
Abstract
In recent times, advances in cancer immunotherapy have yielded impressive, durable clinical responses in patients with varied subtypes of cancer. However, a significant proportion of patients who initially demonstrate encouraging tumor regression develop resistance and progress over time. The identification of novel therapeutic approaches to overcome resistance may result in significantly improved clinical outcomes and remains an area of high scientific priority. This review aims to summarize the current knowledge regarding the role of both tumor-intrinsic and tumor-extrinsic factors in the development of resistance to cancer immunotherapy and to discuss current and possible future therapeutic strategies targeting these mechanisms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
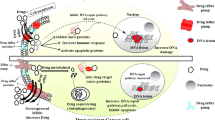
Recent advances in cancer immunotherapy, including the introduction of immune checkpoint inhibitors (ICI), have resulted in a paradigm shift in the treatment landscape of advanced cancer [1,2,3]. To date, these agents have displayed greatest activity in metastatic melanoma (MM) [4,5,6,7,8], advanced non-small-cell lung cancer (NSCLC) [9,10,11], urothelial cancers [12,13,14], head and neck squamous carcinoma [15,16,17], renal cell carcinoma [18], classical Hodgkin’s lymphoma [19,20,21], Merkel cell carcinoma [22, 23], and solid tumors characterized by microsatellite instability and mismatch repair deficiency [16, 24]. Despite the impressive results of the last decade, the ever-increasing number of immunotherapy-based clinical trials has highlighted the emergence of a significant proportion of patients that acquire resistance to immunotherapy (Fig. 1). Approximately 30% of MM patients treated with ICI who obtain an initial objective response will progress within 3 years [1, 2]. The potential mechanisms underlying acquired resistance are numerous and not completely understood, overlapping at least in part with mechanisms associated with primary resistance. In this review, we discuss the most comprehensively described mechanisms associated with acquired resistance and emerging approaches serving to restore effective immunosurveillance.

Graphical representation of acquired resistance to cancer immunotherapy. PD, progressive disease; SD, stable disease; PrlSD, prolonged stable disease; PR, partial response; CR, complete response; LtPR, long-term partial response; LtCR, long-term complete response
Current cancer immunotherapies and the immune checkpoints
Effective activation of tumor-specific T cells is the result of a set of complex interactions occurring at the immunological synapse, consisting of (i) recognition of a major histocompatibility complex (MHC)-bound tumor epitope via the T cell receptor (TCR); (ii) effective co-stimulation, i.e., interaction of receptors/ligands such as CD28 on T cells and B7 on antigen-presenting cells [25]; (iii) a relative lack of co-inhibition, i.e., interaction between negative regulators of T cells (immune checkpoints) such as cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) or programmed cell death protein 1 (PD-1) and their ligands [26].
The physiological role of immune checkpoints is to maintain self-tolerance and to control inflammation, yet they also represent one of the most common mechanisms of cancer-mediated immune evasion [26, 27]. Indeed, the primary goal of ICI therapy is to restore and maintain the activity of tumor-specific T cells by blocking immune checkpoint-mediated suppression [28]. CTLA-4 is a competitive antagonist of CD28 and can suppress T cell responses when expressed on effector T cells [29]. Ipilimumab, an anti-CTLA-4 fully human monoclonal antibody, was the first checkpoint inhibitor approved by the US Food and Drug Administration (FDA), as a monotherapy for the treatment of advanced melanoma in 2011 [8, 30]. Long-term survival data from clinical trials of ipilimumab has demonstrated durable disease control lasting over 5 years in approximately 20% of patients [31]. Another checkpoint inhibitor expressed by activated T cells is PD-1. The expression of its ligands, PD-L1/PD-L2, varies by tumor type and can be constitutive or induced by activated T cell-produced IFN gamma (IFNγ). Clinical targeting of this protein and its ligands via ICI has further improved the response rate of patients with metastatic cancer [5,6,7, 32]. Two anti-PD-1 human monoclonal antibodies, nivolumab and pembrolizumab, and three new human monoclonal antibodies directed against PD-L1, durvalumab, atezolizumab, and avelumab, have received FDA approval for the treatment of selected tumor types [1, 33, 34].
Other types of immunotherapy, thus far evaluated predominantly in clinical trials, include (i) adoptive transfer of T cells, either expanded from the tumor-microenvironment (tumor-infiltrating lymphocytes, or TILs [35]) or genetically modified (i.e., chimeric-antigen receptor or CAR-T cells [36, 37] and TCR-engineered T cells [38, 39]); (ii) therapeutic cancer vaccines, including those stimulating the adaptive immune system to generate an immune response against patient-specific tumor neoantigens [40, 41]; and (iii) oncolytic virotherapy, in particular T-VEC, alone or in combination with ICI [42, 43].
Mechanisms of acquired immune resistance
Most human tumors develop in an immunologically competent environment; thus, some level of immune escape and resistance is intrinsic to any established malignancy [3]. Acquired resistance to cancer immunotherapy can develop via genetic and epigenetic heritable traits that pre-exist in the tumor prior to treatment and then emerge following immune selection. However, acquired resistance can also arise from de novo alterations at a single-cell level [2]. When under attack, cancer cells and other components of the tumor microenvironment (TME) can alter their transcriptome in response to interactions with immune cells and their products [2, 44]. Genomic instability can further boost the emergence of immune-resistant cancer cell clones.
Tumor cells can protect themselves and evade the immune system through intrinsic resistance mechanisms such as loss/downregulation of target antigen expression, defective antigen presentation, insensitivity to immune effector molecules, upregulation of alternative immune checkpoints and epigenetic alterations or through extrinsic resistance mechanisms mediated by non-cancerous cells in the TME, such as tumor-associated macrophages (TAMs), regulatory T cells (Tregs), and myeloid-derived suppressor cells (MDSCs) [45,46,47,48,49]. As described, all such mechanisms can exist prior to cancer immunotherapy exposure and overlap with primary immune resistance. It is therefore most likely that the co-existence of multiple primary and/or acquired immune evasion mechanisms within the same tumor leads to acquired resistance in the clinical setting. In the next sections, we will discuss the current knowledge on the major resistance mechanisms associated with acquired resistance to cancer immunotherapy which have been identified to date in solid tumors.
Tumor-intrinsic factors
Defective presentation of tumor-antigens
Tumor cell recognition by the adaptive immune system represents an essential step in obtaining tumor regression. For an antigen on tumor cells to be recognized by T cells, it must be processed and presented in association with MHC molecules [50]. Therefore, immune evasion can be mediated via defects in the antigen presentation pathway. These defects can be categorized into two groups: (i) loss of antigenic molecules (either at a DNA, RNA, or post-translational level) or (ii) impaired antigen processing and presentation ability.
Cancer neoantigens, i.e. those arising from somatic tumor mutations, have been shown to be critical targets for eliciting immune responses and immune-mediated tumor killing [25]. In a pioneering study, Brown et al. [51] analyzed RNA-sequencing data to predict potentially immunogenic mutations in 515 patients from six different cancer types. The predicted immunogenic mutational counts demonstrated a strong correlation with increased overall survival, providing initial evidence of the importance of neoantigens in human cancer immune recognition. Using large-scale genomic data sets generated from biopsies of 18 different subtypes of solid tumor, Rooney et al. [52] demonstrated that the number of predicted neoantigens was positively associated with in situ immunological activity (“cytolytic activity”) across numerous tumor types. Moreover, neoantigen depletion in certain tumor types supported the hypothesis of an immune-mediated pressure to eliminate immunogenic neoantigens. More recently, Rizvi et al. [53] reported that a higher somatic nonsynonymous mutational burden correlates with improved clinical outcomes following ICI therapy in patients with NSCLC. Similar results were obtained by Snyder et al. [54] and van Allen et al. [55] in advanced melanoma. In a recent publication by Riaz et al. [56], the genomic changes in tumors from 68 patients with advanced melanoma, who progressed or were naïve to anti-CTLA-4 therapy, were assessed before and after anti-PD-1 therapy. A reduction in mutation and neoantigen load was observed in association with a response to anti-PD-1 therapy independent of prior immune therapy exposure, suggesting clonal evolution as result of treatment-dependent immunoediting. These studies provided clear evidence that cancer neoantigens represent important targets for the human immune system and appear to play an important role in tumor elimination following treatment with ICI.
Loss of antigenic molecules
Recently, Anagnostou et al. [57] demonstrated that loss of expression of 7 to 18 putative mutation-associated neoantigens can result in acquired resistance to ICI therapy. The analyses were conducted on matched pre-treatment and resistant tumors from patients with NSCLC undergoing ICI therapy and demonstrated that neoantigen loss occurred via the elimination of tumor subclones and chromosomal loss of truncal alterations. This was the first study demonstrating that an evolving landscape of mutations can be linked to acquired resistance to ICI. Moreover, the majority of eliminated mutations were typically highly expressed and encoded for neoantigens that were predicted to have high MHC binding affinity or to induce stronger TCR mediated responses. A separate study, conducted by Verdegaal et al. [58], suggested that the dynamic interactions between T cells and cancer cells can induce T cell-mediated neoantigen immunoediting, resulting in partial or total neoantigen loss. In contrast, in a separate study investigating a single patient case in which a complete response to adoptive T cell therapy (ACT) was achieved, acquired resistance occurred without loss of target neoantigens [4]. However, in a mouse model of ACT against murine melanoma, tumor progression after initial regression was associated with a loss of the gene encoding the target tumor antigen [59]. These data demonstrate that downregulation/loss of neoantigens may occur in patients treated with immunotherapy, yet this is not a universal mechanism of acquired immune resistance.
It is likely that proinflammatory cytokines can contribute to immune escape by inducing loss of antigen expression [44]. In a mouse model of ACT [60], melanoma has been shown to acquire resistance through TNFα-induced epithelial-to-mesenchymal de-differentiation. This process, resulting in a loss of melanocytic antigens, causes a switch to a less immunogenic tumor phenotype that can more easily evade immune surveillance. Other cytokines produced by tumor-infiltrating cells, such as IL-6 or TGFβ, have been shown to induce epithelial-to-mesenchymal transition in mouse models of several types of cancer, indicating that inflammation may promote acquired resistance in numerous types of histologically varied cancer [61, 62]. Further work is needed to determine the role of this process in clinical practice.
Impaired antigen processing and presentation machinery
Clinical evidence of defective antigen presentation induced by immunotherapy was recently reported by Tran et al. [63]. This group reported the case of a patient with metastatic colorectal cancer treated with an infusion of HLA-C*08:02-restricted TILs targeting mutant KRAS G12D. Loss of the chromosome 6 haplotype encoding the HLA-C*08:02 class I MHC molecule caused the progression of a single lesion 9 months after ACT. Lack of surface expression of class I MHC molecules can also be induced by alterations in genes encoding components of the antigen presentation machinery. Mutations in the β2-microglobulin (B2M) gene have been shown to mediate acquired resistance to IL-2 or ACT [64]. B2M is associated with MHC class I and is essential for its folding and transport to the T cell surface [47, 65]. Recently, Zaretsky et al. [66] reported a case of late acquired resistance to anti-PD-1 therapy in a patient with MM. In this study, loss of MHC surface expression was associated with the acquisition of a new homozygous truncating mutation in the B2M gene. Further studies [67, 68] have supported the role of B2M loss in the development of anti-PD-1 therapy resistance in melanoma and lung cancer. Chang et al. [69] demonstrated a combination of structural and epigenetic defects in MHC class I antigen processing and presentation machinery in MM after immunotherapy. These defects included loss of tapasin (a MHC class I antigen processing molecule) due to a germline frameshift in TAPBP exon 3 in association with somatic loss of the other allele, selective epigenetic silencing of the HLA-A3 antigen, and loss of one HLA haplotype. An additional case, documented by our group [70], provided further evidence of alterations to the MHC class I antigen processing and presentation machinery as a mechanism of acquired resistance after T cell-based immunotherapy in MM. To summarize, these mechanisms allow previously antigenic mutations to become effectively invisible to the immune system, due to the loss of antigen presentation ability.
Insensitivity to immune effector molecules
It has been demonstrated that immunotherapy can induce resistance to T cell-mediated tumor regression by altering the normally vulnerable pathways targeted by T cells [65]; the IFNγ pathway is emerging as a key mediator of this process [71, 72]. After secretion by activated T cells, IFNγ binds to its receptor (IFNGR) on cancer cells and recruits both Janus kinase 1 and 2 (JAK1/JAK2). This recruitment can subsequently induce phosphorylation, dimerization, and activation of the transcription factor STAT1, whose translocation to the nucleus activates the transcription of IFNγ-inducible genes [72]. The process culminates in tumor cell growth inhibition, induction of apoptosis, further T cell infiltration, and upregulation of MHC class I and PD-L1 [66, 71,72,73]. In the previously described study by Zaretsky et al. [66], whole-exome sequencing was used to compare the DNA of paired baseline and relapsing lesions from four MM patients who exhibited disease progression after an initial response to anti-PD-1. The core aim of the analyses was to identify the genetic basis of this change in phenotype, and it resulted in the detection of truncating loss-of-function mutations in both JAK1 and JAK2, with an accompanying loss of heterozygosity, in two of the patients. This inactivation of JAK1 and JAK2 presumably caused the acquired insensitivity to IFNγ and impacted upon class I expression, immune surveillance and tumor cell proliferation. It has been previously suggested that a lack of T cell infiltrate may be caused by JAK1/JAK2 mutations, as the IFNγ pathway regulates the expression of chemokines, such as CXCL9, CXCL10, and CXCL11, with potent chemoattractant effects on T cells [72]. To further support this, both CD8+ T cell infiltration and PD-L1 expression were present in the baseline biopsies, whereas relapses were associated with the restriction of CD8+ T cells and PD-L1 expression to the tumor margin. In the context of anti–PD-1 therapy, a lack of effective interferon signaling becomes a highly advantageous immune escape strategy, as the potential protection afforded to tumors via PD-L1 is nullified by the treatment. Interferon insensitivity through other mechanisms, such as the expression of negative regulators or epigenetic silencing of JAKs, may have a similar impact and has already been documented in other forms of cancer [74,75,76].
Further studies are required in order to identify additional IFNγ-resistance mechanisms. Genomic alterations affecting IFNγ-pathway genes, such as the loss of IFNGR1, IFNGR2, IRF1, IFIT1, IFIT2, IFIT3, MTAP, miR31 or amplification of the suppressor genes SOCS1 and PIAS4, have already been reported as potential mechanisms of primary resistance to multiple ICI therapies [77]. Discerning the role of these defects as drivers of acquired resistance to immunotherapy is immensely important for developing strategies to overcome such obstacles.
Upregulation of immune-suppressive pathways
Activation of alternative immunosuppressive pathways in the TME may promote resistance to immunotherapy through impact upon T cell function [45, 65]. Numerous alternative immune checkpoints, such as T cell immunoglobulin and mucin-3 (TIM-3), lymphocyte activation gene 3 (LAG-3), V-domain Ig-containing suppressor of T cell activation (VISTA), and indoleamine-2,3-dioxygenase (IDO), have been characterized in recent years. These are often upregulated during ICI therapy with anti-PD1/PD-L1 or anti-CTLA-4 antibodies due to either the activation of the IFNγ-pathway or miscellaneous cellular signals [78, 79]. Thommen et al. [80] reported that acquired resistance to anti-PD1 therapy in patients affected by NSCLC was associated with the co-expression of PD-1, TIM-3, CTLA-4, LAG-3, and B and T lymphocyte attenuator (BTLA). Their research demonstrated that expression of these five molecules on the surface of effector T cells was associated with reduced proliferation, migration, and cytokine production. Upregulation of these, as well as multiple other inhibitory checkpoints, has been documented in response to ICI therapy in a diverse range of pre-clinical cancer models [79, 81,82,83,84]. Recently, TIM-3 upregulation was observed in lung adenocarcinoma patients who developed acquired resistance to anti-PD1 treatment [83]. In addition, a recent study from Balko et al. [85] illustrated that LAG-3 upregulation post-relapse in anti-PD1 treated tumors was associated with MHC II expression, indicating that MHC II+ tumors might derive increased benefit from anti-LAG3 therapy. Combinational therapy utilizing anti-PD-1 antibodies in association with LAG-3 inhibitors [86] or TIM-3 inhibitors [87] has already demonstrated notable efficacy in pre-clinical models and a phase I clinical trial. Indeed a study evaluating the combination of PD-1 blockade with LAG-3 blockade in patients with melanoma previously treated with anti-PD1/anti-PD-L1 monotherapy, reported an objective response rate of 16% [88]. Currently, there are numerous ongoing clinical trials evaluating the possibility of incorporating novel agents targeting alternative immune checkpoints, both as a monotherapy and in combination.
A recent pre-clinical study by Bertrand et al. [89] suggested that TNFα expression in an inflamed TME correlates positively with PD-L1 and TIM-3 expression upon anti-PD1 therapy in melanoma mouse models. Indeed, this study provides the first evidence that the TNFR1-dependent TNFα signaling pathway may be an immune evasion mechanism conferring resistance to anti-PD1 therapy. In addition, it was demonstrated that TNFα impairs the accumulation of CD8+ TILs and triggers activation-induced cell death of CD8+ T cells in mouse melanoma. Accordingly, TNFα blockade prevented PD-L1 and TIM3 expression, and anti-PD1-induced TIL death. Consequently, this study provides a rationale for the combination of anti-PD1 and anti-TNFα antibodies as a novel potential combinational therapy for the treatment of melanoma and other cancers. A phase 1 clinical trial testing this association is ongoing (NCT03293784).
Preliminary studies have identified a potential role of specific oncogenic signaling pathways in the acquisition of resistance to immunotherapy. Activation of the β-catenin signaling pathway has been shown to correlate with an absence of CD103+ dendritic cells and T cell exclusion from the TME. This pathway appears to be activated in at least 10% of human cancers [90]. The activation of another pathway, the PI3-kinase pathway, can inhibit T cell infiltration through loss of the tumor suppressor PTEN and may represent an additional mechanism of immune escape [64].
Epigenetic alterations
Emerging evidence indicates that epigenetic modification may play an important role in antigen processing and presentation; in the proliferation, differentiation and function of T cells; and in the acquisition of a memory T cell phenotype [45, 91]. Additionally, epigenetic silencing of the genes encoding CXCL9 and CXCL10 has been proposed to interfere with T cell migration into tumors [92]. Changes in the chromatin landscape could therefore be at the basis of T cell exhaustion, mediating primary resistance or contributing to relapse during ICI therapy. Epigenetic targeting drugs, such as those targeting DNA methylation, histone deacetylation, or histone methylation; have demonstrated promising activity in the pre-clinical setting [91]. Ongoing clinical trials are testing various combinations of demethylating agents, hypomethylating agents, and histone deacetylase inhibitors; in association with ICI in patients with acquired resistance to immunotherapy [47, 91]. Further results are needed to assess the role of these epigenetic modulators in clinical practice.
Tumor-extrinsic factors
Aside from tumor cell-mediated mechanisms of immune suppression, there are numerous factors operating in the TME that can impact upon the efficacy of immunotherapy agents. A subset of T cells that is commonly associated with cancer progression is Tregs [93]. Tregs are known to suppress effector T cell function via the secretion of inhibitory cytokines such as IL-10 and TGFβ, or by direct cell-to-cell contact [45]. IL-10 can affect antigen presentation by reducing the expression of MHC class II and co-stimulatory molecules on dendritic cells, thereby preventing the activation of effector T cells [94]. Pre-clinical studies conducted in mouse models of cancer demonstrate that in addition to expanding effector T cells in secondary lymphoid organs, anti-CTLA-4 antibodies also act to deplete intra-tumoral Tregs via antibody-dependent cell-mediated cytotoxicity (ADCC) [95,96,97]. This differential activity results from higher expression of CTLA-4 on Tregs relative to effector T cell subsets and enrichment of Fc gamma receptor (FcγR)-expressing cell subsets within capacity for ADCC in the TME.
Despite its potentially depleting isotype, the contribution of ADCC and role of FcγRs in the activity of anti-CTLA-4 has remained unclear until recently. Two clinical studies have previously identified a reduction in tumor-infiltrating Tregs post anti-CTLA-4 therapy [98, 99]. Moreover, in vitro studies demonstrate that ipilimumab depletes CTLA-4-expressing Tregs in the presence of FcγR-expressing monocytes and NK cells, consistent with the predicted binding affinity for activatory FcγRs [98, 100]. Most recently, a role for Fc effector function in the activity of human anti-CTLA-4 antibodies has been confirmed [101]. Importantly, this study suggested that Treg depletion only appears relevant in the context of an inflamed TME, explaining the relatively modest response rates to anti-CTLA-4 compared to the pre-clinical setting. Further clinical studies targeting Tregs in combination with ICI are already ongoing [93]. Additionally, there is some indication that epigenetic modulation can be responsible for the accumulation of Tregs in cancers [91].
The presence of other suppressive cells, such as MDSCs and TAMs, in the TME has been associated with resistance to multiple facets of immunotherapy, including ACT, ICI, and dendritic cell vaccines. Furthermore, MDSCs have been shown to play a role in promoting angiogenesis, metastasis, and immune cell suppression. Combinational strategies targeting this cell subset in association with other immunotherapy agents may enhance the clinical benefits derived by cancer patients [45]. In a recent study, M2-polarized TAMs were shown to directly interfere with anti-PD1 therapy by removing anti-PD1 antibodies from the surface of PD-1+ T cells [102]. Previous clinical studies documented a connection between high TAM infiltration and poor prognosis [103]. Studies testing the blockade of macrophage colony-stimulating factor receptor (CSF-1R) in combination with either ICI or ACT reported improved tumor regression, indicating that a reduction of TAMs may restore immunotherapy efficacy. Clinical trials evaluating the efficacy and safety of CSF-1R blockade in association with ICI across various solid tumor types are currently ongoing [45].
Future directions and conclusions
Acquired resistance to cancer immunotherapy is often the result of multiple immune escape mechanisms. Efforts to identify universal mechanisms of acquired resistance are unlikely to be successful. Future efforts should focus upon developing biomarker-guided strategies to counteract immune resistance mechanisms on an individual patient basis, or alternatively, combination therapies targeting multiple pathways simultaneously to prevent the development of acquired resistance. Potential strategies may include the introduction of drugs inhibiting immune-suppressive cells or proteins, blockade of pathways downregulating anti-tumor immunity, modulation of epigenetic mechanisms, and combination of checkpoint inhibition [5, 7, 88] with immune pathway stimulators, such as the stimulator of interferon gene (STING) which promotes STAT activation in a JAK2-independent manner [66].
In conclusion, although patients treated with cancer immunotherapy have the potential to derive substantial benefit, the emergence of acquired resistance poses a significant challenge for a considerable proportion of patients. An improved understanding of the mechanisms underlying acquired resistance and the identification of relevant biomarkers in each individual is required for the development of novel therapeutic strategies and improved personalized cancer immunotherapies [44]. Further studies in larger cohorts will help elucidate alternative escape pathways. Current efforts aim to promote long-lasting disease control for the majority, rather than a select few.
References
Sharma P, Allison JP (2015) The future of immune checkpoint therapy. Science 348(6230):56–61. https://doi.org/10.1126/science.aaa8172
Restifo NP, Smyth MJ, Snyder A (2016) Acquired resistance to immunotherapy and future challenges. Nat Rev Cancer 16(2):121–126. https://doi.org/10.1038/nrc.2016.2
Restifo NP, Dudley ME, Rosenberg SA (2012) Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol 12(4):269–281. https://doi.org/10.1038/nri3191
Ribas A, Hamid O, Daud A, Hodi FS, Wolchok JD, Kefford R, Joshua AM, Patnaik A, Hwu WJ, Weber JS, Gangadhar TC, Hersey P, Dronca R, Joseph RW, Zarour H, Chmielowski B, Lawrence DP, Algazi A, Rizvi NA, Hoffner B, Mateus C, Gergich K, Lindia JA, Giannotti M, Li XN, Ebbinghaus S, Kang SP, Robert C (2016) Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA 315(15):1600–1609. https://doi.org/10.1001/jama.2016.4059
Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, Ferrucci PF, Hill A, Wagstaff J, Carlino MS, Haanen JB, Maio M, Marquez-Rodas I, McArthur GA, Ascierto PA, Long GV, Callahan MK, Postow MA, Grossmann K, Sznol M, Dreno B, Bastholt L, Yang A, Rollin LM, Horak C, Hodi FS, Wolchok JD (2015) Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 373(1):23–34. https://doi.org/10.1056/NEJMoa1504030
Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, Larkin J, Lorigan P, Neyns B, Blank CU, Hamid O, Mateus C, Shapira-Frommer R, Kosh M, Zhou H, Ibrahim N, Ebbinghaus S, Ribas A, investigators K (2015) Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 372(26):2521–2532. https://doi.org/10.1056/NEJMoa1503093
Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS, Shaheen M, Ernstoff MS, Minor D, Salama AK, Taylor M, Ott PA, Rollin LM, Horak C, Gagnier P, Wolchok JD, Hodi FS (2015) Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 372(21):2006–2017. https://doi.org/10.1056/NEJMoa1414428
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363(8):711–723. https://doi.org/10.1056/NEJMoa1003466
Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, Gottfried M, Peled N, Tafreshi A, Cuffe S, O’Brien M, Rao S, Hotta K, Leiby MA, Lubiniecki GM, Shentu Y, Rangwala R, Brahmer JR, Investigators K (2016) Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med 375(19):1823–1833. https://doi.org/10.1056/NEJMoa1606774
Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, Barlesi F, Kohlhaufl M, Arrieta O, Burgio MA, Fayette J, Lena H, Poddubskaya E, Gerber DE, Gettinger SN, Rudin CM, Rizvi N, Crino L, Blumenschein GR Jr, Antonia SJ, Dorange C, Harbison CT, Graf Finckenstein F, Brahmer JR (2015) Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med 373(17):1627–1639. https://doi.org/10.1056/NEJMoa1507643
Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, Waterhouse D, Ready N, Gainor J, Aren Frontera O, Havel L, Steins M, Garassino MC, Aerts JG, Domine M, Paz-Ares L, Reck M, Baudelet C, Harbison CT, Lestini B, Spigel DR (2015) Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med 373(2):123–135. https://doi.org/10.1056/NEJMoa1504627
Sharma P, Retz M, Siefker-Radtke A, Baron A, Necchi A, Bedke J, Plimack ER, Vaena D, Grimm MO, Bracarda S, Arranz JA, Pal S, Ohyama C, Saci A, Qu X, Lambert A, Krishnan S, Azrilevich A, Galsky MD (2017) Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol 18(3):312–322. https://doi.org/10.1016/S1470-2045(17)30065-7
Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, Dawson N, O’Donnell PH, Balmanoukian A, Loriot Y, Srinivas S, Retz MM, Grivas P, Joseph RW, Galsky MD, Fleming MT, Petrylak DP, Perez-Gracia JL, Burris HA, Castellano D, Canil C, Bellmunt J, Bajorin D, Nickles D, Bourgon R, Frampton GM, Cui N, Mariathasan S, Abidoye O, Fine GD, Dreicer R (2016) Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 387(10031):1909–1920. https://doi.org/10.1016/S0140-6736(16)00561-4
Powles T, O’Donnell PH, Massard C, Arkenau HT, Friedlander TW, Hoimes CJ, Lee JL, Ong M, Sridhar SS, Vogelzang NJ, Fishman MN, Zhang J, Srinivas S, Parikh J, Antal J, Jin X, Gupta AK, Ben Y, Hahn NM (2017) Efficacy and safety of durvalumab in locally advanced or metastatic urothelial carcinoma: updated results from a phase 1/2 open-label study. JAMA Oncol 3(9):e172411. https://doi.org/10.1001/jamaoncol.2017.2411
Bauml J, Seiwert TY, Pfister DG, Worden F, Liu SV, Gilbert J, Saba NF, Weiss J, Wirth L, Sukari A, Kang H, Gibson MK, Massarelli E, Powell S, Meister A, Shu X, Cheng JD, Haddad R (2017) Pembrolizumab for platinum- and cetuximab-refractory head and neck cancer: results from a single-arm, phase II study. J Clin Oncol 35(14):1542–1549. https://doi.org/10.1200/JCO.2016.70.1524
Seiwert TY, Burtness B, Mehra R, Weiss J, Berger R, Eder JP, Heath K, McClanahan T, Lunceford J, Gause C, Cheng JD, Chow LQ (2016) Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol 17(7):956–965. https://doi.org/10.1016/S1470-2045(16)30066-3
Harrington KJ, Ferris RL, Blumenschein G Jr, Colevas AD, Fayette J, Licitra L, Kasper S, Even C, Vokes EE, Worden F, Saba NF, Kiyota N, Haddad R, Tahara M, Grunwald V, Shaw JW, Monga M, Lynch M, Taylor F, DeRosa M, Morrissey L, Cocks K, Gillison ML, Guigay J (2017) Nivolumab versus standard, single-agent therapy of investigator’s choice in recurrent or metastatic squamous cell carcinoma of the head and neck (CheckMate 141): health-related quality-of-life results from a randomised, phase 3 trial. Lancet Oncol 18(8):1104–1115. https://doi.org/10.1016/S1470-2045(17)30421-7
Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER, Castellano D, Choueiri TK, Gurney H, Donskov F, Bono P, Wagstaff J, Gauler TC, Ueda T, Tomita Y, Schutz FA, Kollmannsberger C, Larkin J, Ravaud A, Simon JS, Xu LA, Waxman IM, Sharma P, CheckMate I (2015) Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 373(19):1803–1813. https://doi.org/10.1056/NEJMoa1510665
Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, Schuster SJ, Millenson MM, Cattry D, Freeman GJ, Rodig SJ, Chapuy B, Ligon AH, Zhu L, Grosso JF, Kim SY, Timmerman JM, Shipp MA, Armand P (2015) PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med 372(4):311–319. https://doi.org/10.1056/NEJMoa1411087
Armand P, Shipp MA, Ribrag V, Michot JM, Zinzani PL, Kuruvilla J, Snyder ES, Ricart AD, Balakumaran A, Rose S, Moskowitz CH (2016) Programmed death-1 blockade with pembrolizumab in patients with classical Hodgkin lymphoma after brentuximab vedotin failure. J Clin Oncol 34(31):3733–3739. https://doi.org/10.1200/JCO.2016.67.3467
Chen R, Zinzani PL, Fanale MA, Armand P, Johnson NA, Brice P, Radford J, Ribrag V, Molin D, Vassilakopoulos TP, Tomita A, von Tresckow B, Shipp MA, Zhang Y, Ricart AD, Balakumaran A, Moskowitz CH, Keynote (2017) Phase II study of the efficacy and safety of pembrolizumab for relapsed/refractory classic Hodgkin lymphoma. J Clin Oncol 35(19):2125–2132. https://doi.org/10.1200/JCO.2016.72.1316
Kaufman HL, Russell J, Hamid O, Bhatia S, Terheyden P, D’Angelo SP, Shih KC, Lebbe C, Linette GP, Milella M, Brownell I, Lewis KD, Lorch JH, Chin K, Mahnke L, von Heydebreck A, Cuillerot JM, Nghiem P (2016) Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol 17(10):1374–1385. https://doi.org/10.1016/S1470-2045(16)30364-3
Nghiem PT, Bhatia S, Lipson EJ, Kudchadkar RR, Miller NJ, Annamalai L, Berry S, Chartash EK, Daud A, Fling SP, Friedlander PA, Kluger HM, Kohrt HE, Lundgren L, Margolin K, Mitchell A, Olencki T, Pardoll DM, Reddy SA, Shantha EM, Sharfman WH, Sharon E, Shemanski LR, Shinohara MM, Sunshine JC, Taube JM, Thompson JA, Townson SM, Yearley JH, Topalian SL, Cheever MA (2016) PD-1 blockade with pembrolizumab in advanced Merkel-cell carcinoma. N Engl J Med 374(26):2542–2552. https://doi.org/10.1056/NEJMoa1603702
Alley EW, Lopez J, Santoro A, Morosky A, Saraf S, Piperdi B, van Brummelen E (2017) Clinical safety and activity of pembrolizumab in patients with malignant pleural mesothelioma (KEYNOTE-028): preliminary results from a non-randomised, open-label, phase 1b trial. Lancet Oncol 18(5):623–630. https://doi.org/10.1016/S1470-2045(17)30169-9
Schumacher TN, Schreiber RD (2015) Neoantigens in cancer immunotherapy. Science 348(6230):69–74. https://doi.org/10.1126/science.aaa4971
Pardoll DM (2012) The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12(4):252–264. https://doi.org/10.1038/nrc3239
Keir ME, Butte MJ, Freeman GJ, Sharpe AH (2008) PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 26:677–704. https://doi.org/10.1146/annurev.immunol.26.021607.090331
Quezada SA, Peggs KS (2013) Exploiting CTLA-4, PD-1 and PD-L1 to reactivate the host immune response against cancer. Br J Cancer 108(8):1560–1565. https://doi.org/10.1038/bjc.2013.117
Krummel MF, Allison JP (1995) CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med 182(2):459–465
Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, Lebbe C, Baurain JF, Testori A, Grob JJ, Davidson N, Richards J, Maio M, Hauschild A, Miller WH Jr, Gascon P, Lotem M, Harmankaya K, Ibrahim R, Francis S, Chen TT, Humphrey R, Hoos A, Wolchok JD (2011) Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 364(26):2517–2526. https://doi.org/10.1056/NEJMoa1104621
Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, Patt D, Chen TT, Berman DM, Wolchok JD (2015) Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 33(17):1889–1894. https://doi.org/10.1200/JCO.2014.56.2736
Schachter J, Ribas A, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, Larkin J, Lorigan P, Neyns B, Blank C, Petrella TM, Hamid O, Zhou H, Ebbinghaus S, Ibrahim N, Robert C (2017) Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 390(10105):1853–1862. https://doi.org/10.1016/S0140-6736(17)31601-X
FDA (2018) Novel drug approvals for 2016. FDA website https://wwwfdagov/Drugs/DevelopmentApprovalProcess/DrugInnovation/ucm483775htm Accessed 02 May 2018
FDA (2018) Novel drug approvals for 2017. FDA Website https://wwwfdagov/Drugs/DevelopmentApprovalProcess/DrugInnovation/ucm537040htm Accessed 02 May 2018
Geukes Foppen MH, Donia M, Svane IM, Haanen JB (2015) Tumor-infiltrating lymphocytes for the treatment of metastatic cancer. Mol Oncol 9(10):1918–1935. https://doi.org/10.1016/j.molonc.2015.10.018
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, Shen A, Teachey DT, Levine BL, June CH, Porter DL, Grupp SA (2014) Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 371(16):1507–1517. https://doi.org/10.1056/NEJMoa1407222
Han S, Latchoumanin O, Wu G, Zhou G, Hebbard L, George J, Qiao L (2017) Recent clinical trials utilizing chimeric antigen receptor T cells therapies against solid tumors. Cancer Lett 390:188–200. https://doi.org/10.1016/j.canlet.2016.12.037
Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, Wunderlich JR, Nahvi AV, Helman LJ, Mackall CL, Kammula US, Hughes MS, Restifo NP, Raffeld M, Lee CC, Levy CL, Li YF, El-Gamil M, Schwarz SL, Laurencot C, Rosenberg SA (2011) Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol 29(7):917–924. https://doi.org/10.1200/JCO.2010.32.2537
Karpanen T, Olweus J (2015) T-cell receptor gene therapy—ready to go viral? Mol Oncol 9(10):2019–2042. https://doi.org/10.1016/j.molonc.2015.10.006
Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M, Bukur V, Tadmor AD, Luxemburger U, Schrors B, Omokoko T, Vormehr M, Albrecht C, Paruzynski A, Kuhn AN, Buck J, Heesch S, Schreeb KH, Muller F, Ortseifer I, Vogler I, Godehardt E, Attig S, Rae R, Breitkreuz A, Tolliver C, Suchan M, Martic G, Hohberger A, Sorn P, Diekmann J, Ciesla J, Waksmann O, Bruck AK, Witt M, Zillgen M, Rothermel A, Kasemann B, Langer D, Bolte S, Diken M, Kreiter S, Nemecek R, Gebhardt C, Grabbe S, Holler C, Utikal J, Huber C, Loquai C, Tureci O (2017) Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547(7662):222–226. https://doi.org/10.1038/nature23003
Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, Chen C, Olive O, Carter TA, Li S, Lieb DJ, Eisenhaure T, Gjini E, Stevens J, Lane WJ, Javeri I, Nellaiappan K, Salazar AM, Daley H, Seaman M, Buchbinder EI, Yoon CH, Harden M, Lennon N, Gabriel S, Rodig SJ, Barouch DH, Aster JC, Getz G, Wucherpfennig K, Neuberg D, Ritz J, Lander ES, Fritsch EF, Hacohen N, Wu CJ (2018) Corrigendum: an immunogenic personal neoantigen vaccine for patients with melanoma. Nature 555(7696):402. https://doi.org/10.1038/nature25145
Burke EE, Zager JS (2018) Pharmacokinetic drug evaluation of talimogene laherparepvec for the treatment of advanced melanoma. Expert Opin Drug Metab Toxicol 14(4):469–473. https://doi.org/10.1080/17425255.2018.1455825
Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, Olszanski AJ, Malvehy J, Cebon J, Fernandez E, Kirkwood JM, Gajewski TF, Chen L, Gorski KS, Anderson AA, Diede SJ, Lassman ME, Gansert J, Hodi FS, Long GV (2017) Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell 170(6):1109–1119 e1110. https://doi.org/10.1016/j.cell.2017.08.027
Ribas A (2015) Adaptive immune resistance: how cancer protects from immune attack. Cancer Discov 5(9):915–919. https://doi.org/10.1158/2159-8290.CD-15-0563
Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A (2017) Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168(4):707–723. https://doi.org/10.1016/j.cell.2017.01.017
Pitt JM, Vetizou M, Daillere R, Roberti MP, Yamazaki T, Routy B, Lepage P, Boneca IG, Chamaillard M, Kroemer G, Zitvogel L (2016) Resistance mechanisms to immune-checkpoint blockade in cancer: tumor-intrinsic and -extrinsic factors. Immunity 44(6):1255–1269. https://doi.org/10.1016/j.immuni.2016.06.001
Syn NL, Teng MWL, Mok TSK, Soo RA (2017) De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol 18(12):e731–e741. https://doi.org/10.1016/S1470-2045(17)30607-1
Jenkins RW, Barbie DA, Flaherty KT (2018) Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer 118(1):9–16. https://doi.org/10.1038/bjc.2017.434
O’Donnell JS, Long GV, Scolyer RA, Teng MW, Smyth MJ (2017) Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev 52:71–81. https://doi.org/10.1016/j.ctrv.2016.11.007
Ward JP, Gubin MM, Schreiber RD (2016) The role of neoantigens in naturally occurring and therapeutically induced immune responses to cancer. Adv Immunol 130:25–74. https://doi.org/10.1016/bs.ai.2016.01.001
Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, Holt RA (2014) Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res 24(5):743–750. https://doi.org/10.1101/gr.165985.113
Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N (2015) Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160(1–2):48–61. https://doi.org/10.1016/j.cell.2014.12.033
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, Ibrahim F, Bruggeman C, Gasmi B, Zappasodi R, Maeda Y, Sander C, Garon EB, Merghoub T, Wolchok JD, Schumacher TN, Chan TA (2015) Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348(6230):124–128. https://doi.org/10.1126/science.aaa1348
Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, Hollmann TJ, Bruggeman C, Kannan K, Li Y, Elipenahli C, Liu C, Harbison CT, Wang L, Ribas A, Wolchok JD, Chan TA (2014) Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 371(23):2189–2199. https://doi.org/10.1056/NEJMoa1406498
Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MHG, Goldinger SM, Utikal J, Hassel JC, Weide B, Kaehler KC, Loquai C, Mohr P, Gutzmer R, Dummer R, Gabriel S, Wu CJ, Schadendorf D, Garraway LA (2015) Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350(6257):207–211. https://doi.org/10.1126/science.aad0095
Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, Hodi FS, Martin-Algarra S, Mandal R, Sharfman WH, Bhatia S, Hwu WJ, Gajewski TF, Slingluff CL Jr, Chowell D, Kendall SM, Chang H, Shah R, Kuo F, Morris LGT, Sidhom JW, Schneck JP, Horak CE, Weinhold N, Chan TA (2017) Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 171(4):934–949 e915. https://doi.org/10.1016/j.cell.2017.09.028
Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, Zhang T, Adleff V, Phallen J, Wali N, Hruban C, Guthrie VB, Rodgers K, Naidoo J, Kang H, Sharfman W, Georgiades C, Verde F, Illei P, Li QK, Gabrielson E, Brock MV, Zahnow CA, Baylin SB, Scharpf RB, Brahmer JR, Karchin R, Pardoll DM, Velculescu VE (2017) Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov 7(3):264–276. https://doi.org/10.1158/2159-8290.CD-16-0828
Verdegaal EM, de Miranda NF, Visser M, Harryvan T, van Buuren MM, Andersen RS, Hadrup SR, van der Minne CE, Schotte R, Spits H, Haanen JB, Kapiteijn EH, Schumacher TN, van der Burg SH (2016) Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature 536(7614):91–95. https://doi.org/10.1038/nature18945
Kaluza KM, Thompson JM, Kottke TJ, Flynn Gilmer HC, Knutson DL, Vile RG (2012) Adoptive T cell therapy promotes the emergence of genomically altered tumor escape variants. Int J Cancer 131(4):844–854. https://doi.org/10.1002/ijc.26447
Landsberg J, Kohlmeyer J, Renn M, Bald T, Rogava M, Cron M, Fatho M, Lennerz V, Wolfel T, Holzel M, Tuting T (2012) Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 490(7420):412–416. https://doi.org/10.1038/nature11538
Knutson KL, Lu H, Stone B, Reiman JM, Behrens MD, Prosperi CM, Gad EA, Smorlesi A, Disis ML (2006) Immunoediting of cancers may lead to epithelial to mesenchymal transition. J Immunol 177(3):1526–1533
Santisteban M, Reiman JM, Asiedu MK, Behrens MD, Nassar A, Kalli KR, Haluska P, Ingle JN, Hartmann LC, Manjili MH, Radisky DC, Ferrone S, Knutson KL (2009) Immune-induced epithelial to mesenchymal transition in vivo generates breast cancer stem cells. Cancer Res 69(7):2887–2895. https://doi.org/10.1158/0008-5472.CAN-08-3343
Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, Pasetto A, Zheng Z, Ray S, Groh EM, Kriley IR, Rosenberg SA (2016) T-cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med 375(23):2255–2262. https://doi.org/10.1056/NEJMoa1609279
Haanen J (2017) Cancer immunotherapy: escape of response. [Article released in occasion of ESMO Immuno-Oncology Congress 2017]
O’Donnell JS, Smyth MJ, Teng MW (2016) Acquired resistance to anti-PD1 therapy: checkmate to checkpoint blockade? Genome Med 8(1):111. https://doi.org/10.1186/s13073-016-0365-1
Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, Saco J, Homet Moreno B, Mezzadra R, Chmielowski B, Ruchalski K, Shintaku IP, Sanchez PJ, Puig-Saus C, Cherry G, Seja E, Kong X, Pang J, Berent-Maoz B, Comin-Anduix B, Graeber TG, Tumeh PC, Schumacher TN, Lo RS, Ribas A (2016) Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med 375(9):819–829. https://doi.org/10.1056/NEJMoa1604958
Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane JP, Bjorgaard SL, Hammond MR, Vitzthum H, Blackmon SM, Frederick DT, Hazar-Rethinam M, Nadres BA, Van Seventer EE, Shukla SA, Yizhak K, Ray JP, Rosebrock D, Livitz D, Adalsteinsson V, Getz G, Duncan LM, Li B, Corcoran RB, Lawrence DP, Stemmer-Rachamimov A, Boland GM, Landau DA, Flaherty KT, Sullivan RJ, Hacohen N (2017) Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun 8(1):1136. https://doi.org/10.1038/s41467-017-01062-w
Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, Wurtz A, Dong W, Cai G, Melnick MA, Du VY, Schlessinger J, Goldberg SB, Chiang A, Sanmamed MF, Melero I, Agorreta J, Montuenga LM, Lifton R, Ferrone S, Kavathas P, Rimm DL, Kaech SM, Schalper K, Herbst RS, Politi K (2017) Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov 7(12):1420–1435. https://doi.org/10.1158/2159-8290.CD-17-0593
Chang CC, Pirozzi G, Wen SH, Chung IH, Chiu BL, Errico S, Luongo M, Lombardi ML, Ferrone S (2015) Multiple structural and epigenetic defects in the human leukocyte antigen class I antigen presentation pathway in a recurrent metastatic melanoma following immunotherapy. J Biol Chem 290(44):26562–26575. https://doi.org/10.1074/jbc.M115.676130
Donia M, Harbst K, van Buuren M, Kvistborg P, Lindberg MF, Andersen R, Idorn M, Munir Ahmad S, Ellebaek E, Mueller A, Fagone P, Nicoletti F, Libra M, Lauss M, Hadrup SR, Schmidt H, Andersen MH, Thor Straten P, Nilsson JA, Schumacher TN, Seliger B, Jonsson G, Svane IM (2017) Acquired immune resistance follows complete tumor regression without loss of target antigens or IFNgamma signaling. Cancer Res 77(17):4562–4566. https://doi.org/10.1158/0008-5472.CAN-16-3172
Sucker A, Zhao F, Pieper N, Heeke C, Maltaner R, Stadtler N, Real B, Bielefeld N, Howe S, Weide B, Gutzmer R, Utikal J, Loquai C, Gogas H, Klein-Hitpass L, Zeschnigk M, Westendorf AM, Trilling M, Horn S, Schilling B, Schadendorf D, Griewank KG, Paschen A (2017) Acquired IFNgamma resistance impairs anti-tumor immunity and gives rise to T-cell-resistant melanoma lesions. Nat Commun 8:15440. https://doi.org/10.1038/ncomms15440
Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, Grasso CS, Hugo W, Sandoval S, Torrejon DY, Palaskas N, Rodriguez GA, Parisi G, Azhdam A, Chmielowski B, Cherry G, Seja E, Berent-Maoz B, Shintaku IP, Le DT, Pardoll DM, Diaz LA Jr, Tumeh PC, Graeber TG, Lo RS, Comin-Anduix B, Ribas A (2017) Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov 7(2):188–201. https://doi.org/10.1158/2159-8290.CD-16-1223
Bach EA, Aguet M, Schreiber RD (1997) The IFN gamma receptor: a paradigm for cytokine receptor signaling. Annu Rev Immunol 15:563–591. https://doi.org/10.1146/annurev.immunol.15.1.563
Fish EN, Platanias LC (2014) Interferon receptor signaling in malignancy: a network of cellular pathways defining biological outcomes. Mol Cancer Res 12(12):1691–1703. https://doi.org/10.1158/1541-7786.MCR-14-0450
Dunn GP, Sheehan KC, Old LJ, Schreiber RD (2005) IFN unresponsiveness in LNCaP cells due to the lack of JAK1 gene expression. Cancer Res 65(8):3447–3453. https://doi.org/10.1158/0008-5472.CAN-04-4316
Kaplan DH, Shankaran V, Dighe AS, Stockert E, Aguet M, Old LJ, Schreiber RD (1998) Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci U S A 95(13):7556–7561
Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, Chen PL, Hwu P, Allison JP, Futreal A, Wargo JA, Sharma P (2016) Loss of IFN-gamma pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell 167(2):397–404 e399. https://doi.org/10.1016/j.cell.2016.08.069
Topalian SL, Drake CG, Pardoll DM (2015) Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 27(4):450–461. https://doi.org/10.1016/j.ccell.2015.03.001
Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, Cucolo L, Lee DSM, Pauken KE, Huang AC, Gangadhar TC, Amaravadi RK, Schuchter LM, Feldman MD, Ishwaran H, Vonderheide RH, Maity A, Wherry EJ, Minn AJ (2016) Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell 167(6):1540–1554 e1512. https://doi.org/10.1016/j.cell.2016.11.022
Thommen D, Uhlenbrock F, Herzig P, Prince SS, Moersig W, Lardinois D, Zippelius A (2016) 66P Highly exhausted PD-1hi T cell subsets in human NSCLC are co-defined by the predominant expression of distinct inhibitory receptors and correlate with clinical outcome. J Thorac Oncol 11(4 Suppl):S83. https://doi.org/10.1016/S1556-0864(16)30179-4
Gao J, Ward JF, Pettaway CA, Shi LZ, Subudhi SK, Vence LM, Zhao H, Chen J, Chen H, Efstathiou E, Troncoso P, Allison JP, Logothetis CJ, Wistuba II, Sepulveda MA, Sun J, Wargo J, Blando J, Sharma P (2017) VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat Med 23(5):551–555. https://doi.org/10.1038/nm.4308
Huang RY, Francois A, McGray AR, Miliotto A, Odunsi K (2017) Compensatory upregulation of PD-1, LAG-3, and CTLA-4 limits the efficacy of single-agent checkpoint blockade in metastatic ovarian cancer. Oncoimmunology 6(1):e1249561. https://doi.org/10.1080/2162402X.2016.1249561
Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, Gandhi L, Redig AJ, Rodig SJ, Asahina H, Jones RE, Kulkarni MM, Kuraguchi M, Palakurthi S, Fecci PE, Johnson BE, Janne PA, Engelman JA, Gangadharan SP, Costa DB, Freeman GJ, Bueno R, Hodi FS, Dranoff G, Wong KK, Hammerman PS (2016) Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun 7:10501. https://doi.org/10.1038/ncomms10501
Shayan G, Srivastava R, Li J, Schmitt N, Kane LP, Ferris RL (2017) Adaptive resistance to anti-PD1 therapy by Tim-3 upregulation is mediated by the PI3K-Akt pathway in head and neck cancer. Oncoimmunology 6(1):e1261779. https://doi.org/10.1080/2162402X.2016.1261779
Balko JM JD, Wang DY, Ericsson-Gonzalez P, Nixon M, Salgado R, Sanchez V, Schreeder D, Kim JY, Bordeaux J, Sanders M, Davis RS (2018) MHC-II expression to drive a unique pattern of adaptive resistance to antitumor immunity through receptor checkpoint engagement. J Clin Oncol 36, 2018 (suppl 5S; abstr 180) Poster from ASCO-SITC Clinical Immuno-Oncology Symposum 2018
Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, Bettini ML, Gravano DM, Vogel P, Liu CL, Tangsombatvisit S, Grosso JF, Netto G, Smeltzer MP, Chaux A, Utz PJ, Workman CJ, Pardoll DM, Korman AJ, Drake CG, Vignali DA (2012) Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res 72(4):917–927. https://doi.org/10.1158/0008-5472.CAN-11-1620
Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC (2010) Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med 207(10):2187–2194. https://doi.org/10.1084/jem.20100643
Ascierto PA, Melero I, Bhatia S, Bono P, Sanborn RE, Lipson EJ, Callahan MK, Gajewski T, Gomez-Roca CA, Hodi FS, Curigliano G, Nyakas M, Preusser M, Koguchi Y, Maurer M, Clynes R, Mitra P, Suryawanshi S, Muñoz-Couselo E (2017) Initial efficacy of anti-lymphocyte activation gene-3 (anti–LAG-3; BMS-986016) in combination with nivolumab (nivo) in pts with melanoma (MEL) previously treated with anti–PD-1/PD-L1 therapy. J Clin Oncol 35(15_suppl):9520–9520. https://doi.org/10.1200/JCO.2017.35.15_suppl.9520
Bertrand F, Montfort A, Marcheteau E, Imbert C, Gilhodes J, Filleron T, Rochaix P, Andrieu-Abadie N, Levade T, Meyer N, Colacios C, Segui B (2017) TNFalpha blockade overcomes resistance to anti-PD-1 in experimental melanoma. Nat Commun 8(1):2256. https://doi.org/10.1038/s41467-017-02358-7
Luke JJ, Bao R, Spranger S, Sweis RF, Gajewski T (2016) Correlation of WNT/β-catenin pathway activation with immune exclusion across most human cancers. J Clin Oncol 34(15_suppl):3004–3004. https://doi.org/10.1200/JCO.2016.34.15_suppl.3004
Gallagher SJ, Shklovskaya E, Hersey P (2017) Epigenetic modulation in cancer immunotherapy. Curr Opin Pharmacol 35:48–56. https://doi.org/10.1016/j.coph.2017.05.006
Nagarsheth N, Peng D, Kryczek I, Wu K, Li W, Zhao E, Zhao L, Wei S, Frankel T, Vatan L, Szeliga W, Dou Y, Owens S, Marquez V, Tao K, Huang E, Wang G, Zou W (2016) PRC2 epigenetically silences Th1-type chemokines to suppress effector T-cell trafficking in colon cancer. Cancer Res 76(2):275–282. https://doi.org/10.1158/0008-5472.CAN-15-1938
Shitara K, Nishikawa H (2018) Regulatory T cells: a potential target in cancer immunotherapy. Ann N Y Acad Sci 1417:104–115. https://doi.org/10.1111/nyas.13625
Commeren DL, Van Soest PL, Karimi K, Lowenberg B, Cornelissen JJ, Braakman E (2003) Paradoxical effects of interleukin-10 on the maturation of murine myeloid dendritic cells. Immunology 110(2):188–196
Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, Korman AJ (2013) Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res 1(1):32–42. https://doi.org/10.1158/2326-6066.CIR-13-0013
Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, Peggs KS, Ravetch JV, Allison JP, Quezada SA (2013) Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med 210(9):1695–1710. https://doi.org/10.1084/jem.20130579
Bulliard Y, Jolicoeur R, Windman M, Rue SM, Ettenberg S, Knee DA, Wilson NS, Dranoff G, Brogdon JL (2013) Activating Fc gamma receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J Exp Med 210(9):1685–1693. https://doi.org/10.1084/jem.20130573
Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, Michielin O, Weide B, Romero P, Speiser DE (2015) Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci U S A 112(19):6140–6145. https://doi.org/10.1073/pnas.1417320112
Tarhini AA, Edington H, Butterfield LH, Lin Y, Shuai Y, Tawbi H, Sander C, Yin Y, Holtzman M, Johnson J, Rao UN, Kirkwood JM (2014) Immune monitoring of the circulation and the tumor microenvironment in patients with regionally advanced melanoma receiving neoadjuvant ipilimumab. PLoS One 9(2):e87705. https://doi.org/10.1371/journal.pone.0087705
Jie HB, Schuler PJ, Lee SC, Srivastava RM, Argiris A, Ferrone S, Whiteside TL, Ferris RL (2015) CTLA-4(+) regulatory T cells increased in cetuximab-treated head and neck cancer patients suppress NK cell cytotoxicity and correlate with poor prognosis. Cancer Res 75(11):2200–2210. https://doi.org/10.1158/0008-5472.CAN-14-2788
Arce Vargas F, Furness AJS, Litchfield K, Joshi K, Rosenthal R, Ghorani E, Solomon I, Lesko MH, Ruef N, Roddie C, Henry JY, Spain L, Ben Aissa A, Georgiou A, Wong YNS, Smith M, Strauss D, Hayes A, Nicol D, O'Brien T, Martensson L, Ljungars A, Teige I, Frendeus B, Melanoma TR, Renal TR, consortia TRL, Pule M, Marafioti T, Gore M, Larkin J, Turajlic S, Swanton C, Peggs KS, Quezada SA (2018) Fc effector function contributes to the activity of human anti-CTLA-4 antibodies. Cancer Cell 33(4):649–663.e4. https://doi.org/10.1016/j.ccell.2018.02.010
Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS, Miller MA, Carlson JC, Freeman GJ, Anthony RM, Weissleder R, Pittet MJ (2017) In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci Transl Med 9(389):eaal3604. https://doi.org/10.1126/scitranslmed.aal3604
Hu W, Li X, Zhang C, Yang Y, Jiang J, Wu C (2016) Tumor-associated macrophages in cancers. Clin Transl Oncol 18(3):251–258. https://doi.org/10.1007/s12094-015-1373-0
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Marco Donia has received honoraria for lectures from Bristol-Myers Squibb, Merck, Astra Zeneca, and Genzyme, as well as financial support for attending symposia from Bristol-Myers Squibb, Merck, Novartis, Pfizer, and Roche. The other authors declare that they have no conflict of interest.
Additional information
This article is a contribution to the special issue on Anti-cancer Immunotherapy: Breakthroughs and Future Strategies - Guest Editor: Mads Hald Andersen
Rights and permissions
About this article

Cite this article
Draghi, A., Chamberlain, C.A., Furness, A. et al. Acquired resistance to cancer immunotherapy. Semin Immunopathol 41, 31–40 (2019). https://doi.org/10.1007/s00281-018-0692-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-018-0692-y