
Abstract
Auricularia polytricha is one of the most widely cultivated edible mushrooms in China. Many advances have been made to A. polytricha, but there is still no proteomic information of this species. Our current understanding was based upon the translated information of its transcriptome or other relative species. This study presented the proteomic information of fruiting-body proteins by shotgun liquid chromatography and tandem mass spectrometry (LC–MS/MS), which identified 15,508 peptides corresponding to 1850 high-confidence proteins. Of these, 1383 were annotated across the GO subcategories with 829 (44.81%) involved in biological process, 908 (49.08%) in molecular function, and 406 (21.95%) in cellular components. Among these high-confidence proteins, 132 proteins were annotated as carbohydrate-active enzymes, of which 51 were secreted enzymes. Moreover, a number of commercially important enzymes were detected, functioning as auxiliary activity (AA) family 5 glyoxal oxidase, AA5 galactose oxidase, glycoside hydrolase (GH) family 20 hexosaminidase, and GH47 alpha-mannosidase. To the best of our knowledge, this is the first study to characterize A. polytricha proteome, and also fills the gap of our knowledge on the under-developed mushroom species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A. polytricha (Mont.) Sacc is known as hairy wood ear in China [30], and the fourth most important cultivated black fungus used by humans worldwide [6]. Many published studies have examined the benefits of A. polytricha [6]. Its polysaccharide was shown to be potential agents against dementia, hypercholesterolemia, mutagenicity, tumor, oxidation, and DNA damage [3, 7, 38, 46, 51, 53]. As a dietary supplement, it contributed a beneficial effect on postprandial levels of blood sugar [41]. As an alternative biosorbent, its fruiting bodies or powdered mycelia could be employed to aid detoxification of effluents contaminated by Cr, Cd, Cu, Pb, Zn, or emulsified oil [13, 16, 29, 42, 52]. It has drawn great attention due to its various biological activities [6].
An enormous effort has been undertaken to develop methods for cultivation [1], molecular markers [48], pathogen identification [30], and transcriptomic exploration [54]. Despite these advances, no studies were made to characterize the proteome of A. polytricha. Our current understanding was dependent upon translated information from its transcriptome or other closely relative species. There is an urgent need to broaden our knowledge of its proteome.
To date, proteomic technique has become an effective tool to characterize protein profiles. Among current proteomic methods, the shotgun proteomic approach possesses the virtues of high efficiency, and time and labor savings. It is suitable for use as a high throughput technology for the identification of proteins samples in comparison with the two-dimensional electrophoresis (2-DE) combined with mass spectrometry (MS) [35]. The proteomic analyses have been performed on mushroom species such as Ganoderma lucidum [45], Armillaria mellea [8], Grifola frondosa [14], Sparassis crispa [15], Antrodia cinnamomea [21], Termitomyces heimii [31], Lignosus rhinocerotis [43], and Agaricus bisporus [26]. These may contribute to understanding the molecular mechanism of mycelial growth, fruiting-body formation, spore germination, and so on.
In this study, proteomic analysis of fruiting-body proteins (FBPs) in A. polytricha was performed using the shotgun approach based on liquid chromatography and tandem mass spectrometry (LC–MS/MS). As an initial foray into A. polytricha proteome, it should facilitate future analysis of the localization, structure, and function of interested proteins in some biological context.
Materials and Methods
A. Polytricha Strains and Culture Conditions
Strain huanger 10 of A. polytricha was preserved in Soil and Fertilizer Institute, Sichuan Academy of Agricultural Sciences, China. The mycelia of strain huanger 10 were grown on potato dextrose agar (PDA) medium for one week at room temperature [40], and cultivated on packs of growth medium consisting of 33% sawdust, 30% corncob, 20% rice bran, 10% cottonseed hulls, 2% corn flour, 4% lime, and 1% plaster powder in the dark at 25 °C. The fruiting bodies were cultivated as reported by Zhou [54], and sampled at stage of initial maturity from Shifang City in China. GN fluid medium contained glucose 10 g/L, (NH4)2SO4 6 g/L, NaCl 1 g/L, K2HPO4 1 g/L, MgSO4 0.5 g/L [24]. Cellulose medium was GN medium supplemented with 10 g/L microcrystalline cellulose, and used to investigate whether tested proteins could be induced by cellulose.
Protein Extraction
The fruiting-body specimen of strain huanger 10 (1.0 g) was finely ground. The powders were precipitated in a 10% (w/v) trichloroacetic acid/acetone solution containing 65 mM dithiothreitol (DTT) at −20 °C for 1 h. The solution was centrifuged at 10,000×g for 45 min. The supernatant was discarded, and the precipitate was vacuum-dried and solubilized in 1/10 volumes of SDT buffer (4% SDS, 100 mM DTT, and 150 mM Tris-HCl, pH 8.0). The suspensions were incubated in boiling water for 5 min followed by ultrasonication (80 w, 10 s ultrasonic at a time, every 15 s, and 10 times) [12] and incubation at 100 °C for 5 min. The crude extract was clarified by centrifugation at 13,000×g at 25 °C for 10 min [39]. The protein concentration was determined by the BCA Protein Assay Reagent (Promega, USA). The supernatants were stored at −80 °C before use.
In-Solution Digestion
Protein digestion was performed following the FASP procedure described by Wisniewski and colleagues [39]. 200 μg of proteins was mixed with 200 μL of UA buffer (8 M Urea, 150 mM Tris-HCl pH 8.0), loaded into the filter devices, and centrifuged at 14,000×g for 15 min. The concentrates were diluted in the device with 100 μL of UA buffer and centrifuged again. Then this substance was mixed with 100 μL of 50 mM IAA in UA buffer (600 rpm, 1 min) and incubated in darkness at room temperature for 30 min followed by centrifugation at 14,000×g for 10 min, after which the concentrate was diluted with 100 μL of UA buffer and concentrated, and this step repeated twice. Next, this concentrate was diluted with 100 μL of 25 mM NH4HCO3 and concentrated again. This step was repeated twice. The resulting concentrate was diluted to 40 µL of 25 mM NH4HCO3 containing 2 µg of trypsin (600 rpm, 1 min), and incubated at 37 °C for 16–18 h. Lastly, peptides were collected by centrifugation of the filter units at 14,000×g for 10 min, and their quantities were determined by OD280.
Off-Line High pH Reversed-Phase Fractionation
By high pH reversed-phase chromatography, the digested peptides were fractionated with AKTA Purifier system (GE Healthcare). When reconstituted in buffer A (NH3·H2O, pH 9.0), the peptide was loaded onto an XBridge Peptide BEH 4.6 mm × 150 mm C18 Column (300 Å, 5 µm, Waters) and eluted at a flow rate of 0.8 mL/min with a gradient of 0–4% buffer B (NH3·H2O in ACN, pH 9.0) during 0–15 min, 4–40% buffer B during 15–42 min, 40–90% buffer B during 42–47 min, 90% buffer B during 47–52 min, and buffer B was reset to 90% after 52 min. When monitored by absorbance at 214 nm [25], the elution was collected every 1 min. The collected fractions were dried by vacuum centrifugation.
Liquid Chromatography and Tandem Mass Spectrometry
Experiments were performed on a Q Exactive mass spectrometer coupled to Easy nLC (Thermo Fisher Scientific). Samples were loaded onto a the C18 reversed-phase column (75 μm i.d., 150 mm length) packed in-house with RP-C18 5 μm resin in mobile phase A (0.1% Formic acid in HPLC-grade water) and separated with a linear gradient of 4–100% mobile phase B (0.1% Formic acid in 84% acetonitrile) at a flow rate of 250 nL/min over 60 min [9, 22]. With a data-dependent top 10 method, MS data were acquired dynamically choosing the most abundant precursor ions from the survey scan (300–1800 m/z) for HCD fragmentation. The target value is determined based on predictive Automatic Gain Control (pAGC), with the following dynamic exclusion duration was 20 s. Survey scans were acquired at a resolution of 70,000 at m/z 200 and resolution for HCD spectra was set to 17,500 at m/z 200. Normalized collision energy was 27 eV and the underfill ratio was defined as 0.1%. The instrument was run with peptide recognition mode enabled.
Bioinformatic Analysis
The acquired MS/MS spectra were searched using MASCOT engine (Matrix Science, London, UK; version 2.2) [10] against combined protein database including the sequences of NCBI Auriculariales proteins (14 October 2015, 47,231 sequences) and putative proteins translated from A. polytricha transcriptome (33,573 sequences) (Supplemental dataset). For protein identification, the options were as follows: Peptide mass tolerance = 20 ppm, MS/MS tolerance = 0.1 Da, Enzyme = Trypsin, Missed cleavage = 2 [50], Fixed modification: Carbamidomethyl (C), Variable modification: Oxidation (M), False discovery rate (FDR) <0.01 at peptide and protein level.
With unique peptides ≥2, high-confidence proteins were chosen for further analysis [35]. Using the NCBI BLAST+ client software (ncbi-blast-2.2.28+-win32.exe), these sequences were locally searched against SwissProt database to find homologue sequences from which the functional annotation can be transferred to the studied sequences.
Using E-value less than 1e−3 for each query sequence, the top 10 blast hits were retrieved and loaded into Blast2GO1 (Version 2.7.2) for GO2 mapping and annotation. Annotation configurations with E-value filter of 1e-6, default gradual EC weights, Gene ontology (GO) weight of 5, and annotation cutoff of 55 were chosen. With more permissive parameters, un-annotated sequences were then re-annotated. The sequences without BLAST hits and un-annotated sequences were then selected to go through an InterProScan3 against EBI databases to retrieve functional annotations of protein motifs and merge the InterProScan GO terms to the annotation set [10]. Subsequently, the studied proteins were blasted against the Kyoto Encyclopedia of Genes and Genomes (KEGG) GENES (fungi, database updated: October 28, 2015) to retrieve their KOs and mapped to pathways in KEGG4.
The identified proteins were annotated using dbCAN for carbohydrate-active enzyme (CAZyme) families (based on CAZyDB 07/15/2016) [2, 44]. And secreted proteins were predicted by the SignalP 4.1 server [32, 33].
Quantitative Real-Time PCR (qRT-PCR) Analysis
In order to validate the proteins identified by this study, qRT-PCR assay was performed. Strain huanger 10 was cultured in cellulose medium and GN fluid medium (as control) [24]. GH37 trehalase (protein ID: comp21191_c0_seq1, intracellular proteins, Treh), GH3 beta-glucosidase (protein ID: comp15213_c0_seq2, intracellular proteins, beta-Glu(N)), and GH3 beta-glucosidase (protein ID: comp21080_c0_seq1, extracellular proteins, beta-Glu(Y)) were chosen as tested proteins, and glyceraldehyde phosphate dehydrogenase (Gapdh) gene as the endogenous control. The reverse transcriptions were performed using an HiScript 1st Strand cDNA Synthesis Kit from Vazyme Biotech Co., Ltd. Their reverse transcripts from each biological replicate were used as templates. Gene-specific primers based on selected proteins are listed in Table 1. PCR protocol: 95 °C 40 s, 1 cycle; 95 °C 20 s, 60 °C 20 s, plate read, 45 cycles. The linear amount of the target gene expression to the calibrator was calculated by \(2^{{ - \Delta \Delta C_{\text{T}} }}\) [49].
Results
Identification of FBPs of A. Polytricha
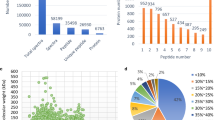
SDS-PAGE with silver staining indicated that proteins were successfully extracted from fruiting bodies (Fig. 1). BCA protein assay showed that its concentration was 2.3 μg/μL. The fruiting-body sample was digested with the FASP method and identified using the shotgun LC–MS/MS proteomic technique. According to the criteria of high-confidence, candidate proteins were chosen for the following GO and KEGG pathway analysis. The results demonstrated that 15,508 peptides were identified from fruiting-body samples corresponding to 1850 high-confidence proteins (Table S1), of which 221 proteins (27.8%) are putative or uncharacterized proteins. 1383 proteins were annotated across the GO subcategories with 829 (44.81%) involved in biological process, 908 (49.08%) in molecular function, and 406 (21.95%) in cellular components (Table 2). Among these GO classifications, the rich cellular component was cell (GO: 0005623) accounting for 24.5%. The rich molecular function was catalytic activity (GO: 0003824) accounting for 45.7%. The rich biological process was metabolic process (GO: 0008152) accounting for 50.6% (Fig. 2, Table S2). So many proteins were assigned to cell (GO: 0005623), catalytic activity (GO: 0003824), and metabolic process (GO: 0008152), reflecting that A. Polytricha FBPs continued exuberant metabolic activities resulting in cell growth.

Separation of fruiting-body proteins of Auricularia polytricha by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Lane M, protein marker (15–170 kDa); Lane A, the extracted fruiting-body proteins of A. polytricha

Gene ontology (GO) categories of the identified fruiting-body proteins of Auricularia polytricha. Y-axis (left) represents the protein number, and Y-axis (right) represents percentages of proteins identified. The proteins were categorized based on GO annotation, and the number of each category is displayed based on biological process, cellular components, and molecular functions (Color figure online)
KEGG Pathway Analysis
The sequences of 1850 high-confidence proteins were searched against the KEGG reference pathway database. The result showed that these proteins were assigned to 309 pathways according to KEGG pathway taxonomy (Table S3). In general, the pathways were classified into six categories: cellular processes (255 proteins, 22 subcategories), environmental information processing (273, 30), genetic information processing (370, 22), human diseases (620, 65), metabolism (816, 99), and organismal systems (466, 71), in which protein numbers of metabolism and human diseases were more than others (Fig. 3). Among these pathways, the top 3 proteins were involved in ribosome (70), RNA transport (51), and protein processing in endoplasmic reticulum (50). All the three pathways fell into the category of genetic information processing (Table S4), suggesting the significance of genetic information processing during initial maturity of A. polytricha.

Number of annotated proteins and their pathways, in which the top 3 proteins were involved in metabolism (816), human diseases (620), and organismal systems (466) (Color figure online)
Analysis of CAZymes
The CAZyme database annotation of A. polytricha identified a total of 132 CAZymes, which were grouped into carbohydrate-binding modules (CBMs, 10 proteins), auxiliary activities (AAs, 16), glycoside hydrolases (GHs, 77), carbohydrate esterases (CEs, 9), polysaccharide lyases (PLs, 3), and glycosyltransferases (GTs, 24) (Table S5). The top proteins were assigned to GH families, in which nine were classified to GH3 family, a preponderance of proteins in GH subfamilies. Among them, eight GH3 proteins were annotated as beta-glucosidase, and one as beta-xylosidase.
Lignin Oxidases and Hemicellulases
Degradation of cellulose is restricted by lignin and hemicelluloses, which are heterogeneous polysaccharides formed by various types of sugar units, mainly including mannans and xylans [18]. This study identified lignin oxidases such as AA2 peroxidase, AA5 galactose oxidase, AA5 glyoxal oxidase, and also detected hemicellulases including CE1 Acetyl xylan esterase, GH27 alpha-galactosidase, GH2 alpha-l-arabinofuranosidase, GH29 alpha-l-fucosidase, GH47 alpha-mannosidase, GH31 alpha-xylosidase, GH35 beta-galactosidase, GH3 beta-xylosidase, GH52 beta-xylosidase, CE1 carboxylesterase, and GH5 endo-1,4-beta-mannanase.
Cellulases
Nineteen cellulose-degrading proteins were detected in A. polytricha proteome. These proteins included GH3 beta-glucosidase, GH94 cellobiose phosphorylase, GH16 endo-1,3(4)-beta-glucanase, GH5 endo-beta-1,4-glucanase, GH17 glucan 1,3-beta-glucosidase, and GH74 oligoxyloglucan reducing end-specific cellobiohydrolase.
Saccharide-Degrading Proteins
By searching against the CAZyme database, saccharide-degrading proteins were annotated. These enzymes included GH33 sialidase, GH37 trehalase, GH38 alpha-mannosidase, GH63 alpha-glucosidase, GH76 alpha-1,6-mannanase, GH88 D-4,5-unsaturated beta-glucuronyl hydrolase, GH92 alpha-1,2-mannosidase, GH13/CBM48 alpha-amylase, GH109 alpha-N-acetylgalactosaminidase, GH15/CBM20 glucoamylase, GH31 alpha-1,4-glucosidase, GH72/CBM43 beta-1,3-glucanosyltransglycosylase, GH81 endo-beta-1,3-glucanase, PL14 alginate lyase, and PL8 chondroitin ABC lyase.
CAZymes Secreted by A. Polytricha
Among A. polytricha CAZymes, 51 secreted proteins were predicted by the SignalP 4.1 server [33, 34] accounting for 38.6% (51/132) (Table S5). Enzymes secreted by A. polytricha can be classified into AA, GH, CE, PL, CBM, and GT families. These secretomic proteins included two lignin oxidases, 12 hemicellulases, six cellulases, 14 saccharide-degrading proteins, three pectin-degrading proteins (GH43 endo-1,5-alpha-l-arabinanase, PL3 pectate lyase, GH79 beta-glucuronidase), and four chitin-degrading proteins (CE4 chitin deacetylase, GH18/CBM5 chitinase, GH20 beta-hexosaminidase). These secreted enzymes would contribute to the degradation of lignocellulosic substrate.
Proteins Involved in TCA Cycle
The citrate cycle (TCA cycle) is an important aerobic pathway involved in the conversion of carbohydrates, fats, and proteins to form energy [4]. The TCA cycle plays a central role in all living cells because energy production is essential [47]. In this study, 21 proteins are assigned to the citrate cycle (Table S4), including citrate synthase, ATP citrate (pro-S)-lyase, isocitrate dehydrogenase, 2-oxoglutarate dehydrogenase, pyruvate carboxylase, and so on (Table S3). So many proteins were involved in TCA cycle, suggesting the significance of TCA cycle in stage of initial maturity.
In summary, 1850 high-confidence proteins identified by this study confirmed the existence of many putative proteins translated from A. Polytricha transcriptome. This greatly enriched A. Polytricha proteomic database.
Validation of Selected Proteins by qRT-PCR
The qRT-PCR results showed that three tested genes were all detected both in GN fluid medium and cellulose medium, and were upregulated 1.10- to 1.59-fold when cultivated in cellulose medium compared with GN fluid medium. GH3 beta-glucosidase (protein ID: comp21080_c0_seq1) was annotated as secreted enzyme. Its expression level increased up to 1.59-fold over control. GH37 trehalase (protein ID: comp21191_c0_seq1) and GH3 beta-glucosidase (protein ID: comp15213_c0_seq2) were predicted as intracellular proteins, and had 1.10- and 1.30-fold increase (Fig. 4, Table S6). It shows that secreted GH3 beta-glucosidase is more prone to be induced by cellulose and high expression.

qPCR validation of selected proteins from fruiting bodies of Auricularia polytricha at the mRNA level. Samples were normalized with the reference gene Gapdh. Vertical lines represent ± S.D. Specimens of strain huanger 10 and huanger 10c harvested from GN fluid medium and cellulose medium, respectively. Treh(N) represents GH37 trehalase (protein ID: comp21191_c0_seq1, intracellular proteins). Beta-Glu(N) represents GH3 beta-glucosidase (protein ID: comp15213_c0_seq2, intracellular proteins). Beta-Glu(Y) represents GH3 beta-glucosidase (protein ID: comp21080_c0_seq1, extracellular proteins) (Color figure online)
Discussion
15508 peptides were identified in the tested sample corresponding to 1850 high-confidence proteins. With FDR <0.01 at peptide and two or more unique peptides, the high-confidence proteins were chosen for further analysis, which made the proteins identified more reliable [36]. And qRT-PCR assay provided further evidence to validate these proteomics data.
A. polytricha is saprophytic and cultivated on an extensive range of wood substrates [5] which includes cellulose, hemicellulose, and lignin (lignocellulosic complex) [19]. It is indicated that numerous lignocellulases are essential to A. polytricha. This study annotated 105 lignocellulases which are more than 61 ones identified in Ganoderma lucidum [45]. Proteins assigned to GHs (77) were the most numerous in A. polytricha. This result is in accordance with data described by Ospina-Giraldo et al., who found out that most homologs belonged to the GH families by scanning the genomes of Phytophthora infestans, P. sojae, and P. ramorum [28], and also in accordance with the data from Ganoderma lucidum proteome [45], reflecting GHs are a widespread group of enzymes. Additionally, so many identified lignocellulases may also enrich our knowledge of such proteins.
In comparison with A. polytricha transcriptome [54], GO terms of the rich cellular component, molecular function, and biological process are in agreement, except that the rich cellular component is also cell part in A. polytricha transcriptome. The general agreement on GO terms validates the two studies. In descending order of the top 3 proteins involved in metabolism, the KEGG pathways were carbohydrate metabolism (263 proteins), amino acid metabolism (157 proteins), and energy metabolism (90 proteins) in A. polytricha proteome (Table S7), while amino acid metabolism (265 ESTs), carbohydrate metabolism (248 ESTs), and energy metabolism (193 ESTs) in A. polytricha transcriptome. The different order of pathways may be resulted from the discrepancy between transcriptome and proteome, or resulted from different developmental stages, in which mature fruiting bodies were used as experimental materials in transcriptomic analysis [54], and initial maturity fruiting bodies were used in this study.
51 secreted CAZymes were predicted by the SignalP 4.1 server, such as GH3 beta-glucosidase, AA11 lytic polysaccharide monooxygenase, GH18 chitinase, GH3 beta-xylosidase, AA3 cellobiose dehydrogenase, GH37 trehalase, PL3 pectate lyase, GH81 endo-beta-1,3-glucanase, CE4 chitin deacetylase. These secreted proteins fall into AA, GH, CE, PL, CBM, and GT families. Numerous secreted CAZymes were detected, and may be able to provide an explanation of phenomenon that A. polytricha could be cultivated on an extensive range of wood substrates.
Enzymes of higher fungi have a potential for use in the industry [11]. Alginate lyases have been expected to become potential enzymes in the bioenergy generation from alginate [27]. Alpha-Glucosidases are involved in diverse physiological processes including carbohydrate assimilation [17]. AA5 glyoxal oxidase and AA5 galactose oxidase belong to the copper radical oxidases family [20], and are involved in lignin degradation. GH2 alpha-l-arabinofuranosidase and xylan degrading enzymes have been used in the food, feed, and pulp industries as well as conversion of hemicellulosic biomass to biofuels and chemicals [34]. GH2 alpha-l-arabinofuranosidase is more stable at higher temperatures, and has great potential to be used in industrial processes [34]. GH20 hexosaminidases are found in crustaceans, insects, and fungi, participating in the degradation of chitin [37]. GH88 D-4,5-unsaturated beta-glycosidases have been implicated in the degradation of unsaturated oligosaccharides. GH47 alpha-mannosidases are thought to participate in post-translational modification of secreted proteins [23]. Our assay detected the above functional proteins. This information may help shed some light on those who are prepared to apply biologically active proteins for industry.
Direct proteomic information of A. polytricha was unavailable previously, thus we got this information depending on the translated sequences of transcriptome of A. polytricha or other related species. This study firstly presented the proteomic information for researchers based on LC–MS/MS, and would give helpful information for the further development of such species.
References
Abd Razak DL, Abdullah N, Khir Johari NM, Sabaratnam V (2013) Comparative study of mycelia growth and sporophore yield of Auricularia polytricha (Mont.) Sacc on selected palm oil wastes as fruiting substrate. Appl Microbiol Biotechnol 97(7):3207–3213. doi:10.1007/s00253-012-4135-8
Bengtsson O, Arntzen MO, Mathiesen G, Skaugen M, Eijsink VG (2016) A novel proteomics sample preparation method for secretome analysis of Hypocrea jecorina growing on insoluble substrates. J Proteom 131:104–112. doi:10.1016/j.jprot.2015.10.017
Bennett L, Sheean P, Zabaras D, Head R (2013) Heat-stable components of wood ear mushroom, Auricularia polytricha (higher Basidiomycetes), inhibit in vitro activity of beta secretase (BACE1). Int J Med Mushrooms 15(3):233–249
Cetica P, Pintos L, Dalvit G, Beconi M (2003) Involvement of enzymes of amino acid metabolism and tricarboxylic acid cycle in bovine oocyte maturation in vitro. Reproduction 126(6):753–763
Chang ST, Quimio TH (1982) Tropical mushrooms, biological nature and cultivation methods, 1st edn. The Chinese University Press, Hong Kong
Chellappan DK, Ganasen S, Batumalai S, Candasamy M, Krishnappa P, Dua K, Chellian J, Gupta G (2016) The protective action of the aqueous extract of Auricularia polytricha in paracetamol induced hepatotoxicity in rats. Recent Pat Drug Deliv Formul 10(1):72–76
Chiu WC, Yang HH, Chiang SC, Chou YX, Yang HT (2014) Auricularia polytricha aqueous extract supplementation decreases hepatic lipid accumulation and improves antioxidative status in animal model of nonalcoholic fatty liver. Biomedicine (Taipei) 4:12. doi:10.7603/s40681-014-0012-3
Collins C, Keane TM, Turner DJ, O’Keeffe G, Fitzpatrick DA, Doyle S (2013) Genomic and proteomic dissection of the ubiquitous plant pathogen, Armillaria mellea: toward a new infection model system. J Proteome Res 12(6):2552–2570. doi:10.1021/pr301131t
Day J, Gietz RD, Rampitsch C (2015) Proteome changes induced by Pyrenophora tritici-repentis ToxA in both insensitive and sensitive wheat indicate senescence-like signaling. Proteome Sci 13:3
Dong M, Gu J, Zhang L, Chen P, Liu T, Deng J, Lu H, Han L, Zhao B (2014) Data in support of comparative proteomics analysis of superior and inferior spikelets in hybrid rice during grain filling and response of inferior spikelets to drought stress using isobaric tags for relative and absolute quantification. Data Brief 1:51–55. doi:10.1016/j.dib.2014.08.001
Erjavec J, Kos J, Ravnikar M, Dreo T, Sabotic J (2012) Proteins of higher fungi-from forest to application. Trends Biotechnol 30(5):259–273. doi:10.1016/j.tibtech.2012.01.004
Fan S, Meng Y, Song M, Pang C, Wei H, Liu J, Zhan X, Lan J, Feng C, Zhang S, Yu S (2014) Quantitative phosphoproteomics analysis of nitric oxide-responsive phosphoproteins in cotton leaf. PLoS One 9(4):e94261. doi:10.1371/journal.pone.0094261
Galli E, Di Mario F, Rapana P, Lorenzoni P, Angelini R (2003) Copper biosorption by Auricularia polytricha. Lett Appl Microbiol 37(2):133–137
Hohmann L, Sherwood C, Eastham A, Peterson A, Eng JK, Eddes JS, Shteynberg D, Martin DB (2009) Proteomic analyses using Grifola frondosa metalloendoprotease Lys-N. J Proteome Res 8(3):1415–1422. doi:10.1021/pr800774h
Horie K, Rakwal R, Hirano M, Shibato J, Nam HW, Kim YS, Kouzuma Y, Agrawal GK, Masuo Y, Yonekura M (2008) Proteomics of two cultivated mushrooms Sparassis crispa and Hericium erinaceum provides insight into their numerous functional protein components and diversity. J Proteome Res 7(5):1819–1835. doi:10.1021/pr070369o
Huang H, Cao L, Wan Y, Zhang R, Wang W (2012) Biosorption behavior and mechanism of heavy metals by the fruiting body of jelly fungus (Auricularia polytricha) from aqueous solutions. Appl Microbiol Biotechnol 96(3):829–840. doi:10.1007/s00253-011-3846-6
Jiang J, Kuo CL, Wu L, Franke C, Kallemeijn WW, Florea BI, van Meel E, van der Marel GA, Codee JD, Boot RG, Davies GJ, Overkleeft HS, Aerts JM (2016) Detection of active mammalian GH31 alpha-glucosidases in health and disease using in-class, broad-spectrum activity-based probes. ACS Cent Sci 2(5):351–358. doi:10.1021/acscentsci.6b00057
Katsimpouras C, Dimarogona M, Petropoulos P, Christakopoulos P, Topakas E (2016) A thermostable GH26 endo-beta-mannanase from Myceliophthora thermophila capable of enhancing lignocellulose degradation. Appl Microbiol Biotechnol 100(19):8385–8397. doi:10.1007/s00253-016-7609-2
Kuuskeri J, Hakkinen M, Laine P, Smolander OP, Tamene F, Miettinen S, Nousiainen P, Kemell M, Auvinen P, Lundell T (2016) Time-scale dynamics of proteome and transcriptome of the white-rot fungus Phlebia radiata: growth on spruce wood and decay effect on lignocellulose. Biotechnol Biofuels 9(1):192. doi:10.1186/s13068-016-0608-9
Levasseur A, Drula E, Lombard V, Coutinho PM, Henrissat B (2013) Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol Biofuels 6(1):41. doi:10.1186/1754-6834-6-41
Lin YL, Wen TN, Chang ST, Chu FH (2011) Proteomic analysis of differently cultured endemic medicinal mushroom Antrodia cinnamomea T.T. Chang et W.N. Chou from Taiwan. Int J Med Mushrooms 13(5):473–481
Liu JY, Chang MC, Meng JL, Feng CP, Liu YN (2016) iTRAQ-based comparative proteomics analysis of the fruiting dikaryon and the non-fruiting monokaryon of Flammulina velutipes. Curr Microbiol. doi:10.1007/s00284-016-1164-z
Mahajan S, Master ER (2010) Proteomic characterization of lignocellulose-degrading enzymes secreted by Phanerochaete carnosa grown on spruce and microcrystalline cellulose. Appl Microbiol Biotechnol 86(6):1903–1914. doi:10.1007/s00253-010-2516-4
Merhej J, Richard-Forget F, Barreau C (2011) The pH regulatory factor Pac1 regulates Tri gene expression and trichothecene production in Fusarium graminearum. Fungal Genet Biol 48(3):275–284. doi:10.1016/j.fgb.2010.11.008
Mori T, Guo M, Li X, Mori E (2002) Human malignant cell death by apoptosis-inducing nucleosides from the decidua derived CD57(+)HLA-DR(bright) natural suppressor cell line. J Reprod Immunol 53(1–2):289–303
O’Brien M, Grogan H, Kavanagh K (2014) Proteomic response of Trichoderma aggressivum f europaeum to Agaricus bisporus tissue and mushroom compost. Fungal Biol 118(9–10):785–791. doi:10.1016/j.funbio.2014.06.004
Ogura K, Yamasaki M, Yamada T, Mikami B, Hashimoto W, Murata K (2009) Crystal structure of family 14 polysaccharide lyase with pH-dependent modes of action. J Biol Chem 284(51):35572–35579. doi:10.1074/jbc.M109.068056
Ospina-Giraldo MD, Griffith JG, Laird EW, Mingora C (2010) The CAZyome of Phytophthora spp.: a comprehensive analysis of the gene complement coding for carbohydrate-active enzymes in species of the genus Phytophthora. BMC Genom 11:525. doi:10.1186/1471-2164-11-525
Pan R, Cao L, Huang H, Zhang R, Mo Y (2010) Biosorption of Cd, Cu, Pb, and Zn from aqueous solutions by the fruiting bodies of jelly fungi (Tremella fuciformis and Auricularia polytricha). Appl Microbiol Biotechnol 88(4):997–1005. doi:10.1007/s00253-010-2821-y
Peng W, He X, Wang Y, Zhang Y, Ye X, Jia D, Guo Y, Gan B, Zheng C, Yang Z, Sun Q (2014) A new species of Scytalidium causing slippery scar on cultivated Auricularia polytricha in China. FEMS Microbiol Lett 359(1):72–80. doi:10.1111/1574-6968.12564
Rahmad N, Al-Obaidi JR, Nor Rashid NM, Zean NB, Mohd Yusoff MH, Shaharuddin NS, Mohd Jamil NA, Mohd Saleh N (2014) Comparative proteomic analysis of different developmental stages of the edible mushroom Termitomyces heimii. Biol Res 47:30. doi:10.1186/0717-6287-47-30
Rustiguel CB, Rosa JC, Jorge JA, de Oliveira AH, Guimaraes LH (2016) Secretome analysis of Metarhizium anisopliae under submerged conditions using bombyx mori chrysalis to induce expression of virulence-related proteins. Curr Microbiol 72(2):220–227. doi:10.1007/s00284-015-0943-2
Sermsathanaswadi J, Baramee S, Tachaapaikoon C, Pason P, Ratanakhanokchai K, Kosugi A (2017) The family 22 carbohydrate-binding module of bifunctional xylanase/beta-glucanase Xyn10E from Paenibacillus curdlanolyticus B-6 has an important role in lignocellulose degradation. Enzyme Microb Technol 96:75–84. doi:10.1016/j.enzmictec.2016.09.015
Shi H, Zhang Y, Xu B, Tu M, Wang F (2014) Characterization of a novel GH2 family alpha-l-arabinofuranosidase from hyperthermophilic bacterium Thermotoga thermarum. Biotechnol Lett 36(6):1321–1328. doi:10.1007/s10529-014-1493-6
Sun D, Zhang H, Guo D, Sun A, Wang H (2013) Shotgun proteomic analysis of plasma from dairy cattle suffering from footrot: characterization of potential disease-associated factors. PLoS One 8(2):e55973. doi:10.1371/journal.pone.0055973
Tu CJ, Dai J, Li SJ, Sheng QH, Deng WJ, Xia QC, Zeng R (2005) High-sensitivity analysis of human plasma proteome by immobilized isoelectric focusing fractionation coupled to mass spectrometry identification. J Proteome Res 4(4):1265–1273. doi:10.1021/pr0497529
Val-Cid C, Biarnes X, Faijes M, Planas A (2015) Structural-functional analysis reveals a specific domain organization in family GH20 hexosaminidases. PLoS One 10(5):e0128075. doi:10.1371/journal.pone.0128075
Wang W, Zhang G, Zou J (2013) The interaction of polysaccharide from Auricularia polytricha with quantum dots and the protection of plasmid DNA from damage. Appl Biochem Biotechnol 169(8):2263–2272. doi:10.1007/s12010-013-0135-0
Wisniewski JR, Zougman A, Nagaraj N, Mann M (2009) Universal sample preparation method for proteome analysis. Nat Methods 6(5):359–362. doi:10.1038/nmeth.1322
Wu J, Ji Z, Wang N, Chi F, Xu C, Zhou Z, Zhang J (2016) Identification of conidiogenesis-associated genes in Colletotrichum gloeosporioides by Agrobacterium tumefaciens-mediated transformation. Curr Microbiol 73(6):802–810. doi:10.1007/s00284-016-1131-8
Wu NJ, Chiou FJ, Weng YM, Yu ZR, Wang BJ (2014) In vitro hypoglycemic effects of hot water extract from Auricularia polytricha (wood ear mushroom). Int J Food Sci Nutr 65(4):502–506. doi:10.3109/09637486.2014.886183
Yang X, Guo M, Wu Y, Wu Q, Zhang R (2014) Removal of emulsified oil from water by fruiting bodies of macro-fungus (Auricularia polytricha). PLoS One 9(4):e95162. doi:10.1371/journal.pone.0095162
Yap HY, Fung SY, Ng ST, Tan CS, Tan NH (2015) Shotgun proteomic analysis of tiger milk mushroom (Lignosus rhinocerotis) and the isolation of a cytotoxic fungal serine protease from its sclerotium. J Ethnopharmacol 174:437–451. doi:10.1016/j.jep.2015.08.042
Yin Y, Mao X, Yang J, Chen X, Mao F, Xu Y (2012) dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res 40(Web Server issue):W445–451. doi:10.1093/nar/gks479
Yu GJ, Yin YL, Yu WH, Liu W, Jin YX, Shrestha A, Yang Q, Ye XD, Sun H (2015) Proteome exploration to provide a resource for the investigation of Ganoderma lucidum. PLoS One 10(3):e0119439
Yu J, Sun R, Zhao Z, Wang Y (2014) Auricularia polytricha polysaccharides induce cell cycle arrest and apoptosis in human lung cancer A549 cells. Int J Biol Macromol 68:67–71. doi:10.1016/j.ijbiomac.2014.04.018
Yu L, Wang SF, Zhai QZ, Yao YQ, Jiang F, Lu YX (2015) Exceptional material requirement for reproduction in mouse oocytes. Genet Mol Res 14(4):14356–14365. doi:10.4238/2015.November.13.21
Yu M, Ma B, Luo X, Zheng L, Xu X, Yang Z (2008) Molecular diversity of Auricularia polytricha revealed by inter-simple sequence repeat and sequence-related amplified polymorphism markers. Curr Microbiol 56(3):240–245. doi:10.1007/s00284-007-9067-7
Zhang J, Li C, Tang X, Lu Q, Sa R, Zhang H (2015) Proteome changes in the small intestinal mucosa of broilers (Gallus gallus) induced by high concentrations of atmospheric ammonia. Proteome Sci 13:9
Zhang Z, Wang S, Huang J, Liu L, Lu M, Li M, Sui Y, Xu L, Yan R, Song X, Li X (2015) Proteomic analysis of Eimeria acervulina sporozoite proteins interaction with duodenal epithelial cells by shotgun LC-MS/MS. Mol Biochem Parasitol 202(2):29–33. doi:10.1016/j.molbiopara.2015.09.006
Zhao S, Rong C, Liu Y, Xu F, Wang S, Duan C, Chen J, Wu X (2015) Extraction of a soluble polysaccharide from Auricularia polytricha and evaluation of its anti-hypercholesterolemic effect in rats. Carbohydr Polym 122:39–45. doi:10.1016/j.carbpol.2014.12.041
Zheng S, Huang H, Zhang R, Cao L (2014) Removal of Cr(VI) from aqueous solutions by fruiting bodies of the jelly fungus (Auricularia polytricha). Appl Microbiol Biotechnol 98(20):8729–8736. doi:10.1007/s00253-014-5862-9
Zhou J, Chen Y, Xin M, Luo Q, Gu J, Zhao M, Xu X, Lu X, Song G (2013) Structure analysis and antimutagenic activity of a novel salt-soluble polysaccharide from Auricularia polytricha. J Sci Food Agric 93(13):3225–3230. doi:10.1002/jsfa.6161
Zhou Y, Chen L, Fan X, Bian Y (2014) De novo assembly of Auricularia polytricha transcriptome using Illumina sequencing for gene discovery and SSR marker identification. PLoS One 9(3):e91740. doi:10.1371/journal.pone.0091740
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
No conflict of interest declared.
Additional information
Supplemental dataset
The combined protein database including NCBI Auriculariales sequences (14 October 2015, 47231 sequences) and putative proteins (translated from 33573 open reading frames (ORFs) of Auricularia polytricha transcriptome).
Electronic supplementary material
Below is the link to the electronic supplementary material.
284_2017_1268_MOESM2_ESM.xlsx
Supplementary material 2 (XLSX 35 kb). Second level GO annotation in three categories of cellular component, molecular function and biological process
284_2017_1268_MOESM3_ESM.xlsx
Supplementary material 3 (XLSX 71 kb). Proteins from fruiting bodies of Auricularia polytricha and their KEGG pathways, in which proteins were assigned to 309 KEGG pathways
Rights and permissions
About this article

Cite this article
Jia, D., Wang, B., Li, X. et al. Proteomic Analysis Revealed the Fruiting-Body Protein Profile of Auricularia polytricha . Curr Microbiol 74, 943–951 (2017). https://doi.org/10.1007/s00284-017-1268-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-017-1268-0