
Abstract
Rhodococcus sp. Eu-32 has shown an extended novel dibenzothiophene desulfurization sulfur-specific 4S pathway and could remove significant amounts of organic sulfur from coal. Here, we present the draft genome sequence of Eu-32 with a genome size of approximately 5.61 Mb, containing 5065 protein coding sequences with a G+C content of 65.1%. The Rhodococcus sp. Eu-32 showed ~ 99% identity at the 16S rRNA gene sequence level while < 34% digital DNA–DNA hybridization and < 81% average nucleotide identity values with the genome sequence of most closely related known Rhodococcus species, suggesting that it is taxonomically different from the already reported Rhodococcus species. Among the annotated genes, 90 are involved in the metabolism of sulfur. Comparative genome analysis suggests many commonalities in sulfur metabolism gene sets that may have evolved due to many factors including ecological pressures. Our study and the genome sequence data will be available for further research and will provide insights into potential biotechnological and industrial applications of this bacterium.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sulfur oxide (SOx) emission from the burning of the sulfur loaded fossil fuels causes serious health (bronchial irritation and asthma), environmental (acidic rains), as well as technical problems (corrosion of the machinery) [15, 17]. The process of hydrodesulfurization (HDS) is being used to remove sulfur; however, HDS demands severe operational conditions and cannot remove sulfur from thiophenic compounds such as dibenzothiophene (DBT), benzothiophene (BT), and their alkylated forms [8, 18, 20]. Biodesulfurization (BDS) is thought to be an alternative complementary technique for the refining of petroleum, as it operates at milder conditions, removes sulfur in a selective manner, and is environment friendly [5, 25]. The three main components of any generalized industrial microbial process are microorganisms, substrate, and product. In such processes, microorganisms or their enzymes play a remarkable role as biocatalysts. For an efficient process, the microorganisms should be fast growing, non-pathogenic/eco-friendly, and have high substrate range specificity and conversion efficiency. However, the process of biological removal of sulfur has been limited by some important challenges like low specific activity/conversion rate, narrow substrate range, laborious task of handling various growth controlling parameters, and inhibition of desulfurization biocatalysts/enzymes by accumulation of end product (s) e.g., 2-hydroxybiphenyl [1, 13, 16]. For laboratory and industrial scale applications of the biodesulfurization process, an improved understanding of the biodesulfurizing microorganisms, as one of the key components of the process reaction, would be far more desirable to make the process more efficient. The whole genome sequencing approach has facilitated in understanding new microbial enzymes and processes in an efficient way [19].
Members of the genus Rhodococcus are metabolically versatile, able to degrade/metabolize a range of organic and synthetic (xenobiotic) compounds, and also have the ability to desulfurize the thiophenic sulfur containing compounds present in fossil fuels [24]. The Rhodococcus sp. Eu-32 has the ability to desulfurize the thiophenic sulfur containing compounds like DBT by cleaving the C–S bonds [3]. The Rhodococcus sp. Eu-32 follows a novel extended sulfur-specific 4S pathway and can also efficiently desulfurize the coal [2, 3]. In an effort to understand the genome sequence-based taxonomic position and metabolic versatility of the Eu-32 related to sulfur metabolism, its genome was sequenced and is reported in this paper.
Materials and Methods
Genome Sequencing and Assembly
The Rhodococcus sp. Eu-32 was isolated from the soil sample taken from the roots of a Eucalyptus tree [3]. The genomic DNA extraction and genome sequencing were carried out by the LGC Genomics GmbH, Germany using the Illumina MiSeq V3 platform with 2 × 300 bp paired-end sequencing. The filtered high quality reads were de novo assembled using SPAdes version 3.10.1 (default settings).
Genome Analysis
The genome annotation was carried out using the NCBI Prokaryotic Genome Annotation Pipeline (PGAP; default settings) and Rapid Annotation using Subsystem Technology (RAST; ClassicRAST default settings) server version 2.0 [4]. Sequence and function based genome comparisons were also performed using RAST. The genomic digital DNA–DNA Hybridization (dDDH) values were computed using the Genome-to-Genome Distance Calculator (GGDC) version 2.1 (default settings) [14] by submitting the genome sequences to DSMZ (http://ggdc.dsmz.de). Average Nucleotide Identity (ANI) values were calculated using Kostas Lab web server (http://enve-omics.ce.gatech.edu/ani/); default parameters—minimum length = 700 bp, minimum identity = 70%, and minimum alignments = 50. This ANI calculator estimates the average nucleotide identity between two genomic datasets using both best hits (one-way ANI) and reciprocal best hits (two-way ANI) [10].
Taxonomic Evaluation
The 16S rRNA gene sequences of the closely related Rhodococcus species were downloaded from the National Center for Biotechnology Information (NCBI) and EzBioCloud databases [26]. The retrieved sequences were aligned in MEGA 6 using ClustalW (default parameters—gap opening = 15, and gap extension = 6.66). Phylogenetic tree was constructed using the neighbor-joining and Tamura-Nei methods in the software package MEGA 6 (default settings) [22]. The tree topologies were evaluated by bootstrap analysis with 100 replicates.
Sequence Accession Numbers
For dDDH, ANI and comparative genomic analysis, the nucleotide sequences of the closely related genome-sequenced Rhodococcus strains were retrieved from the NCBI GenBank database. The accession numbers (in parentheses) of the selected genome sequences are as follows: Rhodococcus erythropolis NBRC 15567 (BCRM00000000), Rhodococcus fascians NBRC 12155 = LMG 3623 strain (BCWW00000000), Rhodococcus yunnanensis NBRC 103083 (BCXH00000000), Rhodococcus globerulus NBRC 14531 (BCWX00000000), Rhodococcus jostii NBRC 16295 (BCWY00000000), and Rhodococcus opacus strain NRRL B-24011 (JOIM00000000).
The raw sequence reads generated using the Illumina MiSeq V3 platform with 2 × 300 bp paired-end sequencing have been deposited in the NCBI SRA database under the SRA accession PRJNA472681. Moreover, the Whole Genome Shotgun project of Rhodococcus sp. Eu-32 has also been deposited at DDBJ/ENA/GenBank under the accession QGNK00000000 (BioSample: SAMN09240609 and BioProject: PRJNA472681). The version described in this paper is version QGNK01000000.
Results and Discussion
Assembly and Annotation of the Genome Sequence
The total number of raw reads obtained was 3,127,208. The filtered high-quality reads (2,976,541) were de novo assembled using SPAdes (Version 3.10.1). A total of 83 contigs were obtained, corresponding to 5,612,575 bp, with an average size (N50) of 239,284 bp, G+C content of 65.1%, and genome coverage of 172X (Table 1). The contig with the largest size was 703,717 bp. The PGAP showed that annotated genome contains 5065 protein-encoding genes, total number of RNAs 62 (8 rRNA genes, 51 tRNA genes, 3 ncRNA genes) and 123 pseudo genes. A comparison of the genome assembly statistics between Rhodococcus sp. Eu-32 and other representative Rhodococcus species has been shown in Supplementary Table 1.
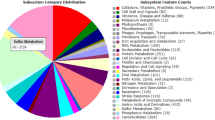
Automatic annotation performed using the RAST server, generated 5344 features potentially assigned to protein coding genes. The RAST annotation indicates that Rhodococcus jostii (score, 514) and Rhodococcus opacus (score, 495) are its closest neighbors (Supplementary Table 2). In RAST genome analysis, a total of 432 subsystems were identified, representing the metabolism of amino acids and derivatives (620 ORFs); carbohydrates (582 ORFs); cofactors/vitamins/prosthetic groups/pigments (383 ORFs) and proteins (271 ORFs) in large quantities. Eighty-eight open reading frames (ORFs) are involved in the metabolism of aromatic compounds, while 90 ORFs are involved in the metabolism of sulfur (Fig. 1).

Genes connected to subsystems and their distribution in different categories in Rhodococcus sp. Eu-32
Phylogenetic Inference and Genome Sequence Based Taxonomic Evaluation
The 16S rRNA gene sequence of Eu-32 was retrieved from the genome sequence of the isolate. The BLASTn search showed that it is most similar (~ 99% identity) to different species of the genus Rhodococcus (fascians, yunnanensis and cercidiphylli). The known Rhodococcus species appear to group into two main clades, as indicated by the phylogenetic analysis based on the 16S rRNA gene sequences (Fig. 2). The Eu-32, together with R. fascians, R. yunnanensis, and R. cercidiphylli comprise one clade, while R. opacus, R. jostii, and R. erythropolis make up the other. The Rhodococcus erythropolis IGTS8, a well known dibenzothiophene desulfurizing bacterium appeared to be phylogenetically distant from the Eu-32 (Fig. 2).

Phylogenetic tree of Rhodococcus species based on 16S rRNA gene sequences. The analysis was carried out by neighboring joining method using MEGA 6
Beyond the 16S rRNA gene sequence based phylogenetic analysis, to gain further insight into the evolutionary relatedness of the Rhodococcus sp. Eu-32, a combination of digital DNA–DNA hybridization (dDDH) and average nucleotide identity (ANI) was utilized. The proposed and generally accepted species boundary for ANI and dDDH values are 95–96% and 70%, respectively [6]. Moreover, for calculating dDDH and ANI, the genome sequence data of the type strains of those species are obtained showing ≥ 98.7% 16S rRNA gene similarity to the strain in question [6]. Among the Rhodococcus species showing ≥ 98.7% 16S rRNA gene similarity to the 16S rRNA gene sequence of Eu-32 (Supplementary Table 3), the genome sequence data of two type strains, i.e., Rhodococcus fascians NBRC 12155 = LMG 3623 strain (BCWW00000000) and Rhodococcus yunnanensis NBRC 103083 (BCXH00000000) were available in the NCBI database. Estimates for dDDH between Rhodococcus sp. Eu-32 (present study) and R. fascians NBRC 12155 and R. yunnanensis NBRC 103083 were 21.40% and 33.10%, respectively, well below the 70% species cutoff [6, 10, 14]. The ANI was calculated using both one-way ANI and two-way ANI between two genomes. The ANI value was 80% for Rhodococcus sp. Eu-32 and R. fascians NBRC 12155 and 80.90% for Rhodococcus sp. Eu-32 and R. yunnanensis NBRC 103083. The ANI values were also well below the 95–96% species cutoff [6, 10, 14].
The Eu-32 shares ~ 99% identity at the 16S rRNA gene sequence level while < 34% dDDH and < 81% ANI values with the most closely related known Rhodococcus species, suggesting that it represents a novel Rhodococcus species. We suggest that the isolate Rhodococcus sp. Eu-32 [3] may be regarded as new species of the genus Rhodococcus.
Sulfur Metabolism
About 0.5–1% of bacterial cell dry weight is comprised of sulfur [12]. Sulfur is needed by the microorganisms for their growth and biological activities [12, 21]. Generally, it is required for the biosynthesis of several essential compounds like amino acids (cysteine and methionine), vitamins (biotin, thiamin), and prosthetic groups. Depending upon the presence of enzymes and metabolic pathway, the microorganisms have the ability to acquire their required sulfur from various sources [21].
A common feature of the Rhodococcus species is their involvement in the sulfur metabolism. To fully determine this capability in isolate Eu-32, the draft genome sequence was analyzed for the presence of genes/pathways specific to the sulfur metabolism (Table 2). Moreover, to gain insight into the similarities and divergences of the sulfur metabolism genes present in Eu-32, we compared the Eu-32 genome sequence with the genome sequences available for the type strains of five Rhodococcus species, i.e., R. erythropolis NBRC 15567, R. fascians NBRC 12155, R. yunnanensis NBRC 103083, R. globerulus NBRC 14531, R. jostii NBRC 16295 and R. opacus NRRL B-24011. All genomes were uploaded to the RAST server for sequence and function based comparison. The Eu-32 genome annotation showed that a total of 90 genes are involved in the metabolism of sulfur (see Fig. 1). These genes were sub-categorized into inorganic sulfur assimilation (~ 27 genes), organic sulfur assimilation (~ 50 genes) and sulfur metabolism with no subcategory (~ 14 genes). The inorganic sulfur analysis showed that Eu-32 genome encoded a gene cluster for sulfite reductase, adenylylsulfate/phosphoadenylyl-sulfate reductase, and sulfate adenylyltransferase (Tables 2, 3). These enzymes are probably necessary for assimilatory reduction of sulfate to sulfite via adenylylsulfate (APS) and 3′-Phosphoadenylyl-sulfate (PAPS), and the subsequent reduction of sulfite to hydrogen sulfide [19]. Besides, the comparison to other sequenced genomes revealed the presence of genes involved in inorganic sulfur assimilation in all strains (Table 3).
The RAST subsystem feature counts indicated that the genome of Eu-32 contained at least 50 organic sulfur metabolizing genes. The genomic analysis showed that organic sulfur metabolism in Eu-32 genome encoded for genes related to tau and ssu operons (Table 3), necessary for the acquisition of sulfur during sulfur limitations, from organosulfonates and taurine respectively [7]. The tau operon encodes for taurine dioxygenase (TauD) that catalyzes the oxygenation of taurine to a hydroxytaurine intermediate, and an ABC-type transporter involved in the cellular uptake of taurine [7]. In RAST comparative genomic analysis, the alkanesulfonate utilization/assimilation genes were detected in all Rhodococcus strains; however, the taurine utilization genes were not detected in R. fascians and R. jostii (Table 3). In ssu operon, the FMN reductase (SsuE) provides reduced flavin to the monooxygenase enzyme (SsuD), and monooxygenase enzyme subsequently catalyzes the desulfonation of organosulfonated compounds via carbon–sulfur bond cleavage [7]. The SsuD and SsuE enzymes were present in all bacterial genomes mentioned in this study; however, the SsuE was not detected in R. fascians (Table 3).
The key genes of dsz operon (dszA, dszB, and dszC), encoding for different enzymes of the dibenzothiophene (DBT) desulfurization 4S pathway were not completely detected in all Rhodococcus genomes (Table 3). The dszC and dszA genes which encode for dibenzothiophene monooxygenase and dibenzothiophene sulfone monooxygenase respectively were present in the genome of Eu-32. However, the dszB gene encoding for desulfinase in DBT desulfurization 4S pathway was not traced (Table 3). The desulfinase belongs to the hydrolases class of the enzymes. It is documented that the Eu-32 has the ability to completely desulfurize the DBT [3] so, it can be suggested that a different hydrolase enzyme other than desulfinase may be involved in the metabolism of DBT in this bacterium.
The Pc gene encoding the ABC-type nitrate/sulfonate/bicarbonate transport systems, periplasmic components, required for the initial step of sulfur oxidation pathway [23] is present in Eu-32, R. yunnanensis, R. jostii, and R. globerulus (Table 3). Similarly, the genes SO (encodes for sulfonate monooxygenase), Prc (encodes for ABC-type nitrate/sulfonate/bicarbonate transport system, permease component), and Ac (encodes for ABC-type nitrate/sulfonate/bicarbonate transport system, ATPase component) were present in the genome of Eu-32, R. yunnanensis, and R. erythropolis. However, the SO was absent in R. globerulus and the Prc was absent in the R. opacus, while the Ac was absent from both R. fascians and R. jostii. Overall, the major routes of sulfur utilization in Rhodococcus are diverse and include inorganic sulfur assimilation, thioredoxin-disulfide reduction, taurine and alkanesulfonate utilization: the Rhodococcus sp. Eu-32 genome has components encoding for all of these pathways.
Further, the KEGG pathway analysis was performed using BlastKOALA (default settings) [11], which revealed that 28 pathways were associated with sulfur metabolism and 20 pathways were involved in the degradation of aromatic compounds. Furthermore, the genome of Eu-32 contains at least 75 oxygenase encoding genes (including monooxygenases and dioxygenases). The monooxygenase enzymes catalyze the carbon–sulfur bond cleavage of a broad range of organosulfonated compounds. The genes responsible for the biosynthesis of trehalose (a substrate for the synthesis of trehalose lipid) were also annotated. Trehalose lipid lowers the interfacial tension and increases pseudosolubility/bioavailability of hydrophobic compounds [9]. At least 11 ORFs were associated with the biosynthesis of trehalose which encode for different enzymes including trehalose-6-phosphate synthetase, trehalose phosphate phosphatase, and trehalohydrolase.
Conclusions
The Eu-32 shares ~ 99% 16S rRNA gene identity while < 34% dDDH and < 81% ANI values with the most closely related known type strains of Rhodococcus species, suggesting that it represents a novel Rhodococcus species. Interestingly, the occurrence of high number of sulfur metabolism genes showed that this bacterium has multiple strategies for the utilization of sulfur in diverse compounds. Comparative genome analysis suggests many commonalities in sulfur metabolism gene sets that may have evolved due to some evolutionary activities including ecological pressures. Moreover, it is equitable to suggest that the trehalose biosynthesis genes may contribute to efficient organosulfur compounds degradation by enhancing their uptake. The draft genome sequence of Rhodococcus sp. Eu-32 will facilitate the further understanding and development of potential applications of this organism.
References
Abin-Fuentes A, Mohamed MES, Wang DI, Prather KL (2013) Exploring the mechanism of biocatalyst inhibition in microbial desulfurization. Appl Environ Microbiol 79(24):7807–7817
Akhtar N, Ghauri MA, Akhtar K (2016) Exploring coal biodesulphurization potential of a novel organic sulphur metabolizing Rhodococcus spp. (Eu-32)—a case study. Geomicrobiol J 33(6):468–472
Akhtar N, Ghauri MA, Anwar MA, Akhtar K (2009) Analysis of the dibenzothiophene metabolic pathway in a newly isolated Rhodococcus spp. FEMS Microbiol Lett 301(1):95–102
Aziz RK, Bartels D, Best AA et al (2008) The RAST server: rapid annotation using subsystem technology. BMC Genomics 9:75
Boniek D, Figueiredo D, dos Santos AFB, de Resende Stoianoff MA (2015) Biodesulfurization: a mini review about the immediate search for the future technology. Clean Technol Environ Policy 17(1):29–37
Chun J, Oren A, Ventosa A et al (2018) Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int J Syst Evol Microbiol 68:461–466
Ellis HR (2011) Mechanism for sulfur acquisition by the alkanesulfonate monooxygenase system. Bioorg Chem 39:178–184
Ferreira P, Sousa SF, Fernandes PA, Ramos M (2017) Improving the catalytic power of the DszD enzyme for the biodesulfurization of crude oil and derivatives. Chem Eur J 23(68):17231–17241
Franzetti A, Gandolfi I, Bestetti G, Smyth TJP, Banat IM (2010) Production and applications of trehalose lipid biosurfactants. Eur J Lipid Sci Technol 112:617–627
Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM (2007) DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol 57:81–91
Kanehisa M, Sato Y, Morishima K (2016) BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol 428:726–731
Kertesz MA (1999) Riding the sulfur cycle-metabolism of sulfonates and sulfate esters in Gram-negative bacteria. FEMS Microbiol Rev 24:135–175
Maass D, Todescato D, Moritz D et al (2015) Desulfurization and denitrogenation of heavy gas oil by Rhodococcus erythropolis ATCC 4277. Bioprocess Biosyst Eng 38(8):1447–1453
Meier-Kolthoff JP, Auch AF, Klenk HP, Göker M (2013) Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform 14:60
Mishra S, Pradhan N, Panda S, Akcil A (2016) Biodegradation of dibenzothiophene and its application in the production of clean coal. Fuel Process Technol 152:325–342
Nuhu AA (2013) Bio-catalytic desulfurization of fossil fuels: a mini review. Rev Environ Sci Biotechnol 12(1):9–23
Porto B, Maass D, Oliveira JV et al (2018) Heavy gas oil biodesulfurization using a low-cost bacterial consortium. J Chem Technol Biotechnol 93(8):2359–2363
Rangra S, Kabra M, Gupta V, Srivastava P (2018) Improved conversion of dibenzothiophene into sulfone by surface display of dibenzothiophene monooxygenase (DszC) in recombinant Escherichia coli. J Biotechnol 287:59–67
Ross DE, Marshall CW, May HD, Norman RS (2016) Comparative genomic analysis of Sulfurospirillum cavolei MES reconstructed from the metagenome of an electro-synthetic microbiome. PLoS ONE 11(3):e0151214
Shavandi M, Soheili M, Zareian S, Akbari N, Khajeh K (2013) The gene cloning, overexpression, purification, and characterization of dibenzothiophene monooxygenase and desulfinase from Gordonia alkanivorans RIPI90A. J Pet Sci Technol 3(2):57–64
Soleimani M, Bassi A, Margaritis A (2007) Biodesulphurization of refractory organic sulfur compounds in fossil fuels. Biotechnol Adv 25(6):570–596
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Urich T, Gomes CM, Kletzin A, Frazao C (2006) X-ray structure of a self-compartmentalizing sulfur cycle metalloenzyme. Science 311:996–1000
Van der Geize R, Dijkhuizen L (2004) Harnessing the catabolic diversity of Rhodococci for environmental and biotechnological applications. Curr Opin Microbiol 7:255–261
Wang J, Butler RR, Wu F et al (2017) Enhancement of microbial biodesulfurization via genetic engineering and adaptive evolution. PLoS ONE 12(1):e0168833
Yoon SH, Ha SM, Kwon S, Lim J, Kim Y et al (2017) Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int J Syst Evol Microbiol 67:1613–1617
Funding
The authors received no specific grant from any funding agency.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Akhtar, N., Ghauri, M.A., Akhtar, K. et al. Comparative Analysis of Draft Genome Sequence of Rhodococcus sp. Eu-32 with Other Rhodococcus Species for Its Taxonomic Status and Sulfur Metabolism Potential. Curr Microbiol 76, 1207–1214 (2019). https://doi.org/10.1007/s00284-019-01737-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-019-01737-1