
Abstract
Immunoglobulin A (IgA) nephropathy (IgAN) is the most common primary glomerulonephritis worldwide. Up to 40% of IgAN patients develop end-stage kidney disease after 15–20 years. Despite the poor prognosis associated with this multifactorial disease, no clear treatment strategy has been identified, primarily due to the lack of understanding of its pathogenesis. Clinical observations indicate that aberrant IgAN immune systems, rather than intrinsic renal abnormalities, may be involved in its pathogenesis. Moreover, nephritogenic IgA and its related immune complexes are considered to be produced not only in the mucosa, but also in systemic immune sites, such as the bone marrow; however, there are numerous challenges to understanding this dynamic and complex immune axis in humans. Thus, several investigators have used experimental animal models. Although there are inter-strain differences in IgA molecules and immune responses between humans and rodents, animal models remain a powerful tool for investigating IgAN’s pathogenesis, and the subsequent development of effective treatments. Here, we introduced some classical models of IgAN with or without genetic manipulation and recent translational approaches with some promising models. This includes humanized mouse models expressing human IgA1 and human IgA Fc receptor (CD89) that develops spontaneously the disease. Pre-clinical studies targeting IgA1 are discussed. Together, animal models are very useful tools to study pathophysiology and to validate new therapeutic approaches for IgAN.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Classical IgAN animal models without genetic manipulation
IgA nephropathy (IgAN) is defined as “primary chronic mesangial proliferative nephritis with predominant deposition of IgA, mainly in the glomerular mesangial region.” In other words, diagnosis of IgAN requires proof of glomerular IgA deposition on a kidney biopsy. However, IgA deposition in the mesangial region is observed in 5–10% of autopsied cases without a history of renal disease, and about 20% of donated kidneys without abnormal urinary findings exhibit similar IgA deposits [1,2,3]. Thus, there is no guarantee that glomerular IgA deposition is always pathological, and we cannot deny the possibility that deposition occurs on a regular basis. Many single-gene manipulation models of IgA deposition have been created before, but IgA deposition and IgAN must be clearly distinguished from one another when being considered. In this and other respects, reductionist methodologies are limited in their scope; the complex system that underlies the pathology of this disease must be dynamically understood and analyzed.
In 1979, Rifai et al. first reported an animal model of IgAN [4]. Injection models were constructed using murine anti-dinitrophenole (DNP) and DNP-conjugated bovine serum albumin (DNP-BSA) to characterize nephritogenic IgA and IgA-containing immune complexes (IC). They confirmed that anti-DNP/DNP-BSA IC can be synthesized in the circulation, and is subsequently localized in the glomerular mesangial region, for which it has a high affinity. Next, they established that persistent mesangial IC deposition requires repeated administration of anti-DNP and DNP-BSA or maintained serum IC levels. They also confirmed that the size of the IgA-IC complex, which involves coupling of the IC with the polymeric form of IgA, is a critical factor for mesangial deposition [5, 6]. Using animal models immunized with a bacterial-derived polysaccharide or chemically modified dextran, Isaacs et al. further elucidated glomerular injury induced by the mesangial deposition of IgA-IC formed in the circulation [7, 8]. These studies emphasized the importance of continuous IgA-IC formation, especially with polymeric IgA, as a driving force for the nephritogenicity of mesangial IgA deposition.
Macroscopic hematuria following an upper respiratory tract infection is the hallmark of IgAN; the pathogenic role of mucosal cells and immune dysregulation on mucosal immunity has been discussed in the pathogenesis of IgAN [9]. Emancipator et al. first demonstrated that circulating IgA-IC generated by repeated mucosal antigen immunization in healthy Balb/c mice induced glomerular injury with mesangial deposits of J-chain-associated polymeric IgA [10]. IgAN patients, especially in Europe, have reported inflammatory bowel disease–associated complications, such as Crohn’s disease and celiac disease, at relatively high frequencies. Indeed, some reports have demonstrated an increase in IgA anti-gliadin, a lectin present in gluten, and altered mucosal processing of gliadin in IgAN patients [11,12,13,14]. Coppo et al. demonstrated renal injury with glomerular deposition of food-specific IgA in murine models that were orally immunized with ovalbumin and gliadin [15]. Furthermore, Pestka et al. reported that food-borne microbial contaminants, such as deoxynivalenol, developed murine IgAN with mesangial IgA deposition, and increased serum levels of IgA/IgA-IC [16,17,18,19]. Impaired oral tolerance has also been discussed as an immune mechanism underlying the pathogenesis of IgAN. Gesualdo et al. described that murine IgAN induced by oral bovine gamma globulin (BGG) administration together with cyclophosphamide and/or estradiol (to block oral tolerance) led to similar mesangial IgA deposits as compared to mice without these drugs but with an aggravated IgAN phenotype due systemic responses to BGG associated with IgG, IgM, and C3 glomerular co-depositions plus hematuria [20]. It is known that mucosal IgA transcytosis and trafficking by polymeric immunoglobulin receptor in mucosal cells are strikingly augmented by estradiol [21, 22].
Models with IgAN caused by specific microbial pathogens have also been reported. As the glomerulonephritis associated with methicillinresistant Staphylococcus aureus (MRSA) infection resembles that observed in human IgAN [23, 24], Koyama et al. generated a murine IgAN model through subcutaneous S. aureus immunization [25]. Based on the clinical findings of high-frequency glomerular deposition in the outer membrane of Haemophilus parainfluenzae (HPI) antigens in IgAN patients [26], Suzuki et al. also demonstrated the development of murine IgAN in models orally immunized with HPI [27]. IgAN-like murine models induced by viral infection with parvo and sendai viruses have also been reported [28,29,30].
Experimental findings in these animal models support the idea that mucosal polymeric IgA and related ICs immunized by mucosal exposure to food and microbial antigens can induce IgAN-like injury in the glomerulus.
Development of a mouse model of spontaneous IgA nephropathy for translational research
-
1)
Generation of spontaneous IgAN model (grouped ddY mice)
To more comprehensively assess the pathogenesis of IgAN, our group focused on developing a spontaneous mouse model. In 1985, Imai et al. reported that some ddY mice widely used in pharmacological and toxicity studies developed chronic mesangial proliferative nephritis with glomerular IgA [31]. However, because these mice were a non-inbred line, significant variance in onset was present, and the line could not serve as a good model. Muso et al. established an inbred line by mating ddY mice with high IgA levels (HIGA mice) [32]. However, while these HIGA mice had high serum IgA levels, the same problem of onset variance remained. To test the possibilities of this model, we carried out time-course renal biopsies of more than 300 ddY mice, and analyzed their onset patterns [33]. We found that one-third of mice exhibited mesangial proliferative nephritis with glomerular IgA and complement C3, and proteinuria by 20 weeks (early onset group). Another third of mice exhibited the same nephropathy by 40 weeks (late onset group), and the last 1/3rd did not exhibit nephropathy at all over the course of their lives (quiescent group) (Fig. 1) [33]. Upon conducting genomic association studies of onset-related genes between the early onset and quiescent groups, we found a candidate gene similar to one discovered on genetic analysis of human familiar IgAN [33]. In response to this, we interbred early onset group mice alone, and successfully established a 100% onset model of spontaneous IgAN (grouped ddY mice: gddY) [34]. In human IgAN, IgA1 with abnormal hinge O-glycosylation, particularly galactose-deficient IgA1 (GdIgA1), is known to be involved in the pathology, and in gddY mice, abnormal glycosylation of the IgA molecule was observed [34]. In this manner, we established a model exceedingly similar to human IgAN phenotypically, genetically, and pathologically, and have used this model to advance translational research on the pathology of this condition. At the same time, various discoveries have further highlighted the validity of this model.
-
2)
Testing the mucosa-bone marrow axis hypothesis

Development of spontaneous IgAN model for translational research
Mucosal infection or the presence of mucosal immune abnormalities has been assumed to play a role in the pathology of the condition. For many years, a mechanism of nephropathy progression involving an immune complex of mucosal antigens and antigen-specific IgA has been investigated with some experimental models above mentioned, and multiple antigens reported, but none of these findings was successfully reproduced on follow-up studies. On the other hand, case reports describing IgAN patients who suffered from leukemia experiencing remission of both nephropathy and leukemia after bone marrow transplantation, and other reports of abnormal bone marrow biopsy findings in IgAN patients have suggested the involvement of the bone marrow in the pathophysiology of the disease. These findings led several groups to propose the “mucosa-bone marrow axis” hypothesis in the first half of the 1980s, which proposed that the main cause of the disease involved mucosae and bone marrow [9, 35, 36]. Unfortunately, lack of a suitable model prevented this hypothesis from being tested. Thus, our group has worked to use the mouse model we established to test the “mucosa-bone marrow axis” hypothesis [9, 37].
To verify the involvement of exogenous antigens, we raised model mice in specific pathogen-free (SPF) and conventional environments. We found that when raising these mice in a conventional environment, only their serum IgA levels increased significantly as they aged, suggesting that exogenous pathogens deeply affect IgA production [38]. At the same time, upon conducting the aforementioned onset-related genome association analysis between the early and late onset groups, MyD88, a pattern recognition receptor and an adapter molecule for Toll-like receptors (TLRs) which plays a central role in innate immunity, was highlighted as a candidate gene for progression [38]. Thus, we analyzed the correlation between the expression of each TLR associated with MyD88 and disease progression/severity and serum IgA levels. We found a strong correlation with TLR9, which recognizes the unmethylated CpG DNA of bacteria and viruses [38]. For further verification, when CpG DNA was nasally and peritoneally sensitized, serum IgA level clearly increased, and urinary protein and nephropathy were exacerbated only after nasal administration [38, 39]. This fact indicates that at the least, aggravation of the innate immune system especially in nasopharyngeal mucosa is deeply involved in the mechanism by which the IgAN model worsens and progresses. Recent study revealed that, although gddY mice in germ-free condition show no glomerular lesions without glomerular IgA, only nasal challenge of CpG DNA, but not fecal transplantation, reconstitutes the murine IgAN, suggesting nasopharyngeal-associated lymphoid tissue (NALT) is more involved in the pathogenesis of this disease than gut-associated lymphoid tissue (GALT) [40].
To further confirm the involvement of TLR9/MyD99 pathways in human IgAN, subjects were divided into two groups according to renal tissue severity, and genetic analysis was performed. A single nucleotide polymorphism (SNP: rs352140) in TLR9 itself, and not in MyD88, showed a very high correlation with disease severity [38]. Since a similar mechanism was hypothesized in humans, we proceeded to verify these findings using the tonsils as the mucosal tissue. It is known that tonsillectomy causes a reduction in IgA levels in IgAN leading to good prognosis [41, 42]. We confirmed that the larger the decrease, the earlier the therapeutic effect of tonsillectomy pulse, and that TLR9 expression in the tonsils was significantly higher in the group of patients whose serum GdIgA1 level had decreased only on tonsillectomy [41, 43]. Thus, it was shown for the first time that abnormal IgA production in humans depends on the degree of innate immune activation in the mucosa and does not necessarily require a specific antigen. This suggests that something as mundane as the common cold can serve as an exacerbating factor for IgAN.
In order to clarify the pathological role of bone marrow in this disease, bone marrow transplantation studies were also carried out. IgAN was reconstituted in normal mice through the transplantation of gddY bone marrow, and conversely, nephropathy in gddY mice disappeared after transplantation of bone marrow from normal mice [44,45,46]. It was therefore shown that the cells responsible for abnormal IgA production in murine IgAN are present not only in the mucosa but also in the bone marrow, as is the case in humans. Furthermore, the discovery that nephropathy could also be reconstituted by adoptive transfer of gddY spleen cells into nude mice showed that responsible cells may be disseminated to systemic lymphoid tissues [46]. It was also found that the severity of these reconstituted IgAN correlated with serum IgA-IgG IC levels rather than serum IgA levels [44].
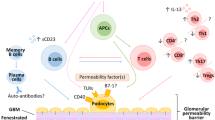
In this manner, abnormalities in the “mucosa-bone marrow axis” were slowly uncovered using the spontaneous IgAN model; however, the cells responsible for and the genetic mechanism of abnormal IgA production remained unknown. In the 2010s, genome-wide association studies (GWAS) for IgAN [47,48,49] revealed some candidate genes involved in mucosal immunity including TNFSF13, which codes for the TNF superfamily ligand member (TNFSF) cytokine A proliferation–inducing ligand (APRIL). It is known that APRIL is produced from dendritic cells and neutrophils and is deeply involved in B cell maturation/differentiation, induction, and IgA class switching. We first tested it in the gddY model, using a neutralizing antibody against APRIL. It was confirmed that antibody administration improved proteinuria and glomerular lesions, and decreased glomerular IgA deposition [50, 51]. Next, we examined human IgAN. It was found that the expression of APRIL and its receptors TACI and BCMA was dramatically increased in patients with IgAN when compared with expression levels in the tonsils of patients with chronic tonsillitis. [52]. Surprisingly, the germinal-center B cells of the tonsils strongly expressed APRIL (APRIL-α), and its positivity rate and degree correlated with decrease in blood GdIgA1 levels after tonsillectomy [52]. We also found that APRIL expression in B cells themselves was induced by continuous stimulation of CpG DNA, a ligand for TLR9, even in tonsil B cells derived from patients with chronic tonsillitis [52]. In consistent with this finding, APRIL and TLR9 expressions in palatine tonsil were highly correlated in IgAN patients [52]. Recent study with gddY model and human samples showed that APRIL and IL-6 synergistically, as well as independently, enhance the synthesis of TLR9-mediated GdIgA1 [53].
The details of the mechanism by which IgG/IgM form the GdIgA1 IC in IgAN are still unclear. Our group and others have reported the mechanism by which amino acid substitutions occur in the variable domain of IgG, giving it affinity for the abnormal glycan portions of IgA in human IgAN patients. We also reported that IC formation is an effector molecule that correlates with renal prognosis, and that IgG is useful as a biomarker [54,55,56,57,58]. In recent years, the soluble scavenger receptor, Apoptosis Inhibitor of Macrophage (AIM), has been found to play an important role in various renal diseases such as acute kidney injury [59]. By editing the genome of gddY mice with CRISPR-Cas9, we have revealed that abnormally glycosylated IgA develops complement activity and subsequent nephritis only after forming IC with IgM/IgG via AIM, and mainly after glomerular deposition [60]. AIM co-stains glomerular IgA deposits in all human IgAN cases, suggesting a similar mechanism is involved in both human and murine IgAN [60]. Most cases die of renal failure in cat are caused by the abnormal binding and dissociation of AIM with IgM [61], and therapeutic applications of this finding are currently being developed. It is possible that therapeutic applications that modulate AIM binding and dissociation will emerge for human IgAN.
Transgenic and humanized mouse models
Since the description of IgAN by professor Jean Berger in the 1960s [62], several groups around the world attempted to reproduce the disease in animals. However, difficulties to obtain a good and reproducible model were faced due to the profound interspecies differences with rodents concerning structure and function of IgA and its main receptor, the FcαR (CD89). While humans display two IgA isotypes, mice and rats have only one which differs from human IgA1 by displaying a shorter hinge region without O-glycan contents. In contrast, rabbits have 13 isotypes of IgA [29]. Another major difference is that mice do not express CD89 whereas rats do express a CD89 homolog [63].
As CD89 is not expressed in mice, two groups developed in the late 1990s transgenic mice expressing CD89 gene either under its own regulatory sequences or under control of the human CD11b promoter. While the first strategy resulted in CD89 expression almost exclusively on neutrophils and no signs of IgAN [64], the mice obtained in the second strategy showed monocyte/macrophage CD89 expression which was associated to late (40 weeks) spontaneous mouse IgA renal deposits with mesangial expansion and macrophage infiltration but without major renal dysfunction except for hematuria [65]. Mouse IgA complexed to soluble (s) CD89 was detected in the serum of these animals that were able to passively transfer the disease to rag2 deficient animals [65]. The late IgAN development of CD89Tg mice was recently explained by the low-affinity interaction between mouse IgA and human CD89 as detected in surface plasmon resonance (Biacore) experiments [66]. Nevertheless, this human CD89 Tg mouse model allowed the demonstration of the role of transmembrane cellular CD89 FcRγ adaptor in disease progression [67]. Glomerular macrophage infiltration is observed following triggering of CD89 by immune complexes resulting in secretion of chemoattractants such as TNF-α and MCP-1. Tg mice expressing a mutated, signaling-incompetent, human FcαR(R209L) that cannot associate with FcRγ developed mesangial IgA deposits but without macrophage infiltration and no proteinuria [67]. These results allow to propose that CD89 triggering by large IgA-immune complexes may induce macrophage recruitment into injured kidneys during IgAN development.
As mouse IgA is not O-glycosylated, a fully humanized mouse model was next generated by backcrossing human IgA1 knock-in (KI) mice [68] with human CD89 transgenic mice, named the α1KICD89Tg mice [66]. These mice express chimeric IgA1 (human α1 chain with mouse light chains) and the human CD89 receptor. They develop an IgAN-like disease much earlier than the CD89 Tg mice due to the stronger affinity of human IgA1 to its CD89 receptor as compared to mouse IgA. As early as 12 weeks of age, these mice present significant hematuria, albuminuria, altered renal function (recent unpublished data revealed an increase in cystatin C plasmatic level) and exhibit serum immune complexes containing human IgA1 and sCD89. Furthermore, histological examination shows mesangial IgA1 and C3 deposits, glomerular macrophage infiltration, and mesangial cell proliferation. Further studies revealed an increased expression of mouse transferrin receptor 1 (CD71) in the α1KICD89Tg mice. CD71 is the main IgA1 receptor at the surface of mesangial cells of IgAN patients and in enterocytes of celiac disease patients [69,70,71]. In α1KICD89Tg mouse model, transglutaminase-2 and CD71 overexpression are involved in IgA1-complex deposition in the mesangium [66]. In contrast, mice expressing IgA1 alone (α1KI mice) display mainly endothelial cell IgA1 deposits associated with some mesangial deposits but do not have functional alteration and fail to display complement deposition in their mesangium [66, 72]. Recently, using α1KI mice it was shown that low-affinity innate-like IgA formed, in the absence of normal antigen-driven maturation, was involved in IgA glomerular deposition [73]. However, mesangial expansion, macrophage infiltration, proteinuria, and hematuria were only observed in α1KICD89Tg mice [66] suggesting a potential pathogenic role of CD89 expression.
The α1KICD89Tg mouse model was used as a pre-clinical model for proof-of-concept of several new therapeutic approaches in IgAN. The first study addressed the role of food antigens notably gluten in the disease development. Indeed, since the 1990s, the detection of anti-gliadin antibodies and an uncontrolled trial with gluten-free diet leading to decreased proteinuria suggested a role for gluten in the physiopathology of IgAN [74]. The α1KICD89Tg mice were recently subjected to gluten-free diet for three generations [75]. This treatment led to a marked decrease in mesangial IgA1 deposits and hematuria, as well as reduced mesangial CD71 and transglutaminase 2 expression. Mice on a gluten-free diet lacked IgA1-sCD89 complexes in serum and kidney eluates. Disease reappeared following refeeding with gluten diet. Gluten diet exacerbated intestinal IgA1 secretion, inflammation, and villous atrophy, and increased serum IgA1 anti-gliadin antibodies, which correlated with proteinuria. A direct mechanism was proposed involving induction of IgA anti-gliadin complexes and a newly discovered interaction of gliadin with sCD89. Interestingly, early treatment of humanized mice from only one generation with a gluten-free diet prevented mesangial IgA1 deposits and hematuria suggesting that a new controlled trial with gluten-free diet may need to be organized for IgAN patients with preserved renal function.
The humanized α1KICD89Tg mouse model was also used to test specific therapies targeting the IgA1 hinge region. α1KICD89Tg mice were treated with recombinant IgA1 protease, a bacterial-derived protein which cleaves human IgA1 in the hinge region [76]. These IgA1 protease-treated animals showed Fcα1 fragments in both serum and urine, decreased levels of IgA1-sCD89 complexes, and marked abolishment of mesangial IgA1 deposits and hematuria. Glomerular deposit partners (sCD89, transferrin receptor, transglutaminase 2, and C3) were also decreased after treatment, as well as CD11b(+) cells, and fibronectin. Anti-IgA1 protease antibodies were found during the treatment but did not alter the protease activity. This pre-clinical study indicated that IgA1 protease could be a new treatment for IgAN patients specially those with rapid deterioration of the renal function. A phase 1b study targeting IgG autoantibodies by an IgG protease (Imlifidase) has been successful and associated with an overall renal survival of 67% at 6 months for patients with Goodpasture syndrome [77] which seems to validate the usage of bacterial recombinant proteases cleaving immunoglobulins in clinics, hoping that IgA proteases will be available in a near future for initial single-shot treatment of severe IgAN patients with rapid deterioration of renal function aiming to clear IgA1 deposits before other therapeutic approaches.
The α1KICD9Tg mice were also essential to study the role of microbiota in IgAN. Gut mucosal involvement has been indicated by genome-wide association studies performed on IgAN patients revealing new loci associated with risk of inflammatory bowel disease or maintenance of intestinal barrier and MALT response to pathogens [47] plus beneficial treatment with corticosteroids targeting the gut mucosa protecting renal function in patients with IgAN [78]. Moreover, analysis of microbiota from IgAN patients indicated that some traits of gut microbiota significantly varied between healthy control subjects, non-progressor and progressor IgAN patients, and that urinary and fecal metabolome consistently differed between groups [79]. As germ-free housing impairs IgA1 production in animals [72], the strategy used was an intervention targeting the gut microbiota by broad antibiotics in 8- or 12-week-old animals, the age when IgA1 reached expected serum levels and IgA1 deposits were clearly detected [80]. Antibiotic treatment efficiently depleted the fecal microbiota and markedly prevented human IgA1 mesangial deposition, glomerular inflammation, and the development of proteinuria. Interestingly, antibiotic treatment did not affect serum levels of human IgA1 and mouse IgG but significantly decreased circulating hIgA1-mIgG autoantibody complexes. Moreover, treatment with broad-spectrum antibiotics reverted established disease (12- to 16-week-old animals). Finally, fecal bacterial load correlated with pathophysiological features of IgAN such as proteinuria and hIgA1-mIgG complexes. To demonstrate the role of microbiota in disease progression, fecal material transfer (FMT) experiments with stools from IgAN patients (progressor versus non-progressor) were performed in antibiotic-pretreated α1KICD9Tg animals. FMT from progressors clearly induced an IgAN phenotype which was associated with BAFF levels [81]. Recently, the α1KICD9Tg mice were also challenged by rifaximin (NORMIX®) treatment, a non-absorbable oral antibiotic, that induces positive modulation of the gut microbiota, favoring the growth of bacteria beneficial to the host [82]. Rifaximin treatment decreased the hIgA1 glomerular deposition, CD11b+ cell infiltration, and urinary protein-to-creatinine ratio, serum levels of hIgA1–sCD89 and mIgG–hIgA1 complexes. Moreover, rifaximin treatment decreased significantly B-cell activating factor (BAFF)-, poly immunoglobulin receptor (pIgR)-, and TNF-mRNA expression. This study suggests rifaximin as a possible approach in the treatment of the disease.
Other transgenic models have been developed, as the uteroglobin antisense-transgenic mice. Uteroglobin is an anti-inflammatory protein secreted by mucosal epithelia, with high affinity for fibronectin, interfering with IgA-fibronectin spontaneous interaction. This model is characterized by microhematuria, albuminuria, and glomerular IgA, C3, and collagen deposits [83]. However, uteroglobin does not seem to be implicated in IgAN pathogenesis in humans [84].
T lymphocytes are commonly found in mononuclear cell infiltrates from biopsies of IgAN patients. To explain how T cells contribute to the pathogenesis of IgAN, a mechanism has been suggested following data obtained with LIGHT transgenic mice [85]. These animals spontaneously develop features similar to those of human IgAN associated with T cell–mediated intestinal inflammation. LIGHT overexpression–induced intestinal inflammation was dependent on its ligand interaction, the lymphotoxin beta receptor (LTbetaR). LIGHT-LTbetaR interaction not only induces IgA synthesis in the intestinal sub mucosa but also increases IgA transcytosis into the gut lumen, causing a major increase in polymeric mouse IgA levels in the serum. These data suggest that dysregulation of LIGHT-LTbetaR pathway may lead to intestinal inflammation and hyper IgA synthesis in mice and that it may become a putative pathogenic factor for IgAN.
More recently, a particular interest in B-cell implication in IgAN led to the generation of two transgenic models for human Bcl-2 (B-cell lymphoma 2) and BAFF. Bcl-2 is usually overexpressed in B cells in autoimmune states, inducing a defect in the regulation of B-cell apoptosis and enhancing the systemic IgA-immune response [86]. BAFF is also a protein of interest in IgAN, involved in antibody class switching and B cell survival. Overexpression of human BAFF has been reported in IgAN patients [87]. Interestingly, BAFF-Tg mice have mesangial deposits of IgA along with high circulating levels of polymeric IgA that are aberrantly glycosylated. It was quite striking that commensal flora was essential for the elevated levels of serum IgA, and that commensal bacteria-reactive IgA antibodies were found in the blood. These data illustrate how excess B cell activation signaling alters the microbiota and are among the first indications of connections between mucosal environments and renal pathology.
Finally, the galactosylation status of IgA1 is essential in the pathophysiological process of IgAN. Murine IgA has N-glycans but not O-glycans, contrary to human IgA1. Nishie et al. found that mice deficient for β-1,4-galactosyltransferase (β4GalT-I), the enzyme responsible for transferring galactose to the terminal N-acetylglucosamine in a beta-1,4 linkage, spontaneously developed IgAN-like lesions with IgA deposition and expanded mesangial matrix. It was associated with high serum IgA levels, increased polymeric IgA forms, albuminuria, hematuria, mesangial matrix expansion, glomerulosclerosis, mesangial IgA, and mesangio-parietal C3 deposits [88]. The authors propose that carbohydrates of serum IgA are involved in the development of IgAN, whether the carbohydrates are O-glycans or N-glycans.
Conclusion
One should state that animal models will never completely replicate human diseases as diseases are often heterogeneous and the human system is quite different from that of rodents notably concerning the molecular feature of human IgA and mucosal immune system. Nevertheless, insights from experimental models with or without genetic manipulation indeed contribute to elucidation of multiple aspects of the pathogenesis of IgAN and facilitate development of IgAN-specific drugs. Transgenic mouse models brought particular answers to specific questions in the pathophysiology of the disease whereas humanized mouse models for IgA1 have been extremely helpful in pre-clinical stages to test new drugs or new diets in attempts to propose new clinical trials for treatment of IgA nephropathy. Translational approaches with appropriate experimental models continue to be critical for future therapies for IgAN.
References
Waldherr R, Rambausek M, Duncker WD, Ritz E (1989) Frequency of mesangial IgA deposits in a non-selected autopsy series. Nephrol Dial Transplant 4:943–946
Suganuma T (1994) Glomerular IgA deposits in an autopsy study. Nihon Jinzo Gakkai Shi 36:813–822
Suzuki K, Honda K, Tanabe K, Toma H, Nihei H, Yamaguchi Y (2003) Incidence of latent mesangial IgA deposition in renal allograft donors in Japan. Kidney Int 63:2286–2294
Rifai A, Small PA, Teague PO, Ayoub EM (1979) Experimental IgA nephropathy. J Exp Med 150:1161–1173
Rifai A, Millard K (1985) Glomerular deposition of immune complexes prepared with monomeric or polymeric IgA. Clin Exp Immunol 60:363–368
Chen A, Wong SS, Rifai A (1988) Glomerular immune deposits in experimental IgA nephropathy: a continuum of circulating and in situ formed immune complexes. Am J Pathol 130:216–222
Isaacs K, Miller F, Lane B (1981) Experimental model for IgA nephropathy. Clin Immunol Immunopathol 20:419–426
Isaacs K, Miller F (1982) Role of antigen size and charge in immune complex glomerulonephritis. I. Active induction of disease with dextran and its derivatives. Lab Investig 47:198–205
Suzuki Y, Tomino Y (2007) The mucosa-bone-marrow axis in IgA nephropathy. Contrib Nephrol 157:70–79
Emancipator SN, Gallo GR, Lamm ME (1983) Experimental IgA nephropathy induced by oral immunization. J Exp Med 157:572–582
Coppo R, Basolo B, Rollino C, Roccatello D, Martina G, Amore A, Bongiorno G, Piccoli G (1986) Mediterranean diet and primary IgA nephropathy. Clin Nephrol 26:72–82
Kumar V, Sieniawska M, Beutner EH, Chorzelski TP (1988) Are immunological markers of gluten-sensitive enteropathy detectable in IgA nephropathy? Lancet 2:1307
Fornasieri A, Sinico RA, Maldifassi P, Bernasconi P, Vegni M, D’Amico G (1987) IgA-antigliadin antibodies in IgA mesangial nephropathy (Berger's disease). Br Med J 295:78–80
Rostoker G, Laurent J, André C, Cholin S, Lagrue G (1988) High levels of IgA antigliadin antibodies in patients who have IgA mesangial glomerulonephritis but not coeliac disease. Lancet 356-357
Coppo R, Mazzucco G, Martina G, Roccatello D, Amore A, Novara R, Bargoni A, Piccoli G, Sena LM (1989) Gluten-induced experimental IgA glomerulopathy. Lab Investig 60:499–506
Yan D, Rumbeiha WK, Pestka JJ (1998) Experimental murine IgA nephropathy following passive administration of vomitoxin-induced IgA monoclonal antibodies. Food Chem Toxicol 36:1095–1106
Pestka JJ (2003) Deoxynivalenol-induced IgA production and IgA nephropathy-aberrant mucosal immune response with systemic repercussions. Toxicol Lett 140-141:287–295
Shi Y, Pestka JJ (2006) Attenuation of mycotoxin-induced IgA nephropathy by eicosapentaenoic acid in the mouse: dose response and relation to IL-6 expression. J Nutr Biochem 17:697–706
Hinoshita F, Suzuki Y, Yokoyama K, Hara S, Yamada A, Ogura Y, Hashimoto H, Tomura S, Marumo F, Ueno Y (1997) Experimental IgA nephropathy induced by a low-dose environmental mycotoxin, nivalenol. Nephron 75:469–478
Gesualdo L, Lamm ME, Emancipator SN (1990) Defective oral tolerance promotes nephritogenesis in experimental IgA nephropathy induced by oral immunization. J Immunol 145:3684–3691
Monteiro RC, Van De Winkel JGJ (2003) IgA Fc receptors. Annu Rev Immunol 21:177–204
Diebel ME, Diebel LN, Liberati DM (2011) Gender dimorphism in the gut: mucosal protection by estrogen stimulation of IgA transcytosis. J Trauma 71:474–479
Koyama A, Kobayashi M, Yamaguchi N, Yamagata K, Takano K, Nakajima M, Irie F, Goto M, Igarashi M, Iitsuka T, Aoki Y, Sakurai H, Sakurayama N, Fukao K (1995) Glomerulonephritis associated with MRSA infection: a possible role of bacterial superantigen. Kidney Int 47:207–216
Satoskar AA, Nadasdy G, Plaza JA, Sedmak D, Shidham G, Hebert L (2006) Nadasdy T:Staphylococcus infection-associated glomerulonephritis mimicking IgA nephropathy. Clin J Am Soc Nephrol 1:1179–1186
Sharmin S, Shimizu Y, Hagiwara M, Hirayama K, Koyama A (2004) Staphylococcus aureus antigens induce IgA-type glomerulonephritis in Balb/c mice. J Nephrol 17:504–511
Suzuki S, Nakatomi Y, Sato H, Tsukada H, Arakawa M (1994) Haemophilus parainfluenzae antigen and antibody in renal biopsy samples and serum of patients with IgA nephropathy. Lancet 343:12–16
Yamamoto C, Suzuki S, Kimura H, Yoshida H, Gejyo F (2002) Experimental nephropathy induced by Haemophilus parainfluenzae antigens. Nephron 90:320–327
Porter DD, Larsen AE, Porter HG (1980) Aleutian disease of mink. Adv Immunol 29:261–286
Portis JL, Coe JE (1979) Deposition of IgA in renal glomeruli of mink affected with Aleutian disease. Am J Pathol 96:227–236
Jessen RH, Emancipator SN, Jacobs GH (1992) Experimental IgA-IgG nephropathy induced by a viral respiratory pathogen. Dependence on antigen form and immune status. Lab Investig 67:379–386
Imai H, Nakamoto Y, Asakura K, Miki K, Yasuda T, Miura A (1985) Spontaneous glomerular IgA deposition in ddY mice: an animal model of IgA nephritis. Kidney Int 27:756–761
Muso E, Yoshida H, Takeuchi E, Yashiro M, Matsushima H, Oyama A, Suyama K, Kawamura T, Kamata T, Miyawaki S, Izui S, Sasayama S (1996) Enhanced production of glomerular extracellular matrix in a new mouse strain of high serum IgA ddY mice. Kidney Int 50:1946–1957
Suzuki H, Suzuki Y, Yamanaka T, Hirose S, Nishimura H, Toei J, Horikoshi S, Tomino Y (2005) Genome-wide association study in IgA nephropathy model identifies susceptibility locus on murine chromosome 10, in a region syntenic to human IGAN1 on chromosome 6q22-23. J Am Soc Nephrol 16:1289–1299
Okazaki K, Suzuki Y, Otsuji M, Suzuki H, Kihara M, Kajiyama T, Hashimoto A, Nishimura H, Brown R, Hall S, Novak J, Izui S, Hirose S, Tomino Y (2012) Establishment of a novel ddY mouse model with early onset IgA nephropathy. J Am Soc Nephrol 23:1364–1374
van den Wall Bake AW, Daha MR, van Es LA (1989) Immunopathogenetic aspects of IgA nephropathy. Nephrologie 10:141–145
de Fijter JW, Eijgenraam JW, Braam CA, Holmgren J, Daha MR, van Es LA, van den Wall Bake AW (1996) Deficient IgA1 immune response to nasal cholera toxin subunit B in primary IgA nephropathy. Kidney Int 50:952–961
Suzuki Y, Suzuki H, Sato D, Kajiyama T, Okazaki K, Hashimoto A, Kihara M, Yamaji K, Satake K, Nakata J, Aizawa M, Novak J, Tomino Y (2011) Reevaluation of the mucosa-bone marrow axis in IgA nephropathy with animal models. Adv Otorhinolaryngol 72:64–67
Suzuki H, Suzuki Y, Narita I, Aizawa M, Kihara M, Yamanaka T, Kanou T, Tsukaguchi H, Novak J, Horikoshi S, Tomino Y (2008) Toll-like receptor 9 affects severity of IgA nephropathy. J Am Soc Nephrol 19:2384–2395
Kajiyama T, Suzuki Y, Kihara M, Suzuki H, Horikoshi S, Tomino Y (2011) Different pathological roles of toll-like receptor 9 on mucosal B cells and dendritic cells in murine IgA nephropathy. Clin Dev Immunol 2011:819646
Kano T, Suzuki H, Makita Y, Fukao Y, Suzuki Y (in press) Nasal-associated lymphoid tissue is the major induction site for nephritogenic IgA in murine IgA nephropathy. Kidney Int 2021
Sato D, Suzuki Y, Kano T, Suzuki H, Matsuoka J, Yokoi H, Horikoshi S, Ikeda K, Tomino Y (2012) Tonsillar TLR9 expression and efficacy of tonsillectomy with steroid pulse therapy in IgA nephropathy patients. Nephrol Dial Transplant 27:1090–1097
Hirano K, Matsuzaki K, Yasuda T, Nishikawa M, Yasuda Y, Koike K, Maruyama S, Yokoo T, Matsuo S, Kawamura T, Suzuki Y (2019) Association between tonsillectomy and outcomes in patients with immunoglobulin A nephropathy. JAMA Netw Open 2:e194772
Suzuki Y, Nakata J, Suzuki H, Sato D, Kano T, Yanagawa H, Matsuzaki K, Horikoshi S, Novak J, Tomino Y (2014) Changes in nephritogenic serum galactose-deficient IgA1 in IgA nephropathy following tonsillectomy and steroid therapy. PLoS One 9:e89707
Suzuki H, Suzuki Y, Aizawa M, Yamanaka T, Kihara M, Pang H, Horikoshi S, Tomino Y (2007) Th1 polarization in murine IgA nephropathy directed by bone marrow-derived cells. Kidney Int 72:319–327
Aizawa M, Suzuki Y, Suzuki H, Pang H, Kihara M, Nakata J, Yamaji K, Horikoshi S, Tomino Y (2014) Uncoupling of glomerular IgA deposition and disease progression in alymphoplasia mice with IgA nephropathy. PLoS One 9:e95365
Nakata J, Suzuki Y, Suzuki H, Sato D, Kano T, Horikoshi S, Novak J, Tomino Y (2013) Experimental evidence of cell dissemination playing a role in pathogenesis of IgA nephropathy in multiple lymphoid organs. Nephrol Dial Transplant 28:320–326
Gharavi AG, Kiryluk K, Choi M, Li Y, Hou P, Xie J, Sanna-Cherchi S, Men CJ, Julian BA, Wyatt RJ, Novak J, He JC, Wang H, Lv J, Zhu L, Wang W, Wang Z, Yasuno K, Gunel M, Mane S, Umlauf S, Tikhonova I, Beerman I, Savoldi S, Magistroni R, Ghiggeri GM, Bodria M, Lugani F, Ravani P, Ponticelli C, Allegri L, Boscutti G, Frasca G, Amore A, Peruzzi L, Coppo R, Izzi C, Viola BF, Prati E, Salvadori M, Mignani R, Gesualdo L, Bertinetto F, Mesiano P, Amoroso A, Scolari F, Chen N, Zhang H, Lifton RP (2011) Genome-wide association study identifies dusceptibility loci for IgA nephropathy. Nat Genet 43:321–327
Kiryluk K, Li Y, Scolari F, Sanna-Cherchi S, Choi M, Verbitsky M, Fasel D, Lata S, Prakash S, Shapiro S, Fischman C, Snyder HJ, Appel G, Izzi C, Viola BF, Dallera N, Del Vecchio L, Barlassina C, Salvi E, Bertinetto FE, Amoroso A, Savoldi S, Rocchietti M, Amore A, Peruzzi L, Coppo R, Salvadori M, Ravani P, Magistroni R, Ghiggeri GM, Caridi G, Bodria M, Lugani F, Allegri L, Delsante M, Maiorana M, Magnano A, Frasca G, Boer E, Boscutti G, Ponticelli C, Mignani R, Marcantoni C, Di Landro D, Santoro D, Pani A, Polci R, Feriozzi S, Chicca S, Galliani M, Gigante M, Gesualdo L, Zamboli P, Battaglia GG, Garozzo M, Maixnerová D, Tesar V, Eitner F, Rauen T, Floege J, Kovacs T, Nagy J, Mucha K, Pączek L, Zaniew M, Mizerska-Wasiak M, Roszkowska-Blaim M, Pawlaczyk K, Gale D, Barratt J, Thibaudin L, Berthoux F, Canaud G, Boland A, Metzger M, Panzer U, Suzuki H, Goto S, Narita I, Caliskan Y, Xie J, Hou P, Chen N, Zhang H, Wyatt RJ, Novak J, Julian BA, Feehally J, Stengel B, Cusi D, Lifton RP, Gharavi AG (2014) Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat Genet 46:1187–1196
Yu XQ, Li M, Zhang H, Low HQ, Wei X, Wang JQ, Sun LD, Sim KS, Li Y, Foo JN, Wang W, Li ZJ, Yin XY, Tang XQ, Fan L, Chen J, Li RS, Wan JX, Liu ZS, Lou TQ, Zhu L, Huang XJ, Zhang XJ, Liu ZH, Liu JJ (2011) A genome-wide association study in Han Chinese identifies multiple susceptibility loci for IgA nephropathy. Nat Genet 44:178–182
Kim YG, Alvarez M, Suzuki H, Hirose S, Izui S, Tomino Y, Bertrand H, Suzuki Y (2015) Pathogenic role of a proliferation-inducing ligand (APRIL) in murine IgA nephropathy. PLoS One 10:e0137044
Myette JR, Kano T, Suzuki H, Sloan SE, Szretter KJ, Ramakrishnan B, Adari H, Deotale KD, Engler F, Shriver Z, Wollacott AM, Suzuki Y, Pereira BJG (2019) A Proliferation Inducing Ligand (APRIL) targeted antibody is a safe and effective treatment of murine IgA nephropathy. Kidney Int 96:104–116
Muto M, Manfroi B, Suzuki H, Joh K, Nagai M, Wakui S, Righini C, Maiguma M, Izui S, Tomino Y, Huard B, Suzuki Y (2017) Toll-like receptor 9 stimulation induces aberrant expression of a proliferation-inducing ligand by tonsillar germinal center B cell in IgA nephropathy. J Am Soc Nephrol 28:1227–1238
Makita Y, Suzuki H, Kano T, Takahata A, Julian BA, Novak J, Suzuki Y (2020) TLR9 activation induces aberrant IgA glycosylation via APRIL-and IL-6-mediated pathways in IgA nephropathy. Kidney Int 97:340–349
Suzuki H, Fan R, Zhang Z, Brown R, Hall S, Julian BA, Chatham WW, Suzuki Y, Wyatt RJ, Moldoveanu Z, Lee JY, Robinson J, Tomana M, Tomino Y, Mestecky J, Novak J (2009) Aberrantly glycosylated IgA1 in IgA nephropathy patients is recognized by IgG antibodies with restricted heterogeneity. J Clin Invest 119:1668–1677
Suzuki Y, Matsuzaki K, Suzuki H, Okazaki K, Yanagawa H, Ieiri N, Sato M, Sato T, Taguma Y, Matsuoka J, Horikoshi S, Novak J, Hotta O, Tomino Y (2014) Serum levels of galactose deficient IgA1 and related immune complex are associated with disease activity of IgA nephropathy. Clin Exp Nephrol 18:770–777
Yanagawa H, Suzuki H, Suzuki Y, Kiryluk K, Gharavi AG, Matsuoka K, Makita Y, Julian BA, Novak J, Tomino Y (2014) A panel of serum biomarkers differentiates IgA nephropathy from other renal diseases. PLoS One 9:e98081
Placzek WJ, Yanagawa H, Makita Y, Renfrow MB, Julian BA, Rizk DV, Suzuki Y, Novak J, Suzuki H (2018) Serum galactose-deficient-IgA1 and IgG autoantibodies correlate in patients with IgA nephropathy. PLoS One 13:e0190967
Suzuki Y, Suzuki H, Makita Y, Takahata A, Takahashi K, Muto M, Sasaki Y, Kelimu A, Matsuzaki K, Yanagawa H, Okazaki K, Tomino Y (2014) Diagnosis and activity assessment of IgA nephropathy: current perspectives on non-invasive testing with aberrantly glycosylated IgA-related biomarkers. Int J Nephrol Renov Dis 7:409–414
Arai S, Kitada K, Yamazaki T, Takai R, Zhang X, Tsugawa Y, Sugisawa R, Matsumoto A, Mori M, Yoshihara Y, Doi K, Maehara N, Kusunoki S, Takahata A, Noiri E, Suzuki Y, Yahagi N, Nishiyama A, Gunaratnam L, Takano T, Miyazaki T (2016) AIM/CD5L enhances intraluminal debris clearance and ameliorates acute kidney injury. Nat Med 22:183–193
Takahata A, Arai S, Hiramoto E, Kitada K, Kato R, Makita Y, Suzuki H, Nakata J, Araki K, Miyazaki T, Suzuki Y (2020) Crucial role of AIM/CD5L in the development of glomerular inflammation in IgA nephropathy. J Am Soc Nephrol 31:2013–2024
Sugisawa R, Hiramoto E, Matsuoka S, Iwai S, Takai R, Yamazaki T, Mori N, Okada Y, Takeda N, Yamamura K, Arai T, Arai S, Miyazaki T (2016) Impact of feline AIM on the susceptibility of cats to renal disease. Sci Rep 6:35251
Berger J, Hinglais N (1968) Intercapillary deposits of IgA-IgG. J Urol Nephrol (Paris) 74(9):694–695
Maruoka T, Nagata T, Kasahara M (2004) Identification of the rat IgA Fc receptor encoded in the leukocyte receptor complex. Immunogenetics. 55(10):712–716
van Egmond M, van Vuuren AJ, Morton HC, van Spriel AB, Shen L, Hofhuis FM, Saito T, Mayadas TN, Verbeek JS, van de Winkel JG (1999) Human immunoglobulin A receptor (FcalphaRI, CD89) function in transgenic mice requires both FcR gamma chain and CR3 (CD11b/CD18). Blood. 93(12):4387–4394
Launay P, Grossetete B, Arcos-Fajardo M, Gaudin E, Torres SP, Beaudoin L, Patey-Mariaud de Serre N, Lehuen A, Monteiro RC (2000) Fca receptor (CD89) mediates the development of Immunoglobulin A (IgA) nephropathy (Berger’s Disease): evidence for pathogenic soluble receptor-IgA complexes in patients and CD89 transgenic mice. J Exp Med 191:1999–2009
Berthelot L, Papista C, Maciel TT, Biarnes-Pelicot M, Tissandie E, Wang PHM, Tamouza H, Jamin A, Bex-Coudrat J, Gestin A, Boumediene A, Arcos-Fajardo M, England P, Pillebout E, Walker F, Daugas E, Vrtosvnik F, Flamant M, Benhamou M, Cogné M, Moura IC, Monteiro RC (2012) Transglutaminase is essential for IgA nephropathy development acting through IgA receptors. J Exp Med 209(4):793–806
Kanamaru Y, Arcos-Fajardo M, Moura IC, Tsuge T, Cohen H, Essig M, Vrtovsnik F, Loirat C, Peuchmaur M, Beaudoin L, Launay P, Lehuen A, Blank U, Monteiro RC (2007) Fc alpha receptor I activation induces leukocyte recruitment and promotes aggravation of glomerulonephritis through the FcR gamma adaptor. Eur J Immunol 37:1116–1128
Duchez S, Amin R, Cogné N, Delpy L, Sirac C, Pascal V, Corthésy B, Cogné M (2010) Premature replacement of mu with alpha immunoglobulin chains impairs lymphopoiesis and mucosal homing but promotes plasma cell maturation. Proc Natl Acad Sci U S A 107(7):3064–3069. https://doi.org/10.1073/pnas.0912393107 Epub 2010 Jan 28
Moura IC, Centelles MN, Arcos-Fajardo M, Malheiros DM, Collawn JF, Cooper MD, Monteiro RC (2001) Identification of the transferrin receptor as a novel immunoglobulin A1 receptor and its enhanced expression on mesangial cells in IgA nephropathy. J Exp Med 194:417–425
Haddad E, Moura IC, Arcos-Fajardo M, Macher M-A, Baudouin V, Alberti C, Loirat C, Monteiro RC, Peuchmaur M (2003) Enhanced expression of the CD71 mesangial IgA1 receptor in Berger’s disease and Henoch-Schönlein nephritis : association between CD71 expression and IgA deposits. J Am Soc Nephrol 14:327–337
Lebreton C, Ménard S, Abed J, Moura IC, Coppo R, Dugave C, Monteiro RC, Fricot A, Traore MG, Griffin M, Cellier C, Malamut G, Cerf-Bensussan N, Heyman M (2012) Interactions among secretory immunoglobulin A, CD71, and transglutaminase-2 affect permeability of intestinal epithelial cells to gliadin peptides. Gastroenterology. 143(3):698–707
Oruc Z, Oblet C, Boumediene A, Druilhe A, Pascal V, Le Rumeur E, Cuvillier A, El Hamel C, Lecardeur S, Leanderson T, Morelle W, Demengeot J, Aldigier JC, Cogné M (2016) IgA structure variations associate with immune stimulations and IgA mesangial deposition. J Am Soc Nephrol 27(9):2748–2761. https://doi.org/10.1681/ASN.2015080911 Epub 2016 Jan 29
Wehbi B, Oblet C, Boyer F, Huard A, Druilhe A, Paraf F, Cogné E, Moreau J, El Makhour Y, Badran B, Van Egmond M, Cogné M, Aldigier JC (2019) Mesangial deposition can strongly involve innate-like IgA molecules lacking affinity maturation. J Am Soc Nephrol 30(7):1238–1249. https://doi.org/10.1681/ASN.2018111089 Epub 2019 Jun 21
Coppo R, Amore A, Roccatello D (1992) Dietary antigens and primary immunoglobulin A nephropathy. J Am Soc Nephrol 2(10 Suppl):S173–S180
Papista C, Lechner S, Ben Mkaddem S, LeStang MB, Abbad L, Bex-Coudrat J, Pillebout E, Chemouny JM, Jablonski M, Flamant M, Daugas E, Vrtovsnik F, Yiangou M, Berthelot L, Monteiro RC (2015) Gluten exacerbates IgA nephropathy in humanized mice through gliadin-CD89 interaction. Kidney Int 88(2):276–285. https://doi.org/10.1038/ki.2015.94 Epub 2015 Mar 25
Lechner SM, Abbad L, Boedec E, Papista C, Le Stang MB, Moal C, Maillard J, Jamin A, Bex-Coudrat J, Wang Y, Li A, Martini PG, Monteiro RC, Berthelot L (2016) IgA1 protease treatment reverses mesangial deposits and hematuria in a model of IgA nephropathy. J Am Soc Nephrol 27(9):2622–2629. https://doi.org/10.1681/ASN.2015080856
Segelmark M, Uhlin F, Sonesson E on behalf of the GOOD-IDES-01 study team. The immunoglobulin G degrading enzyme imlifidase for the treatment of anti-GBM disease – the GOOD-IDES-01 trial. Poster session at ASN in 2020
Fellström BC, Barratt J, Cook H, Coppo R, Feehally J, de Fijter JW, Floege J, Hetzel G, Jardine AG, Locatelli F, Maes BD, Mercer A, Ortiz F, Praga M, Sørensen SS, Tesar V, Del Vecchio L (2017) NEFIGAN Trial Investigators. Targeted-release budesonide versus placebo in patients with IgA nephropathy (NEFIGAN): a double-blind, randomised, placebo-controlled phase 2b trial. Lancet. 389(10084):2117–2127. https://doi.org/10.1016/S0140-6736(17)30550-0 Epub 2017 Mar
De Angelis M, Montemurno E, Piccolo M, Vannini L, Lauriero G, Maranzano V, Gozzi G, Serrazanetti D, Dalfino G, Gobbetti M et al (2014) Microbiota and metabolome associated with immunoglobulin A nephropathy (IgAN). PLoS One 9:e99006. https://doi.org/10.1371/journal.pone.0099006
Chemouny JM, Gleeson PJ, Abbad L, Lauriero G, Boedec E, Le Roux K, Monot C, Bredel M, Bex-Coudrat J, Sannier A, Daugas E, Vrtovsnik F, Gesualdo L, Leclerc M, Berthelot L, Ben Mkaddem S, Lepage P, Monteiro RC (2019) Modulation of the microbiota by oral antibiotics treats immunoglobulin A nephropathy in humanized mice. Nephrol Dial Transplant 34(7):1135–1144. https://doi.org/10.1093/ndt/gfy323
Lauriero G, Abbad L, Vacca M, Calasso M, Chemouny J, Berthelot L, Gesualdo L, De Angelis M, Monteiro RC. Fecal microbiota transplantation modulates renal phenotype in the humanized mouse model of IgA Nephropathy (article in revision)
Di Leo V, Gleeson PJ, Sallustio F, Bounaix C, Da Silva J, Loreto G, Mkaddem SB, Monteiro RC (2021) Rifaximin as a potential treatment for IgA nephropathy in a humanized mice model. J Pers Med 11:309
Zheng F, Kundu GC, Zhang Z, Ward J, DeMayo F, Mukherjee AB (1999) Uteroglobin is essential in preventing immunoglobulin A nephropathy in mice. Nat Med 5(9):1018–1025. https://doi.org/10.1038/12458
Coppo R, Chiesa M, Cirina P, Peruzzi L, Amore A, European IgACE Study Group (2002) In human IgA nephropathy uteroglobin does not play the role inferred from transgenic mice. Am J Kidney Dis 40(3):495–503
Wang J, Anders RA, Wu Q, Peng D, Cho JH, Sun Y, Karaliukas R, Kang HS, Turner JR, Fu YX (2004) Dysregulated LIGHT expression on T cells mediates intestinal inflammation and contributes to IgA nephropathy. J Clin Invest 113:826–835
Marquina R, Díez MA, López-Hoyos M, Buelta L, Kuroki A, Kikuchi S et al (2004) Inhibition of B cell death causes the development of an IgA nephropathy in (New Zealand white x C57BL/6)F(1)-bcl-2 transgenic mice. J Immunol 172(11):7177–7185
McCarthy DD, Kujawa J, Wilson C, Papandile A, Poreci U, Porfilio EA, Ward L, Lawson MAE, Macpherson AJ, McCoy KD, Pei Y, Novak L, Lee JY, Julian BA, Novak J, Ranger A, Gommerman JL, Browning JL (2011) Mice overexpressing BAFF develop a commensal flora-dependent, IgA-associated nephropathy. J Clin Invest 121:3991–4002
Nishie T, Miyaishi O, Azuma H, Kameyama A, Naruse C, Hashimoto N, Yokoyama H, Narimatsu H, Wada T, Asano M (2007) Development of immunoglobulin A nephropathy- like disease in beta-1,4-galactosyltransferase-I-deficient mice. Am J Pathol févr 170(2):447–456
Funding
A part of study with gddY mice was funded by a research grant from the Study Group on IgA Nephropathy, Grant-in-Aid for Progressive Renal Disease Research, Research on Intractable Disease from the Ministry of Health, Labour and Welfare of Japan, and AMED under Grant Number JP21gm0010006; RCM acknowledges funding by ANR and Labex Inflamex (ANR-11-IDEX-0005-02) from French government, Inserm, and Fondation pour la recherche medicale (FRM).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Ethical approval
All experiments with a1KICD89Tg mice were performed in accordance with the French Council of Animal Care guidelines and national ethical guidelines of Paris-Nord Animal Care Committee (Comité d’Éthique Expérimentation Animale Bichat-Debré). The experimental protocol gddY mice for gd was approved by the Ethics Review Committee for Animal Experimentation of Juntendo University Faculty of Medicine.
Informed consent
Not needed for animal studies.
Conflict of interest
Some studies with gddY mice were in collaboration with Kyowa-Kirin Co ltd., Visterra Inc. Some studies of α1KICD89Tg mice were funded by Shire Co.
Additional information
This article is a contribution to the Special issue on: The IgA system, IgA nephropathy and IgA vasculitis - Guest Editors: Jürgen Floege & Jonathan Barratt
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Monteiro, R.C., Suzuki, Y. Are there animal models of IgA nephropathy?. Semin Immunopathol 43, 639–648 (2021). https://doi.org/10.1007/s00281-021-00878-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-021-00878-5