
Abstract
Research deals with the assessment of grafting of cyclosiloxanes onto organically modified montmorillonite (MMT) layers through anionic ring-opening polymerization (AROP) without additional catalyst or initiator. Pristine Na+-MMT was organically modified by systematically varying the hexadecyltrimethylammonium bromide (HDTMABr) loading level. X-ray diffraction and thermogravimetric analysis of differently modified MMTs (OMMTs) showed that HDTMABr cations that exchanged sodium cations were strongly attracted to the silicate platelet surfaces. Part of HDTMABr formed a sandwiched HDTMABr layer between organically modified silicate platelets. The obtained OMMTs were tested as initiator material for AROP of octamethylcyclotetrasiloxane (D4) in the bulk, since the D4 polymerization may be initiated by oxygen anions located on the silicate surfaces. The highest monomer conversion (91 % after 24 h at 80 °C) was observed when OMMT with HDTMABr amount equal to cation exchange capacity was used. At lower amounts of HDTMABr, the MMT was less organically modified and thus less compatible with D4. On the other hand, when the HDTMABr sandwiched layer was thicker, lower rates of polymerization were observed due to the hindered transport of monomer to silicate platelet surfaces. The active function of surface anions in MMT platelets in the initiation process was confirmed experimentally by varying the OMMT/D4 ratio. It was observed that monomer conversion increased with increasing OMMT/D4 ratio. The obtained composites were exfoliated. In the last part of the research, OMMT was added to the formulation for D4 emulsion polymerization, where HDTMABr was also used as an emulsifier. The obtained composite material had a significantly improved thermal stability.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Poly(dimethylsiloxane) (PDMS), the backbone of which consists entirely of silicon–oxygen bonds, is a highly physically, chemically, and thermally stable polymer with an exceptionally low glass transition temperature and low surface energy. Linear PDMS has found application as lubricant, release agent and heat-transfer fluid. Due to the low glass transition, PDMS elastomer shows highly elastic behavior at room temperature and has therefore found use in applications from sealants to nontoxic medical implants. However, the major weakness of PDMS elastomer is its poor mechanical stability, which can be improved by incorporating fillers with high elastic modulus in the PDMS matrix. For this purpose, fumed silica is often used.
Schmidt et al. [1, 2] used melt intercalation/exfoliation technique to produce polysiloxane/layered-silicate nanocomposites based on a variety of polysiloxanes, with respect to both chemical functionality and molecular weight, to determine the origins of layered-silicate dispersion in a generic polysiloxane nanocomposite system. It has been shown [2], in systems based on PDMS and alkylammonium-modified layered-silicate fillers, that there are two key factors controlling layered-silicate dispersion: the presence of the appropriate number of long ammonium-bound alkyl chains and of polar silanol end groups. The ammonium-bound alkyl chains are commonly introduced between silicate layers (silica sheets or platelets) using the ion exchange method for making the hydrophilic layered silicate compatible with hydrophobic polymers [3, 4].
While numerous papers and reviews [5–10] have been published on the subject of layered silicate/polymer composites, only a few detailed studies have focused on polysiloxanes, despite the desirable properties of these materials, their need for reinforcement, and their compatibility with silica-based nanofillers [1]. Maybe, one of the reasons for this was the absence of substantial thermal stability improvement, due to high polysiloxane thermal stability, or even the observed reduced thermal stability of PDMS/OMMT nanocomposite systems [11]. Lewicki et al. [11] have shown that the inclusion of an OMMT into the PDMS matrix has a negative impact on its non-oxidative stability, because a lower-onset degradation temperature was observed. On the other hand, they demonstrated that at clay loadings above 2 %, the physical barrier effects of the nanoclay in the degrading systems become significant and serve to reduce the rate of volatiles evolution and the quantity of volatile species formed. The same research group confirmed also that the PDMS/OMMT nanocomposite elastomers thermally degrade primarily through a depolymerization mechanism that closely correlates with the accepted model of the thermal degradation of linear PDMS.
However, one of the most useful layered silicates often used for polymer composites preparation is montmorillonite [5]. Montmorillonite (MMT) belongs to smectite group of clay minerals, which has a 2:1 type of layer structure. It comprises negatively charged silica sheets held together by charge-balancing counterions such as Mg2+, Na+, and Ca2+. These interlayer cations balance the negative charges that are generated by the isomorphous substitution of Mg2+ and Fe2+ for Al3+ in the octahedral sheet, and Al3+ for Si4+ in the tetrahedral sheet. Besides the interlayer cations, the interlayer space of the smectite group of clay minerals contains large amounts of water molecules. During MMT modification, ammonium-bound alkyl chains replace the interlayer cations.
Nishihama et al. [12] showed that MMTs ion exchanged with inorganic cations and acidic clay can be used as catalysts for the polymerization of tetramethylcyclotetrasiloxane monomer. They concluded that it might be possible to design a catalyst for controlling siloxane polymerization by changing the number of Bronsted acid sites through exchange of the interlayer cations of MMTs.
Ma et al. [13] developed a method that involved in situ polymerization of dimethyldichlorosilane in the presence of OMMT and blending the treated MMT solution with several polymers, which yielded exfoliated/intercalated nanocomposites. The in situ dimethyldichlorosilane polymerization destroyed the strong electrostatic attraction between the silicate layers and the interlayer cations. It was observed that PDMS was grafted onto MMT layer surface via condensation of hydroxyl groups of PDMS and hydroxyl groups on MMT layer surface. They perceived that when the treated MMT solution was blended with other polymers, exfoliated or intercalated nanocomposites were obtained according to the discrepancy of compatibility between polymer and MMT as well as alkyl ammonium and PDMS grafted on the layer surface.
Bruzaud and coworkers [14–16] prepared polysiloxane-g-TiNbO5 nanocomposites, new organic–inorganic hybrid materials, via in situ intercalative polymerization of cyclosiloxanes in the presence of synthetic mineral oxide (TiNbO5), which was modified by tertamethylammonium cations. PDMS/HTiNbO5 nanocomposites were prepared [14] through melt intercalation of the pure mineral, melt intercalation of the organically modified mineral, grafting by a sol–gel process, and grafting by anionic ring-opening polymerization (AROP). Beigbeder et al. [15] stated that the elaboration methods of nanocomposites have much influence on their structure and rheology. They were able to conclude that a more cohesive structure was obtained when the elaboration path started from monomers to chemically graft polymer chains at the surface of the mineral layer. Furthermore, it was found [15] that anionic polymerization is more efficient than polycondensation in terms of nanocomposite structuration.
Messersmith and Giannelis [17, 18] synthesized poly(ε-caprolactone)/clay nanocomposites with exfoliated structures, by the in situ AROP of ε-caprolactone by a coordination/insertion mechanism. Although the exact polymerization mechanism is unknown, it was shown that polymerization also depends on the intercalated cations. AROP of ε-caprolactone [19–25] was catalyzed by metal alkoxides in the presence of organomodified clays that contain hydroxy groups. The hydroxy groups can act as initiators for the AROP to yield surface-grafted polymer chains and lead to an exfoliated morphology [26, 27].
This research deals with the assessment of grafting of cyclosiloxanes onto organically modified montmorillonite layers through AROP without using additional catalyst or initiator. The montmorillonite layers carry anions, which may act as initiator for AROP. The effects of the extent of MMT modification with hexadecyltrimethylammonium bromide (HDTMABr) and OMMT/cyclosiloxane ratio on the octamethylcyclotetrasiloxane (D4) AROP in bulk were considered. The investigated process may be useful for the production of polysiloxane/OMMT nanocomposites by bulk polymerization. At the same time, products of the investigated process could be added as masterbatches to hydrophobic polymers or monomers to produce hydrophobic polymer/PDMS-g-OMMT nanocomposites by melt intercalation or in situ (bulk, solvent, suspension or emulsion) polymerization.
In the last part of the research, OMMT was added to a formulation for D4 emulsion AROP, where HDTMABr was also used as cationic emulsifier. The thermal stability of the obtained composite material was compared to that of PDMS without OMMT obtained by AROP emulsion process.
Experimental
Materials
Siloxane monomer octamethylcyclotetrasiloxane (D4, 98 %, ABCR GmbH&Co, Karlsruhe, Germany) was used as received. Pristine montmorillonite clay Cloisite Na+ (MMT, Southern Clay Products Inc, Texas, USA) applied in this study had a cation exchange capacity (CEC) of 92.6 meq/100 g of clay and a basal spacing 1.17 nm (both reported by the supplier). The cationic emulsifier hexadecyltrimethylammonium bromide (HDTMABr, ≥98 %, Aldrich, Steinheim, Germany) was used for clay modification. Water was deionized previous to use.
For emulsion polymerization recipe, also nonionic emulsifier, secondary alcohol ethoxylate with trade name Tergitol, Type 15-S-9 (p.a., Aldrich), and initiator, potassium hydroxide (p.a., Merck), were used.
Na+-montmorillonite modification and octamethylcyclotetrasiloxane polymerization
Modification of MMT was carried out using the ion exchange method. Different concentrations of HDTMABr were used. The MMT and HDTMABr amounts (Table 1) were dispersed and dissolved, respectively, in 500 mL of deionized water. The dispersion of MMT in HDTMABr solution was vigorously stirred overnight at room temperature. The obtained organically modified MMT (OMMT) particles were separated by centrifugation and washed several times with hot water (cca 60 °C) to remove excess HDTMABr, Br anions, and exchanged Na cations. For Br− indicator, AgNO3 was used. The products were dried in a vacuum oven at 50 °C until a constant sample mass was reached. The OMMT samples were labeled as OMMT/x, where x stands for the amount of HDTMABr used, expressed in terms of CEC (1 CEC equals 92.6 mmol HDTMABr per 100 g MMT or 33.8 g HDTMABr per 100 g MMT).
OMMTs were tested as initiator material for AROP of D4 in the bulk. D4 polymerization was carried out at 80 °C in a 500 mL glass reactor with four necks equipped with a reflux condenser, a mechanical stirrer, a digital thermometer, and a nitrogen gas inlet. An adjustable cooling temperature system was used. The obtained PDMS/OMMT composites were labeled as PD4-OMMT/x/y, where x stands for the amount of HDTMABr in OMMT (expressed in terms of CEC) and y for the mass ratio between OMMT and D4 applied in the polymerization.
To obtain the PDMS/OMMT composite by the emulsion process, 1 wt% of OMMT/1.0 per D4 mass (0.100 g OMMT per 10 g D4) was added to the recipe for AROP of D4 in emulsion. Other amounts of materials employed were: 10 g of D4, 60 g of water, 0.125 g of Tergitol, 1.125 g of HDTMABr, and 0.200 g of KOH. The detailed procedure of the emulsion polymerization is described elsewhere [28].
Characterization methods
The interlayer distance of modified clay was determined applying Bragg’s law equation to X-ray diffraction (XRD) results. The X-ray diffraction was measured with copper Kα1 radiation generated at 45 kV and 40 mA using a PANalytical X’Pert PRO. Approximately, 3 mm-thick and 25 mm-wide flat square samples were prepared. Investigations were carried out in the angle (2\( \theta \)) range of 1.5°–90° with a step of 0.033°.
The thermal stability of modified MMT and PDMS composites was assessed by thermogravimetric analysis (TGA). Measurements were performed in a nitrogen atmosphere with a flow rate of 50 mL min−1 using a Mettler Toledo (Schwerzenbach, Switzerland) TGA/DSC1 instrument. The investigated temperature range was from 25 up to 700 °C and the heating rate 20 K min−1. For measurements, standard 70 µL Al2O3 crucibles were used and the baseline was automatically subtracted. The sample mass was around 10 mg. Before TGA, the unreacted monomer and emulsion water were removed from samples using a vacuum dryer at 50 °C. From TGA results, the OMMT composition and D4 conversion were calculated. The OMMT composition was calculated from residual masses at 350, 500 and 700 °C for MMT and OMMTs. The D4 conversion was calculated from the residual mass at 700 °C, taking into account the thermal stability of OMMT employed in the composite. This was possible because the residual mass of PDMS at 700 °C in nitrogen atmosphere is zero, since in an inert atmosphere PDMS degrades through intermolecular and intramolecular redistribution, intermolecular and intramolecular depolymerization, and unzipping mechanisms, by which volatile cycles are formed. To obtain the polymer mass in a sample, which was previously exposed at 50 °C in a vacuum oven as long as necessary to obtain constant sample mass, the mass of modified clay, calculated from the residual mass at 700 °C, was subtracted from the mass of the sample. During vacuum exposure, all the volatile components, monomer and small oligomeric cycles, were removed from the sample. Thus, by the TGA method only the amount of monomer converted to large, non-volatile polymeric chains was determined and the monomer conversion should be interpreted accordingly.
To confirm D4 polymerization, 29Si NMR spectra of PDMS/OMMT composites were recorded on a Varian Unity Inova 300 NMR Spectrometer equipped with a 29Si probe (5 mm diameter) at 60.19 MHz. The samples were diluted in chloroform containing TMS, which was used as a chemical reference.
Results and discussion
Na+-montmorillonite modification
TGA was used to investigate the thermal stability of HDTMABr, MMT, and OMMTs prepared with different amounts of HDTMABr. By interpreting TGA results, compositions of OMMTs could be deducted. At temperatures below 200 °C, minimal differences between curves of HDTMABr, MMT, and OMMTs (Fig. 1) may be attributed to the mass loss caused by evaporation of water adsorbed on MMT. HDTMABr sample started to decompose at around 200 °C and decomposed completely in one single step. Its residual mass was lower than 2 % at 350 °C, where MMT showed only a 2 % mass loss. The mass loss of MMT increased up to 7.7 % at 700 °C, where the residual mass for HDTMABr was already close to 0 %. For OMMTs, two distinctive steps of mass loss were observed. The first step occurred in the temperature range where pure HDTMABr exhibited an almost 100 % loss of mass. Accordingly, the mass loss up to 350 °C was attributed to degradation of HDTMABr, which was not attracted to and/or in contact with negatively charged MMT platelets. On the other hand, the observed mass loss in the second step, i.e., above 350 °C, was attributed to degradation of HDTMA+ cations that had exchanged sodium cations and were therefore attracted strongly to MMT anionic sites through electrostatic interaction. The observed residual masses for different OMMT samples after the first and the second step are shown in Table 1.

TGA thermograms of MMT, HDTMABr, and MMT modified with different HDTMABr amounts (OMMTs)
According to Zhao et al. [29], who investigated the relationship between the continually expanded interlayer distance of layered silicates and excess intercalation of cationic surfactants, surfactant cations (in our case HDTMA+ cations) enter the interlayer by cation exchange mechanism below the CEC. Then, as soon as the anionic sites are used up, excess surfactant cations enter the interlayer together with their counterions, driven by the hydrophobic interaction between the alkyl chains of the surfactants. With the hydrophobic interaction, excess surfactants would continue to align themselves more orderly in the constrained environment and form a sandwiched surfactant layer with their counterions. As more surfactants enter the interlayer, the sandwiched surfactant layer becomes thicker, leading to the continual increase of the interlayer distance.
However, from our TGA results, it may be deducted that the sandwiched layer was formed already at HDTMABr amounts below the CEC and that the surfactant cations continued to enter the interlayer by cation exchange mechanism also once the sandwiched layer was formed (Fig. 2). It may be observed that the exchanged HDTMABr remained at a similar level regardless of the amount of HDTMABr added, whereas sandwiched HDTMABr linearly increased with the increase in the amount of HDTMABr added as expected. The amount of cationically exchanged HDTMABr was around 16 per 100 g MMT, which is around 60 % of the CEC value. It is necessary to specify that the values for sandwiched HDTMABr levels were calculated from TGA results by subtracting the amount of cationically exchanged HDTMABr, i.e., HDTMA+, from the total HDTMABr amount in OMMT. The total amount of HDTMABr in OMMT was calculated from TGA data at 700 °C taking into account the MMT mass loss at this temperature, while the amount of cationically exchanged HDTMABr was calculated from the mass loss in the interval between 350 and 500 °C.

Amount of HDTMABr that entered the interlayer by cation exchange, the amount of HDTMABr forming the sandwiched layer versus the amount of HDTMABr used for MMT modification
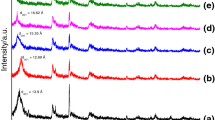
To confirm the interpretation of TGA results and the linearity of the sandwiched HDTMABr according to the added HDTMABr, the interlayer distance for OMMTs was determined using XRD results. The XRD spectra for MMT, OMMT/0.8, OMMT/1.0, and OMMT/1.2 are shown in Fig. 3. The interlayer distances were estimated from the position of the (001) diffraction peak using Bragg’s law equation (Table 1). In Fig. 3, the (001) peak of the original MMT at 7.43°, corresponding to the interlayer distance 1.22 nm, undergoes an obvious shift to a smaller angle as the HDTMABr loading level increases, implying that the interlayer distance monotonically increased with the HDTMABr loading level. The interlayer distance was increased from 1.22 to 2.06 nm when the loading level increased from 0 to 67.6 g HDTMABr per 100 g MMT (Table 1).

XRD spectra for MMT and OMMTs
The increase of the interlayer distance of OMMTs versus amount of sandwiched HDTMABr is shown in Fig. 4, where a linear curve fits the experimental values nearly perfectly. It may be read from the linear equation in Fig. 4 that OMMT without HDTMABr sandwiched layer would have an interlayer thickness of around 1.65 nm (1.22 + 0.43), and that every additional gram of HDTMABr added to 100 g MMT would increase the interlayer distance by approximately 0.01 nm. Of course, the equation assumes constant thickness of the cationically exchanged HDTMABr layer and is valid as long as the structure of the OMMT is stable and its platelets remain aligned. The “sandwich” OMMT structure has been schematically shown by Zhao et al. [29] elsewhere.

The increase of the interlayer distance of OMMTs versus the amount of sandwiched HDTMABr
D4 bulk polymerization in the presence of OMMT
Once the OMMTs structure was known, the OMMTs, which are very often used as nanofiller for polymer reinforcement, were tested as initiator materials for AROP of D4. The oxygen anions of MMT platelets, created by isomorphous substitutions, might act as initiator for AROP. The negative charges of MMT platelets are generated by the substitutions of Mg2+ and Fe2+ for Al3+ in the octahedral sheet, and Al3+ for Si4+ in the tetrahedral sheet. However, the anions in the octahedral sheet are more inaccessible to monomers, unless located on the edge of the platelet.
To confirm D4 polymerization, 29Si NMR spectra of the products obtained after 24 h of polymerization were recorded. NMR spectra showed signals at −19.64 and −22.28 ppm, which were attributed [28, 30] to D4 and the polydimethysiloxane unit of the polymer chain, respectively. In the NMR spectra of samples obtained after conversion determination, the D4 signal was absent, which proved that the monomer was efficiently removed. The XRD spectra of samples containing 10 % of OMMTs did not show signals of ordered clay platelets, which clearly confirmed that D4 polymerization occurred in clay galleries. On the other hand, the spectrum of PD4-MMT/0.0/0.1 (when 10 % of unmodified MMT was employed) did show a diffraction signal corresponding to the unmodified MMT peak at 7.5°.
The effect of MMT modification on monomer conversion was investigated first. TGA curves for samples PD4-OMMT/1.0/0.1 and PD4-OMMT/1.2/0.1 obtained after 24 h of polymerization together with curves for respective OMMTs and the curve for pure D4 are shown in Fig. 5. D4 evaporated completely when 160 °C were reached. The residual mass after PDMS degradation in an inert atmosphere, such as nitrogen atmosphere, is zero, because PDMS degrades through mechanisms by which volatile cycles are formed. Such mechanisms are intermolecular and intramolecular redestributions, intermolecular and intramolecular depolymerization, and unzipping mechanism [31–37]. According to the results shown in Fig. 5, the PDMS/MMT organic part of the composite decomposed completely at around 600 °C, whereas 7 % of PD4-OMMT/1.0/0.1 and 18 % of PD4-OMMT/1.2/0.1 residuals remained.

TGA thermograms of D4 monomer, modified MMTs (OMMT/0.1 and OMMT/1.2), and their polysiloxane/OMMT composites (PD4-OMMT/1.0/0.1 and PD4-OMMT/1.2/0.1) obtained after 24 h of polymerization at 80 °C
The calculated D4 conversions for all investigated PDMS/OMMT samples are shown in Table 2. The highest conversion was obtained for sample PD4-OMMT/1.0/0.1, which had around 60 % Na+ cations exchanged and an interlayer thickness of 1.76 nm. Approximately, half of the HDTMABr in the galleries was cationically exchanged (Fig. 2). When OMMTs with higher or lower amount of HDTMABr were used, the conversion was lower. At lower amounts of HDTMABr in OMMTs, the HDTMABr sandwiched layer was thinner and the galleries were less hydrophobic. Thus the OMMTs were less compatible with D4 and the entrance of the hydrophobic monomer into the interlayer spacing was hampered. As expected, when unmodified MMT was employed (sample PD4-MMT/0.0/0.1) no D4 conversion was detected. The result is in accordance with results obtained by Nishihama et al. [12], who observed that the yield of polysiloxane was strongly dependent on the cationic species in montmorillonites. They showed that montmorillonites with Na+ in the interlayer exhibit negligible ability for siloxane polymerization in both yield and mean molecular weight of the products. However, on the other hand, at higher amounts of HDTMABr, the thick HDTMABr sandwiched layer enhanced the entrance of hydrophobic monomer into the interlayer spacing, but hindered the transport of monomer to silicate platelet surface where initiation occurs.
In the AROP process under investigation, the D4 monomer and MMT oxygen anions coupled with HDTMA+ counterions are involved in the initiation reaction. After the initiation reaction, the active center (anion) for propagation is transferred to the end of the growing PDMS chain, where it remains associated with the HDTMA+. Since the active anions are always associated with counterions, it is possible that the high HDTMABr amount in the interlayer spacing deterred not only the initiation reaction, due to hindered transport of monomer to silicate platelet surface, but also the propagation reaction due to the steric hindrance caused by HDTMA+ and HDTMABr orientation around active anion sites on polymer chain ends. However, backbiting reactions, typical for AROP of cyclosiloxanes, are the main reasons for low equilibrium conversions [28, 38, 39]. By backbiting reactions, small oligomeric cyclosiloxanes are formed from a growing polymer chain carrying an active anion. The reactions for AROP of D4 in the presence of initiator anions are shown and explained in detail elsewhere [28].
When the effect of OMMT/D4 ratio on the polymerization was investigated, the OMMT/1.0 was employed and the monomer amount was varied. Samples PD4-OMMT/1.0/0.050, PD4-OMMT/1.0/0.075, and PD4-OMMT/1.0/0.1 had mass ratios between OMMT and D4 0.050, 0.075, and 0.100, respectively. It may be concluded from the results shown in Table 2 that the monomer conversion increased with increasing OMMT amount. This proved that the OMMT, if organically modified in the proper extent, acts as initiator for AROP of cyclosiloxanes. Furthermore, the clear effect of sandwiched layer thickness on the monomer conversion strongly indicated that initiation of D4 cycles occurred predominantly in clay galleries. The proposed initiation of AROP of D4 by MMT anions is schematically shown in Fig. 6.

Grafting of PDMS on OMMT initiated by MMT anions: a before initiation and b after initiation
D4 emulsion polymerization in the presence of OMMT
In the last part of our research, OMMT/1.0 was included in the recipe for D4 AROP in emulsion [28]. Stable emulsions were obtained. Samples of PDMS emulsion without OMMT and PDMS/OMMT emulsions were dried to constant mass to obtain samples without unreacted monomer and water. From residual masses, monomer conversions after 4 h of polymerization were calculated, which were found to be 74 and 67 % for PDMS and PDMS/OMMT emulsions, respectively. The mechanism of AROP in emulsion for the system without OMMT has been described in detail in our previous publications [28, 38, 39]. According to the description, the higher conversion for PDMS emulsion may be explained by backbiting reactions, which in the absence of OMMT occur only at the particle surface [39], where ionic active sites are located. On the other hand, in the case of PDMS/OMMT emulsion, backbiting reactions are possible also between clay platelets, i.e., in the particle interior. Another possible reason for lower conversion for PDMS/OMMT system could be that MMT platelets hinder the penetration of terminated polymer chains (of a critical degree of polymerization) into the particle interior and diffusion of fresh monomer to particle surface, where polymerization is faster. The polymerization rate in polymer particles, which is caused by MMT anions, must be considerably slower than that on the particle surface, which is caused by hydroxyl anions.
TGA results for dried samples of PDMS emulsion without OMMT and PDMS/OMMT emulsion are shown in Fig. 7. In both TGA curves, the first degradation step corresponded to HDTMABr decomposition. The second step in the PDMS curve (without OMMT) was due to PDMS degradation—in one single step until temperature around 415 °C was reached. The constant residual mass at higher temperatures may be attributed to inorganic salts present in the sample. The salts originated from KOH, HDTMABr, and hydrochloric acid. The latter was used to stop the AROP reaction after 4 h. On the other hand, the TGA curve of dried sample of PDMS/OMMT (Fig. 7) showed four mass loss steps. It may be assumed that the grafted PDMS, which was initiated by MMT anions, degraded in the last step with offset around 600 °C. According to the results shown above (Figs. 1, 5), between the first and the last step non-grafted PDMS and HDTMA+ cations, which were attracted strongly to MMT, decomposed. The formation of polysiloxane chains between clay platelets was indicated by XRD spectra, which showed no signals.

TGA thermograms of dried emulsions with OMMT/1.0 (PDMS/OMMT) and without OMMT/1.0 (PDMS)
Lewicki et al. [11], who studied the thermal degradation behavior of PDMS/OMMT nanocomposites, prepared by blending PDMS with OMMT and using ultrasonic processing, showed that the physical barrier effects of the nanoclay in the degrading system become significant and serve to reduce the rate of volatiles evolution and the quantity of volatile species formed at OMMT loadings above 2 wt% on total resin mass. For composites obtained by our process, the improved thermal stability was observed already at OMMT loading of 1 wt% of OMMT/1.0. Moreover, the thermal stability of PDMS was significantly improved (for around 180 °C), considering the offset temperature of the last step in the PDMS/OMMT thermogram. We believe this considerable improvement was due to the PDMS grafting onto OMMT platelets, which is possible due to the fact that MMT oxygen anions initiate D4 AROP.
Conclusions
It was shown that organically modified montmorillonite (OMMT) initiates AROP of cyclosiloxanes such as octamethylcyclotetrasiloxane without an additional initiator or catalyst. In the process, PDMS is grafted to the OMMT surface. The extent of montmorillonite modification with alkylammonium surfactants drastically affects grafting and polymerization of cyclosiloxane if the OMMT contains a sandwiched layer of cationic surfactant between OMMT platelets. A thicker sandwiched layer of surfactant hinders the transport of monomer to silicate platelet surface, where initiation occurs. On the other hand, if the OMMT is not sufficiently organically modified, it is less compatible with D4 and the entrance of hydrophobic monomer between MMT platelets is hampered.
The described process could be used for polysiloxane/OMMT nanocomposites production by in situ bulk polymerization with or without an additional initiator. Also, products of the investigated process could be used for polymer/PDMS-g-OMMT nanocomposites production by melt intercalation or even by emulsion polymerization. Consequently, the products may be suitable for different applications, such as from lubricants, release agents, and heat-transfer fluids with improved rheology to elastomers with improved barrier properties, mechanical stability, and thermal stability. The improved thermal stability of PDMS/PDMS-g-OMMT nanocomposites has been proved for an emulsion system.
References
Schmidt DF, Giannelis EP (2009) Silicate dispersion and mechanical reinforcement in polysiloxane/layered silicate nanocomposites. Chem Mater 22:167–174. doi:10.1021/cm9026978
Schmidt DF, Clément F, Giannelis EP (2006) On the origins of silicate dispersion in polysiloxane/layered-silicate nanocomposites. Adv Funct Mater 16:417–425. doi:10.1002/adfm.200500008
Fukushima Y, Inagaki S (1987) Synthesis of an intercalated compound of montmorillonite and 6-polyamide. J Incl Phenom 5:473–482. doi:10.1007/BF00664105
Usuki A, Kojima Y, Kawasumi M, Okada A, Fukushima Y, Kurauchi T, Kamigaito O (1993) Synthesis of nylon 6-clay hybrid. J Mater Res 8:1179–1184. doi:10.1557/JMR.1993.1179
LeBaron PC, Wang Z, Pinnavaia TJ (1999) Polymer-layered silicate nanocomposites: an overview. Appl Clay Sci 15:11–29. doi:10.1016/S0169-1317(99)00017-4
Alexandre M, Dubois P (2000) Polymer-layered silicate nanocomposites: preparation, properties and uses of a new class of materials. Mat Sci Eng R 28:1–63. doi:10.1016/S0927-796X(00)00012-7
Okamoto M (2006) Recent advances in polymer/layered silicate nanocomposites: an overview from science to technology. Mater Sci Technol 22:756–779. doi:10.1179/174328406x101319
Sinha Ray S, Okamoto M (2003) Polymer/layered silicate nanocomposites: a review from preparation to processing. Prog Polym Sci 28:1539–1641. doi:10.1016/j.progpolymsci.2003.08.002
Giannelis EP (1996) Polymer layered silicate nanocomposites. Adv Mater 8:29–35. doi:10.1002/adma.19960080104
Tasdelen MA, Kreutzer J, Yagci Y (2010) In situ synthesis of polymer/clay nanocomposites by living and controlled/living polymerization. Macromol Chem Phys 211:279–285. doi:10.1002/macp.200900590
Lewicki JP, Liggat JJ, Patel M (2009) The thermal degradation behaviour of polydimethylsiloxane/montmorillonite nanocomposites. Polym Degrad Stab 94:1548–1557. doi:10.1016/j.polymdegradstab.2009.04.030
Nishihama S, Yamada H, Nakazawa H (1997) Polymerization of tetramethylcyclotetrasiloxane monomer by ion-exchanged montmorillonite catalysts. Clay Min 32:645–651. doi:10.1180/claymin.1997.032.4.14
Ma J, Xu J, Ren J-H, Yu Z-Z, Mai Y-W (2003) A new approach to polymer/montmorillonite nanocomposites. Polymer 44:4619–4624. doi:10.1016/S0032-3861(03)00362-8
Bruzaud S, Levesque G (2002) Polysiloxane-g-TiNbO5 nanocomposites: synthesis via in situ intercalative polymerization and preliminary characterization. Chem Mater 14:2421–2426. doi:10.1021/cm021108b
Beigbeder A, Bruzaud S, Mederic P, Aubry T, Grohens Y (2005) Rheological characterization of polydimethylsiloxane/HTiNbO5 nanocomposites prepared by different routes. Polymer 46:12279–12286. doi:10.1016/j.polymer.2005.10.086
Lebrun L, Bruzaud S, Grohens Y, Langevin D (2006) Elaboration and characterisation of PDMS-HTiNbO5 nanocomposite membranes. Eur Polym J 42:1975–1985. doi:10.1016/j.eurpolymj.2006.03.016
Messersmith PB, Giannelis EP (1993) Polymer-layered silicate nanocomposites: in situ intercalative polymerization of ε-caprolactone in layered silicates. Chem Mater 5:1064–1066. doi:10.1021/cm00032a005
Messersmith PB, Giannelis EP (1995) Synthesis and barrier properties of poly(ε-caprolactone)-layered silicate nanocomposites. J Polym Sci Part A Polym Chem 33:1047–1057. doi:10.1002/pola.1995.080330707
Lepoittevin B, Pantoustier N, Alexandre M, Calberg C, Jerome R, Dubois P (2002) Polyester layered silicate nanohybrids by controlled grafting polymerization. J Mater Chem 12:3528–3532. doi:10.1039/B205787E
Viville P, Lazzaroni R, Pollet E, Alexandre M, Dubois P (2004) Controlled polymer grafting on single clay nanoplatelets. J Am Chem Soc 126:9007–9012. doi:10.1021/ja048657y
Pantoustier N, Lepoittevin B, Alexandre M, Dubois P, Kubies D, Calberg C, Jérôme R (2002) Biodegradable polyester layered silicate nanocomposites based on poly(ε-caprolactone). Polym Eng Sci 42:1928–1937. doi:10.1002/pen.11085
Lepoittevin B, Pantoustier N, Devalckenaere M, Alexandre M, Calberg C, Jerome R, Henrist C, Rulmont A, Dubois P (2003) Polymer/layered silicate nanocomposites by combined intercalative polymerization and melt intercalation: a masterbatch process. Polymer 44:2033–2040. doi:10.1016/s0032-3861(03)00076-4
Lepoittevin B, Pantoustier N, Devalckenaere M, Alexandre M, Kubies D, Calberg C, Jerome R, Dubois P (2002) Poly(ε-caprolactone)/clay nanocomposites by in situ intercalative polymerization catalyzed by dibutyltin dimethoxide. Macromolecules 35:8385–8390. doi:10.1021/ma020300w
Liao LQ, Zhang C, Gong SQ (2007) Preparation of poly(ε-caprolactone)/clay nanocomposites by microwave-assisted in situ ring-opening polymerization. Macromol Rapid Commun 28:1148–1154. doi:10.1002/marc.200700063
Tarkin-Tas E, Goswami SK, Nayak BR, Mathias LJ (2008) Highly exfoliated poly(ε-caprolactone)/organomontmorillonite nanocomposites prepared by in situ polymerization. J Appl Polym Sci 107:976–984. doi:10.1002/app.26964
Ganachaud F, Boileau S (2009) Siloxane-containing polymers. In: Dubois P, Coulembier O, Raquez JM (eds) Handbook of ring-opening polymerization. Wiley-VCH GmbH & Co, KGaA, Germany
Jones RG, Ando W, Chojnowski J (2001) Silicon-containing polymers: the science and technology of their synthesis and applications. Springer, The Netherlands
Mohorič I, Šebenik U (2011) Anionic ring-opening polymerization of octamethylcyclotetrasiloxane in emulsion above critical micelle concentration. Polymer 52:1234–1240. doi:10.1016/j.polymer.2011.01.025
Zhao Z, Tang T, Qin Y, Huang B (2003) Relationship between the continually expanded interlayer distance of layered silicates and excess intercalation of cationic surfactants. Langmuir 19:9260–9265. doi:10.1021/la030056h
LaRochelle RW, Cargioli JD, Williams EA (1976) Viscosity-molecular weight correlations of disiloxanols by 29Si Fourier transform nuclear magnetic resonance. Macromolecules 9:85–88. doi:10.1021/ma60049a016
Lewis RN (1948) Methylphenylpolysiloxanes. J Am Chem Soc 70:1115–1117. doi:10.1021/ja01183a073
Grassie N, Macfarlane IG (1978) Thermal degradation of polysiloxanes.1. Poly(dimethylsiloxane). Eur Polym J 14:875–884. doi:10.1016/0014-3057(78)90084-8
Thomas TH, Kendrick TC (1969) Thermal analysis of polydimethylsiloxanes. I. Thermal degradation in controlled atmospheres. J Polymer Sci 2 Polymer Phys 7:537. doi:10.1002/pol.1969.160070308
Lewis CW (1959) The pyrolysis of dimethylpolysiloxanes. II. J Polym Sci 37:425–429. doi:10.1002/pol.1959.1203713212
Camino G, Lomakin SM, Lazzari M (2001) Polydimethylsiloxane thermal degradation—Part 1. Kinetic aspects. Polymer 42:2395–2402. doi:10.1016/s0032-3861(00)00652-2
Camino G, Lomakin SM, Lageard M (2002) Thermal polydimethylsiloxane degradation. Part 2. The degradation mechanisms. Polymer 43:2011–2015. doi:10.1016/s0032-3861(01)00785-6
Mohorič I, Krajnc M, Šebenik U (2009) Model-free kinetics analysis of thermal degradation of polysiloxane lubricant. Chem Biochem Eng Q 23:493–496
Mohorič I, Šebenik U (2011) Semibatch anionic ring-opening polymerization of octamethylcyclotetrasiloxane in emulsion. Polymer 52:4423–4428. doi:10.1016/j.polymer.2011.07.045
Mohorič I, Šebenik U (2013) Semibatch anionic ring-opening polymerization of octamethylcyclotetrasiloxane in emulsions: effect of the amount of seed polymer particles. Polym Int 62:1022–1028. doi:10.1002/pi.4386
Acknowledgments
The financial support of this work by the Slovenian Ministry of Higher Education, Science and Technology (Grant P2-0191) is gratefully acknowledged. The authors would like to express their gratitude to Dr. Ines Mohorič for the TGA analysis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ručigaj, A., Krajnc, M. & Šebenik, U. Polymerization of octamethylcyclotetrasiloxane between montmorillonite nanoplatelets initiated by surface anions. Polym. Bull. 72, 1863–1878 (2015). https://doi.org/10.1007/s00289-015-1377-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-015-1377-5