydrate using DSC, TGA and DSC-FTIR microspectroscopy. Asian J Pharm Sci 13(3):212–219
Arrieta MP, López J, López D, Kenny J, Peponi L (2015) Development of flexible materials based on plasticized electrospun PLA–PHB blends: structural, thermal, mechanical and disintegration properties. Eur Polym J 73:433–446
Funding
The authors received no specific funding for this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Modarresi-Saryazdi, S.M., Rahmani, S. & Zahedi, P. Hydrophilic modification and cross-linking of polystyrene using the synthesized N,N′-(hexane-1,6-diyl)diacrylamide. Polym. Bull. 78, 1379–1391 (2021). https://doi.org/10.1007/s00289-020-03161-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-020-03161-z