
Abstract
Fibromyalgia is a high impact chronic pain disorder with a well-defined and robust clinical phenotype. Key features include widespread pain and tenderness, high levels of sleep disturbance, fatigue, cognitive dysfunction and emotional distress. Abnormal processing of pain and other sensory input occurs in the brain, spinal cord and periphery and is related to the processes of central and peripheral sensitization. As such, fibromyalgia is deemed to be one of the central sensitivity syndromes. There is increasing evidence of neurogenically derived inflammatory mechanisms occurring in the peripheral tissues, spinal cord and brain in fibromyalgia. These involve a variety of neuropeptides, chemokines and cytokines with activation of both the innate and adaptive immune systems. This process results in several of the peripheral clinical features of fibromyalgia, such as swelling and dysesthesia, and may influence central symptoms, such as fatigue and changes in cognition. In turn, emotional and stress-related physiological mechanisms are seen as upstream drivers of neurogenic inflammation in fibromyalgia.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Overview
Fibromyalgia (also known as fibromyalgia syndrome) is a common chronic pain syndrome, with a well-defined clinical phenotype, that affects between 2 and 8% of adults [1]. It has a very high personal and societal impact [2, 3]. Although there is no clinically useful biomarker to aid diagnosis or monitor progress, numerous abnormalities have been found in the functioning of the pain-related nervous system, both central and peripheral. Many findings in fibromyalgia initially appear disparate, and are difficult to reconcile without an understanding of the functional neuroplasticity of the nervous system.
The relationship between the sensory input to the brain and the subsequent neural output to the body is highly relevant to the clinical features of fibromyalgia. The activated pain-related nervous system that characterises fibromyalgia is dependent on modulation by numerous neural networks, involving neurotransmitters, hormones, neuropeptides, cytokines, chemokines and a variety of other molecules. These neural processes are integrated across the brain, spinal cord and the peripheral tissues, and include within them the process of neurogenic inflammation. The aim of this review is to examine the role of neurogenic inflammation in fibromyalgia with a focus on identifying the ways this process modulates the symptoms that characterise this disorder. Better understanding of this process will aid management of this significant pain disorder.
Clinical features of fibromyalgia
Fibromyalgia presents as a robust clinical phenotype [3, 4]. The patient complains of widespread pain affecting all quadrants of the body, as well as spinal regions. The pain is often described as burning or aching in character and fluctuates in intensity and varies in location over time. The onset of fibromyalgia is often characterised by an identified triggering event that frequently associates with psychological distress [5, 6]. The widespread pain is usually associated with high levels of fatigue, poor quality sleep and cognitive dysfunction, manifesting as poor concentration and memory. Muscular dysfunction can cause abnormal co-contraction of muscle groups during activity, with resultant stiffness, weakness, restricted gait or difficulties in taking a deep breath.
Sensations of non-neuroanatomical pins and needles and numbness, and soft-tissue swelling, particularly in the fingers, hands and feet, may mimic peripheral or entrapment neuropathy or an inflammatory arthritis, respectively. The majority of patients demonstrate the sign of dermatographia, particularly over the upper back, where a brisk wheal and flare response is noted after gentle mechanical stimulation [7].
In addition to the widespread pain, there is also widespread abnormal tenderness to gentle pressure, termed allodynia. This characteristic finding in fibromyalgia is reflective of widespread lowering of pain threshold, a feature that affects all tissues that have been examined, including skin, muscles, entheses, periosteum, fat pads and bones [8]. Emotional distress is also a key feature of fibromyalgia and further delineation as to how this process contributes to fibromyalgia is evolving [9].
Classification and diagnostic criteria of fibromyalgia
Although the clinical phenotype of fibromyalgia has been described in the literature over a long period of time, diagnostic criteria for fibromyalgia are more recent. Useful criteria were published in the 1970s and refined in the early 1980s [10]. Later, the criteria of the American College of Rheumatology, focussing on the association between widespread pain and widespread abnormal tenderness, were promulgated as classification criteria for clinical studies [11]. These criteria, were subsequently revised in 2011 and 2012 to incorporate the association between widespread pain and other key features, including poor quality sleep, high levels of fatigue, poor cognition and other associated features which included depression, headache and bowel pain [12, 13]. These criteria deleted the necessity for widespread tenderness due to the inconsistent use of this clinical sign in everyday practice. Later, the criteria were further refined to focus on a tighter group of patients with widespread pain but still using the same framework of the 2011 criteria [14]. These 2016 criteria are deemed to be validated for both diagnosis and classification and can be used as self-report by the patient. There is a strong correlation between the three sets of criteria and in this review, it is accepted that criteria in one era are equivalent to those in another era for the purposes of describing various associations and functional abnormalities in fibromyalgia.
It is noted that the evolution of criteria link critical symptoms in the central nervous system (sleep, fatigue and cognition) to common and characteristic features in the body (widespread pain and tenderness) [10]. These criteria are relevant to the discussion of neurogenic inflammation given that they link central and peripheral components of the condition. Additionally, current criteria identify fibromyalgia as a spectrum disorder, one that better fits with concepts of neuromodulation and neuroplasticity.
Co-morbid conditions
Fibromyalgia is commonly associated with other disorders that might include headache, migraine, temporomandibular joint dysfunction, pelvic pain, complex regional pain syndrome, restless legs syndrome, irritable bowel or bladder syndrome, hypotension and hypersensitivity to chemicals, light or noise [15]. These conditions have been termed central sensitivity syndromes due to shared pathophysiological processes [16]. These disorders will be further discussed later.
Central mechanisms in fibromyalgia
There are a number of changes in the brain in fibromyalgia [3]. Networks involved in modulating the brain’s influence on the spinal cord are disturbed in fibromyalgia. Connectivity between the default–mode network and pain-inhibitory centres is decreased, while connectivity is increased between this network and the insula [17, 18]. There are higher levels of the excitatory neurotransmitter glutamate in the posterior insular in patients with fibromyalgia compared to controls [19]. These regions interact with areas involved with modulation of sensory input and neuroendocrine output relevant to fibromyalgia to be discussed later.
Central neuroinflammation in fibromyalgia
There is evidence of central neuroinflammation in fibromyalgia. A number of neuropeptides that cause neuroinflammation have been found to be elevated in the cerebrospinal fluid (CSF) of patients with fibromyalgia. For instance, substance P (SP) is several times higher in the CSF of patients with fibromyalgia compared to controls [20, 21]. Additionally, brain-derived neurotrophic factor and nerve growth factor are elevated [22, 23], while studies on calcitonin gene-related protein (CGRP) are more limited [24].
SP and the SP-structurally-related hemokinin-1 (HK-1) and corticotrophin-releasing hormone (CRH) levels have also been shown to be significantly elevated in the blood of 84 female fibromyalgia patients compared to a healthy control group of 15 females and 5 males, although earlier studies of blood levels of SP showed no changes compared to controls [25, 26]. There is a significant association between the levels of SP and HK-1 but not with CRH.
The underlying cause of the elevated blood and CSF neuropeptide levels in fibromyalgia remains unclear. Neuropeptides are distributed widely through the brain, spinal cord and peripheral nervous system. The dorsal root ganglia cell bodies of C-fibres manufacture neuropeptides that are then transported both proximally to the spinal cord and distally to the C-afferent terminals in the periphery. Elevated CSF and blood levels could relate to increased production within the brain, the spinal cord, the periphery, or a combination of all three.
The finding of elevation of the cytokine IL-8, but not IL-1β, in the CSF of fibromyalgia patients, compared to healthy controls, implies that in this location, it is derived from glial cells within the central nervous system [27, 28]. This cytokine is co-localised with the translocator protein (TSPO) in glial cells, which is the rate-limiting step in serotonin synthesis and hence modulates synaptic transmission. Genetic polymorphisms of TSPO associate with symptom severity and cerebral pain processing in fibromyalgia, and interact with the serotonin transporter gene [28]. Other proinflammatory chemicals are also present in the CSF of patients with fibromyalgia (further discussed later) [29].
Taken together elevated levels of neuropeptides, other proinflammatory chemicals and a specific proinflammatory cytokine imply that neuroinflammatory processes are present in the central nervous system in fibromyalgia. The consequences of brain neuroinflammation are unclear in fibromyalgia. The relationship between this process and the key symptoms of fatigue and cognitive dysfunction require further study.
Stress and neurogenic inflammation in fibromyalgia
The sympathetic nervous system (SNS) and the hypothalamic pituitary adrenal (HPA) axis comprise the main neurotransmitter and neuroendocrine response systems to stress [30], and both systems are activated in fibromyalgia [9]. These systems may account for some of the symptoms seen in fibromyalgia, such as palpitations or increased anxiety, or fatigue. However, other stress-related neurogenically mediated pathways are active in fibromyalgia. These relate in particular to those that involve neuropeptides, which may play an intermediary role in translating stress responses to biological effects. For instance, the neuropeptide substance P is evolutionarily conserved and basic to the whole-organism stress response [31]. It is not only elevated in the CSF of patients with fibromyalgia [20, 21] but also in patients affected by a variety of stressful situations [32]. Although not specifically studied in fibromyalgia, it is noted that emotional distress can activate neuroinflammation [33]. The effect of emotional distress and the stress response on the central brain-related events that link to neuroinflammation in fibromyalgia requires further clarification.
Chronic stress may also influence the function of other organ systems that can in turn interact with the central nervous system (CNS). For instance, stress will increase gastrointestinal permeability with lipopolysaccharide translocation leading to release of inflammatory mediators in the CNS [34]. Commensal gut microbiota is recognised to modulate neuroinflammation and they also change function in the context of stress [30, 33].
Mindfulness meditation reduces stress and can decrease cutaneous neurogenic inflammatory responses in humans [35]. As will be discussed later, these responses are exaggerated in fibromyalgia.
Taken together, the activation of the stress system, possibly driven by emotional distress, is likely to play a key upstream role in modulating if not initiating neurogenic inflammation in fibromyalgia.
Sleep and neurogenic inflammation in fibromyalgia
Sleep disturbance also links to neuroinflammation. Selective slow-wave sleep disruption in healthy women associates with onset of musculoskeletal pain, fatigue and an increase in neurogenic flare responses [36]. In mice, sleep deprivation results in elevation of IL6 and hippocampal neuroinflammation, as well as learning and memory impairment [37]. Sleep disturbance is a characteristic of fibromyalgia and closely links to fatigue, cognitive dysfunction and pain, with stress being a driver of all of these symptoms [38].
Spinal cord mechanisms
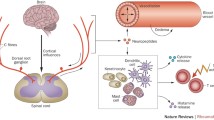
The cell body of the C-fibre, residing in the dorsal root ganglion, manufactures a number of neurotransmitters and neuropeptides. These include glutamate, CGRP, SP, brain-derived neurotrophic factor, CX3CL1 (CX3, chemokine ligand 1, fractalkine) and adenosine triphosphate (ATP) [33]. Many of these, particularly the neuropeptides, are transported distally to the periphery and also proximally to the dorsal horn of the spinal cord. Other neurotransmitters, such as glutamate and ATP, are also manufactured and released at the distal and proximal nerve terminals.
The receptors for these neuroactive substances are found on the neighbouring innate immune cells, the microglia and astrocytes [33]. After activation, the expression of Toll-like receptor 4 (TLR4) is upregulated in microglia leading to production and potential release of a number of potent locally acting chemicals, including excitatory amino acids, nitric oxide, prostaglandins, leukotriene, nerve growth factor and superoxides [39]. Activated microglia and astrocytes may also release proinflammatory cytokines, such as tumour necrosis factor (TNF), interleukin 1(IL1) and IL6 [39, 40]. Thus, the C-fibre may contribute considerable locally acting spinal cord proinflammatory chemicals that result in neuroinflammation within the central nervous system [39].
The C and Aδ fibres input to projection neurons of both the outer and deeper layers of the spinal dorsal horn. At each site, there is considerable modulation of activity of the projection neurons by descending corticospinal neural activity [41,42,43,44]. These pathways arise in the midbrain and brainstem and use both opioid and 5-hydroxytryptaminergic–noradrenergic mechanisms. In fibromyalgia patients, the opioidergic pathways function normally while the 5-hydroxytryptaminergic–noradrenergic mechanisms are attenuated [45, 46]. The inability to activate these endogenous sensory inhibitory mechanisms can enhance reactivity of the neurons, a process termed central sensitisation [47].
There was significant elevation of inflammatory proteins in the CSF of 40 patients with fibromyalgia compared to 10 healthy controls [29]. In particular, high levels of chemokine CX3CL1 (fractalkine) and IL-8 were found. The CSF proteins discriminating for fibromyalgia overlapped with the profile identified in the plasma suggesting that these inflammatory processes are interlinked. It is suggested that in this regard the CSF acts as a “mirror” reflecting processes occurring in the spinal cord [29, 48].
Peripheral neurogenic inflammation in fibromyalgia
The body of evidence for neurogenic inflammation in fibromyalgia in peripheral tissues is larger than that for central neurogenic inflammation, likely related to the ease of access to skin compared to central nervous tissue. Despite this quantity of evidence relating to findings in peripheral tissue, it is not suggested that neurogenic inflammation in fibromyalgia is initiated by peripheral events.
Lewis made one of the first clinical observations of neurogenic inflammation in his description of the triple response [49]. This can be evoked in healthy subjects by direct stimulation of the skin by mechanical, thermal or chemical stimulation. Capsaicin is an example of a potent chemical stimulator of this response [50]. The triple response comprises a classic weal and flare, the weal being due to plasma extravasation and the flare due to redness of the stimulation site and surrounding erythema.
The mechanism behind this cutaneous manifestation of neurogenic inflammation is through the release of proinflammatory peptides from the nerve terminals of peptidergic C-fibres [51, 52]. These predominantly comprise SP, CGRP and neurokinin A, but also may include adrenomedullin, neurokinin B, vasoactive intestinal peptide, neuropeptide Y and gastrin-releasing peptide. In the context of the neurogenic flare response, certain of these peptides act on surrounding blood vessels resulting in vasodilatation and an increase in local blood flow. They also enhance vascular permeability resulting in egress of fluid, as well as certain intravascular proteins and polymorphonuclear leucocytes. CGRP acting on CGRP1 receptors on arterioles is the main cause of neurogenic vasodilatation, while substance P and neurokinin act on neurokinin A1 receptors to increase vascular permeability [53].
The neurogenic flare response, induced by both gentle mechanical stimulation and application of various concentrations of capsaicin, is increased in patients with fibromyalgia compared to healthy controls [54]. The extent of the flare response in patients correlates with the severity of allodynia, suggesting a link between the mechanisms involved in neurogenic inflammation and this common clinical finding in fibromyalgia. Other studies have shown significant associations between flare response and other key clinical features of fibromyalgia. There is significant correlation with slow-wave sleep deprivation, and with increased fatigue and with a decreased pain threshold [36].
There are other clinical findings in fibromyalgia that also likely reflect the process of neurogenic inflammation. Local soft-tissue swelling and fluid retention are commonly reported [55,56,57]. Reticular skin discolouration and livedo reticularis are seen in the majority [58, 59]. Fibronectin, a tissue marker of endothelial activation, is significantly elevated in fibromyalgia patients with Raynaud phenomenon and livedo compared to those without these features and also compared to healthy controls [58].
Hence, the enhanced cutaneous wheal and flare response in fibromyalgia (indicative of increased neurogenic inflammation), through its association with other clinical features, points to the contribution of neurogenic processes to the fibromyalgia phenotype.
Local immune-related effects of neuroinflammation
In the skin, neuropeptides, such as substance P and CGRP, interact with and activate cells of the innate immune system that include mast cells, dendritic cells and keratinocytes. Locally placed mast cells upon activation by these neuropeptides may degranulate and release a variety of neuroactive substances that include histamine, bradykinin, prostaglandins, TNF, 5-hydroxytryptamine and endothelial growth factors. These factors, particularly TNF and histamine, in turn act on nearby sensory nociceptive terminals, such as those on A- δ small myelinated fibres, to increase response sensitivity to lower levels of stimulation of these neurones [60]. This cascade of events leads to further vasodilation, swelling and pain in the involved site. Activated polymodal C-fibres can also directly secrete proinflammatory cytokines that can interact with molecules, such as nitric oxide or prostaglandin E2 that also upregulate responsiveness and activity of other sensory neurones involved in nociception [53].
There is a significant increase in numbers of mast cells, up to 14-fold, in the skin of patients with fibromyalgia, which is consistent with the effects of an increase in neurogenic inflammatory activity [61]. A significant correlation between the number of degranulated or damaged mast cells and the extent of IgG deposition in the skin and vessel walls has been shown [62, 63]. This would favour the explanation that the identified increased deposition of albumin and IgG at the dermoepidermal junction in patients with fibromyalgia [64, 65] relates to the plasma extravasation seen in neurogenic inflammation.
The adaptive immune system, involving T lymphocytes, is also involved in this process. Local actions of neuropeptides lead to activation of T lymphocytes and release of cytokines that further modulate local inflammatory processes [53]. There are several differences in cytokine levels in the plasma or serum of patients with fibromyalgia compared to controls; however, the source of these cytokines remains unclear. The level of cytokines may relate to peripheral mechanisms under discussion, either through direct release from C-fibres or indirectly through enhanced neuropeptide action on local T lymphocytes, or to previously discussed central mechanisms.
A number of small studies of different design and techniques have suggested that a proinflammatory cytokine profile occurs in the blood of patients with fibromyalgia [66,67,68]. These studies suggest that the levels of the proinflammatory cytokines IL-1, IL–6 and IL-8 are elevated. In contrast, TNF levels have usually been found to be normal and anti-inflammatory cytokines IL-4 and IL-10 are lower or normal. One study showed a significant association between both IL-8 and IL-6 and clinical severity scores in fibromyalgia patients suggesting that these cytokines particularly link to a clinically relevant mechanism in fibromyalgia [69]. In contrast, some studies have shown increase in TNF as well as IL6 in serum of fibromyalgia patients [26], while other studies have not confirmed elevations of IL6, IL8 or TNF in fibromyalgia [70, 71]. It is evident that blood levels of cytokines vary between studies and this likely relates to methodology issues, such as differences in body weight, hormone status, physical activity, diurnal variation and medication effects, as much as differences in assay techniques [26, 72].
Other studies also point to the presence of systemic inflammation in fibromyalgia. The levels of 92 inflammation-related proteins (mainly cytokines and chemokines), were recorded, using multiplex proximity extension assay, in the plasma of 17 women with chronic widespread pain (CWP) compared to 21 female healthy controls [70]. Using complex analysis 11 proteins significantly differentiated those with CWP from healthy controls suggesting that CWP was associated with systemic low-grade inflammation [70]. There was a high degree of overlap of these identified biomarkers using the same assay technique with those in a separate cohort of 40 fibromyalgia patients compared to 46 blood donors [29]. This study also showed a similar profile of inflammatory chemokines (such as CX3CC1/fractalkine) and cytokines (such as IL8) in the cerebrospinal fluid (CSF) of the fibromyalgia patients compared to controls. As indicate earlier, this implies the presence of neuroinflammatory mechanisms in fibromyalgia.
Another study showed the cytokine response to mitogenic activators of peripheral blood mononuclear cells from patients with fibromyalgia was lower than those of healthy individuals, suggesting impaired cell-mediated immunity in fibromyalgia [73].
The innate immune system was shown to play a significant role in the chronic pain experienced by 707 patients with chronic multisite pain (who were likely to have fibromyalgia). There was a greater association with lipopolysaccharide-stimulated proinflammatory cytokines, such as TNF and IFNγ, than with several other anti-inflammatory cytokines, suggesting an increased innate immune response [67].
Despite muscle pain being a key feature of fibromyalgia there are few studies that examine cytokines in this tissue. In one study of proinflammatory cytokines released in muscle after repetitive dynamic contractions, it was found that there was no difference between patients with fibromyalgia and healthy controls, and no associations between levels of cytokines and pain or fatigue [74].
Complex regional pain syndrome (CRPS) is a more intense and localised chronic pain syndrome that shares many features with fibromyalgia [72]. In CRPS, there are increased levels of TNF and IL-6 in suction-induced skin blister fluids and blood, while anti-inflammatory cytokines, such as IL-4 and IL-10 are reduced [75,76,77]. There are no similar studies of dermal levels of cytokines in fibromyalgia.
Taken together these observations suggest that proinflammatory cytokines and chemokines are elevated in the blood and CSF of patients with fibromyalgia. While the cause and consequences of these immune–related findings remains unclear it is likely that they relate to the process of neuroinflammation.
Local influences on peripheral neurogenic inflammation
As described above, the small non-myelinated C-fibres and myelinated A δ–fibres are both seen to be involved in peripheral neuroinflammation in fibromyalgia.
Sufficient mechanical, chemical or thermal stimulation of the C-fibre in the periphery will activate transmission of impulses to the spinal cord that after further modulation may result in nociception. Additional to this direct pathway from the periphery to the spinal cord, there is antidromic propagation of the impulse whereby the impulse is reflected at junction points back to the C-fibre terminal resulting in the release of the various proinflammatory substances described above [53].
The C-fibres are thus seen as the driver, with release of proinflammatory cytokines, chemokines and neuropeptides, while the A δ–fibres are seen to primarily respond to several of these stimuli. This leads to increased sensitivity of the neuron to other stimuli with amplification of responsiveness, so called peripheral sensitization.
Systemic influences on peripheral neurogenic inflammation
In addition to local factors, C-fibre function is also influenced by systemic factors.
Activity of the sympathetic nervous system is enhanced in fibromyalgia [78]. Studies have shown reduced heart rate variability (HRV) and also exacerbation of pain after noradrenaline injections in fibromyalgia patients compared to controls [79, 80]. These studies indicate sympathetic hyperactivity in fibromyalgia. The sympathetic nervous system can modulate the peripheral nociceptor neurons as well as the cells of the innate immune system, such as dendritic cells and keratinocytes, through upregulation of the alpha1-adenoreceptors [81, 82].
The links between the peripheral effects of sympathetic hyperactivity and central neuroinflammation are unclear. As noted earlier, there are elevated levels of IL-8 but not IL-1β in the cerebrospinal fluid in fibromyalgia patients compared to healthy controls suggesting central neuroinflammation [27]. However, a correlation between this cytokine and HRV was not found [83].
Local neuroanatomical changes to nociceptors
There are number of abnormalities in morphology, neurophysiology or general function of small myelinated Aδ fibres and non-myelinated C-fibres in around 50% of patients with fibromyalgia compared to healthy controls [84,85,86,87,88,89,90]. The absolute number of non-myelinated fibres in the skin is reduced [84, 85, 87, 88, 90,91,92] and also the mean axon diameter is reduced [90]. Microneurography shows that most patients with fibromyalgia will have changes in structure of C-fibres [93], and there are changes on electron microscopy to both the C-fibre and its associated Schwann cells [94]. In vivo corneal confocal bio-microscopy shows similar findings in the cornea of a sub-group of patients with fibromyalgia [95].
A correlation has been noted between the microRNA (miRNA) miR-let-7d and small nerve fibre density in fibromyalgia. Additionally, miR-let-7d and its downstream target insulin-like growth factor-1 are aberrantly expressed in the skin of fibromyalgia patients who had small fibre impairment [92]. The relationship of this growth factor to the enhanced neuropeptide secretion that occurs in fibromyalgia needs to be clarified.
The cause for the findings detailed above in the peripheral small fibres of patients with fibromyalgia is unclear but may relate to the consequences of local neurogenic inflammation through the effects of inflammatory products on these sensory fibres. These changes in turn may link to certain clinical features seen in fibromyalgia, such as light sensitivity and dysesthesthia [96]. Clinical observations by the authors note significant variability of these symptoms and resolution when there is overall improvement in fibromyalgia, suggesting that there may be no permanent dysfunction in these neural structures.
A rat model of fibromyalgia induced by increasing glutamate levels in the insular showed a subsequent decrease in hind-paw peripheral nerve fibre density [97]. This suggests that the cause of the small fibre abnormalities ultimately relates to changes in the brain, through a “top-down” process. This might imply a similar mechanism in humans.
It has been noted that the enhanced nociceptor activity in muscles and other tissues that results from neurogenic inflammation might further contribute to central sensitization through an increased nociceptive input to the spinal cord [98, 99].
Neuroinflammation in other organ systems in fibromyalgia
The effects of neuroinflammation on other organ systems may explain the common association between the many central sensitivity syndromes detailed previously. For instance, neuroinflammation is a prominent mechanism in complex regional pain syndrome, migraine, irritable bowel and bladder syndromes [72, 100,101,102].
Neurogenic inflammation as the key mechanism in fibromyalgia
Neurogenic inflammation is an important mechanism in fibromyalgia and related disorders. Brain–related mechanisms are the likely source of the cascade of events that leads to neuroinflammation in both the CNS and periphery in fibromyalgia. In clinical practice emotional distress is seen to be critical in initiation, exacerbation and modulation of fibromyalgia symptoms. The link between the psychological factors contributing to emotional distress and the activation of the stress response and other brain biochemical changes requires further study. The release of neuropeptides and the subsequent neuroinflammatory changes in the brain, the spinal cord and the periphery contributes to many of the characteristic symptoms of fibromyalgia.
Better understanding of this top-down neurogenically driven path is required to allow for targeted and personalised approaches to management of fibromyalgia.
References
Branco JC, Bannwarth B, Failde I, Abello Carbonell J, Blotman F, Spaeth M, Saraiva F, Nacci F, Thomas E, Caubere JP, Le Lay K, Taieb C, Matucci-Cerinic M (2010) Prevalence of fibromyalgia: a survey in five European countries. Semin Arthritis Rheum 39(6):448–453. https://doi.org/10.1016/j.semarthrit.2008.12.003
Gerdle B, Bjork J, Coster L, Henriksson K, Henriksson C, Bengtsson A (2008) Prevalence of widespread pain and associations with work status: a population study. BMC Musculoskelet Disord 9:102. https://doi.org/10.1186/1471-2474-9-102
Clauw DJ (2014) Fibromyalgia: a clinical review. JAMA 311(15):1547–1555. https://doi.org/10.1001/jama.2014.3266
Theoharides TC, Tsilioni I, Arbetman L, Panagiotidou S, Stewart JM, Gleason RM, Russell IJ (2015) Fibromyalgia syndrome in need of effective treatments. J Pharmacol Exp Ther 355(2):255–263. https://doi.org/10.1124/jpet.115.227298
Haviland MG, Morton KR, Oda K, Fraser GE (2010) Traumatic experiences, major life stressors, and self-reporting a physician-given fibromyalgia diagnosis. Psychiatry Res 177(3):335–341. https://doi.org/10.1016/j.psychres.2009.08.017
Van Houdenhove B, Egle U, Luyten P (2005) The role of life stress in fibromyalgia. Curr Rheumatol Rep 7(5):365–370
Helme RD, Littlejohn GO, Weinstein C (1987) Neurogenic flare responses in chronic rheumatic pain syndromes. Clin Exp Neurol 23:91–94
Clauw DJ, Arnold LM, McCarberg BH, FibroCollaborative (2011) The science of fibromyalgia. Mayo Clin Proc 86(9):907–911. https://doi.org/10.4065/mcp.2011.0206
Thiagarajah AT, Guymer E, Leech M, Littlejohn G (2014) The relationship between fibromyalgia, stress and depression. Int J Clin Rheum 9(4):371–384
Littlejohn G (2014) Fibromyalgia: honing fibromyalgia diagnosis. Nat Rev Rheumatol 10(5):267–269. https://doi.org/10.1038/nrrheum.2014.48
Wolfe F, Smythe HA, Yunus MB, Bennett RM, Bombardier C, Goldenberg DL, Tugwell P, Campbell SM, Abeles M, Clark P, Fam AG, Farber SJ, Fiechtner JJ, Michael Franklin C, Gatter RA, Hamaty D, Lessard J, Lichtbroun AS, Masi AT, Mccain GA, John Reynolds W, Romano TJ, Jon Russell I, Sheon RP (1990) The American College of Rheumatology 1990 criteria for the classification of fibromyalgia. Report of the multicenter criteria committee. Arthritis Rheum 33(2):160–172
Wolfe F, Clauw DJ, Fitzcharles MA, Goldenberg DL, Katz RS, Mease P, Russell AS, Russell IJ, Winfield JB, Yunus MB (2010) The American College of Rheumatology preliminary diagnostic criteria for fibromyalgia and measurement of symptom severity. Arthritis Care Res 62(5):600–610. https://doi.org/10.1002/acr.20140
Wolfe F, Clauw DJ, Fitzcharles MA, Goldenberg DL, Hauser W, Katz RS, Mease P, Russell AS, Russell IJ, Winfield JB (2011) Fibromyalgia criteria and severity scales for clinical and epidemiological studies: a modification of the ACR preliminary diagnostic criteria for fibromyalgia. J Rheumatol 38(6):1113–1122. https://doi.org/10.3899/jrheum.100594
Wolfe F, Clauw DJ, Fitzcharles MA, Goldenberg DL, Hauser W, Katz RL, Mease PJ, Russell AS, Russell IJ, Walitt B (2016) 2016 revisions to the 2010/2011 fibromyalgia diagnostic criteria. Semin Arthritis Rheum 46(3):319–329. https://doi.org/10.1016/j.semarthrit.2016.08.012
Yunus MB (2015) Editorial review: an update on central sensitivity syndromes and the issues of nosology and psychobiology. Curr Rheumatol Rev 11(2):70–85
Yunus MB (2007) Fibromyalgia and overlapping disorders: the unifying concept of central sensitivity syndromes. Semin Arthritis Rheum 36(6):339–356. https://doi.org/10.1016/j.semarthrit.2006.12.009
Napadow V, Kim J, Clauw DJ, Harris RE (2012) Decreased intrinsic brain connectivity is associated with reduced clinical pain in fibromyalgia. Arthritis Rheum 64(7):2398–2403. https://doi.org/10.1002/art.34412
Jensen KB, Loitoile R, Kosek E, Petzke F, Carville S, Fransson P, Marcus H, Williams SC, Choy E, Mainguy Y, Vitton O, Gracely RH, Gollub R, Ingvar M, Kong J (2012) Patients with fibromyalgia display less functional connectivity in the brain’s pain inhibitory network. Mol Pain 8:32. https://doi.org/10.1186/1744-8069-8-32
Harris RE, Sundgren PC, Craig AD, Kirshenbaum E, Sen A, Napadow V, Clauw DJ (2009) Elevated insular glutamate in fibromyalgia is associated with experimental pain. Arthritis Rheum 60(10):3146–3152. https://doi.org/10.1002/art.24849
Vaeroy H, Helle R, Forre O, Kass E, Terenius L (1988) Elevated CSF levels of substance P and high incidence of Raynaud phenomenon in patients with fibromyalgia: new features for diagnosis. Pain 32(1):21–26
Russell IJ, Orr MD, Littman B, Vipraio GA, Alboukrek D, Michalek JE, Lopez Y, MacKillip F (1994) Elevated cerebrospinal fluid levels of substance P in patients with the fibromyalgia syndrome. Arthritis Rheum 37(11):1593–1601
Giovengo SL, Russell IJ, Larson AA (1999) Increased concentrations of nerve growth factor in cerebrospinal fluid of patients with fibromyalgia. J Rheumatol 26(7):1564–1569
Sarchielli P, Mancini ML, Floridi A, Coppola F, Rossi C, Nardi K, Acciarresi M, Pini LA, Calabresi P (2007) Increased levels of neurotrophins are not specific for chronic migraine: evidence from primary fibromyalgia syndrome. J Pain 8(9):737–745. https://doi.org/10.1016/j.jpain.2007.05.002
Vaeroy H, Sakurada T, Forre O, Kass E, Terenius L (1989) Modulation of pain in fibromyalgia (fibrositis syndrome): cerebrospinal fluid (CSF) investigation of pain related neuropeptides with special reference to calcitonin gene related peptide (CGRP). J Rheumatol Suppl 19:94–97
Reynolds WJ, Chiu B, Inman RD (1988) Plasma substance P levels in fibrositis. J Rheumatol 15(12):1802–1803
Tsilioni I, Russell IJ, Stewart JM, Gleason RM, Theoharides TC (2016) Neuropeptides CRH, SP, HK-1, and inflammatory cytokines IL-6 and TNF are increased in serum of patients with fibromyalgia syndrome, implicating mast cells. J Pharmacol Exp Ther 356(3):664–672. https://doi.org/10.1124/jpet.115.230060
Kadetoff D, Lampa J, Westman M, Andersson M, Kosek E (2012) Evidence of central inflammation in fibromyalgia-increased cerebrospinal fluid interleukin-8 levels. J Neuroimmunol 242(1–2):33–38. https://doi.org/10.1016/j.jneuroim.2011.10.013
Kosek E, Martinsen S, Gerdle B, Mannerkorpi K, Lofgren M, Bileviciute-Ljungar I, Fransson P, Schalling M, Ingvar M, Ernberg M, Jensen KB (2016) The translocator protein gene is associated with symptom severity and cerebral pain processing in fibromyalgia. Brain Behav Immun 58:218–227. https://doi.org/10.1016/j.bbi.2016.07.150
Backryd E, Tanum L, Lind AL, Larsson A, Gordh T (2017) Evidence of both systemic inflammation and neuroinflammation in fibromyalgia patients, as assessed by a multiplex protein panel applied to the cerebrospinal fluid and to plasma. J Pain Res 10:515–525. https://doi.org/10.2147/JPR.S128508
Rea K, Dinan TG, Cryan JF (2016) The microbiome: a key regulator of stress and neuroinflammation. Neurobiol Stress 4:23–33. https://doi.org/10.1016/j.ynstr.2016.03.001
Lyon P, Cohen M, Quintner J (2011) An evolutionary stress-response hypothesis for chronic widespread pain (fibromyalgia syndrome). Pain Med 12(8):1167–1178. https://doi.org/10.1111/j.1526-4637.2011.01168.x
Geracioti TD Jr, Carpenter LL, Owens MJ, Baker DG, Ekhator NN, Horn PS, Strawn JR, Sanacora G, Kinkead B, Price LH, Nemeroff CB (2006) Elevated cerebrospinal fluid substance p concentrations in posttraumatic stress disorder and major depression. Am J Psychiatry 163(4):637–643. https://doi.org/10.1176/appi.ajp.163.4.637
Xanthos DN, Sandkuhler J (2014) Neurogenic neuroinflammation: inflammatory CNS reactions in response to neuronal activity. Nat Rev Neurosci 15(1):43–53. https://doi.org/10.1038/nrn3617
Garate I, Garcia-Bueno B, Madrigal JL, Bravo L, Berrocoso E, Caso JR, Mico JA, Leza JC (2011) Origin and consequences of brain toll-like receptor 4 pathway stimulation in an experimental model of depression. J Neuroinflammation 8:151. https://doi.org/10.1186/1742-2094-8-151
Rosenkranz MA, Davidson RJ, Maccoon DG, Sheridan JF, Kalin NH, Lutz A (2013) A comparison of mindfulness-based stress reduction and an active control in modulation of neurogenic inflammation. Brain Behav Immun 27(1):174–184. https://doi.org/10.1016/j.bbi.2012.10.013
Lentz MJ, Landis CA, Rothermel J, Shaver JL (1999) Effects of selective slow wave sleep disruption on musculoskeletal pain and fatigue in middle aged women. J Rheumatol 26(7):1586–1592
Zhu B, Dong Y, Xu Z, Gompf HS, Ward SA, Xue Z, Miao C, Zhang Y, Chamberlin NL, Xie Z (2012) Sleep disturbance induces neuroinflammation and impairment of learning and memory. Neurobiol Dis 48(3):348–355. https://doi.org/10.1016/j.nbd.2012.06.022
Malin K, Littlejohn GO (2016) Psychological factors mediate key symptoms of fibromyalgia through their influence on stress. Clin Rheumatol 35(9):2353–2357. https://doi.org/10.1007/s10067-016-3315-9
Milligan ED, Watkins LR (2009) Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci 10(1):23–36. https://doi.org/10.1038/nrn2533
Watkins LR, Maier SF (2005) Immune regulation of central nervous system functions: from sickness responses to pathological pain. J Intern Med 257(2):139–155. https://doi.org/10.1111/j.1365-2796.2004.01443.x
Desmeules JA, Cedraschi C, Rapiti E, Baumgartner E, Finckh A, Cohen P, Dayer P, Vischer TL (2003) Neurophysiologic evidence for a central sensitization in patients with fibromyalgia. Arthritis Rheum 48(5):1420–1429. https://doi.org/10.1002/art.10893
Cagnie B, Coppieters I, Denecker S, Six J, Danneels L, Meeus M (2014) Central sensitization in fibromyalgia? A systematic review on structural and functional brain MRI. Semin Arthritis Rheum 44(1):68–75. https://doi.org/10.1016/j.semarthrit.2014.01.001
Sluka KA, Clauw DJ (2016) Neurobiology of fibromyalgia and chronic widespread pain. Neuroscience 338:114–129. https://doi.org/10.1016/j.neuroscience.2016.06.006
Kosek E, Hansson P (1997) Modulatory influence on somatosensory perception from vibration and heterotopic noxious conditioning stimulation (HNCS) in fibromyalgia patients and healthy subjects. Pain 70(1):41–51
Julien N, Goffaux P, Arsenault P, Marchand S (2005) Widespread pain in fibromyalgia is related to a deficit of endogenous pain inhibition. Pain 114(1–2):295–302. https://doi.org/10.1016/j.pain.2004.12.032
Harris RE, Clauw DJ, Scott DJ, McLean SA, Gracely RH, Zubieta JK (2007) Decreased central mu-opioid receptor availability in fibromyalgia. J Neurosci 27(37):10000–10006. https://doi.org/10.1523/JNEUROSCI.2849-07.2007
Woolf CJ (2011) Central sensitization: implications for the diagnosis and treatment of pain. Pain 152(3 Suppl):S2–S15. https://doi.org/10.1016/j.pain.2010.09.030
Roche S, Gabelle A, Lehmann S (2008) Clinical proteomics of the cerebrospinal fluid: towards the discovery of new biomarkers. Proteomics Clin Appl 2(3):428–436. https://doi.org/10.1002/prca.200780040
Lewis T (1927) The blood vessels of the human skin and their responses. Shaw and Sons, London
Wallengren J, Moller H (1986) The effect of capsaicin on some experimental inflammations in human skin. Acta Derm Venereol 66(5):375–380
Holzer P (1998) Neurogenic vasodilatation and plasma leakage in the skin. Gen Pharmacol 30(1):5–11
Schmelz M, Michael K, Weidner C, Schmidt R, Torebjork HE, Handwerker HO (2000) Which nerve fibers mediate the axon reflex flare in human skin? Neuroreport 11(3):645–648
Chiu IM, von Hehn CA, Woolf CJ (2012) Neurogenic inflammation and the peripheral nervous system in host defense and immunopathology. Nat Neurosci 15(8):1063–1067. https://doi.org/10.1038/nn.3144
Littlejohn GO, Weinstein C, Helme RD (1987) Increased neurogenic inflammation in fibrositis syndrome. J Rheumatol 14(5):1022–1025
Yunus M, Masi AT, Calabro JJ, Miller KA, Feigenbaum SL (1981) Primary fibromyalgia (fibrositis): clinical study of 50 patients with matched normal controls. Semin Arthritis Rheum 11(1):151–171
Hauser W, Hayo S, Biewer W, Gesmann M, Kuhn-Becker H, Petzke F, von Wilmoswky H, Langhorst J (2010) Diagnosis of fibromyalgia syndrome—a comparison of Association of the Medical Scientific Societies in Germany, survey, and American College of Rheumatology criteria. Clin J Pain 26(6):505–511. https://doi.org/10.1097/AJP.0b013e3181d92a6c
Littlejohn G, Granges G (1995) The relationship between vertebral dysfunction and clinical features of fibromyalgia syndrome. J Orthopedic Rheum 8:97–105
Pay S, Calguneri M, Caliskaner Z, Dinc A, Apras S, Ertenli I, Kiraz S, Cobankara V (2000) Evaluation of vascular injury with proinflammatory cytokines, thrombomodulin and fibronectin in patients with primary fibromyalgia. Nagoya J Med Sci 63(3–4):115–122
Caro XJ (1984) Immunofluorescent detection of IgG at the dermal-epidermal junction in patients with apparent primary fibrositis syndrome. Arthritis Rheum 27(10):1174–1179
Birklein F, Schmelz M (2008) Neuropeptides, neurogenic inflammation and complex regional pain syndrome (CRPS). Neurosci Lett 437(3):199–202. https://doi.org/10.1016/j.neulet.2008.03.081
Blanco I, Beritze N, Arguelles M, Carcaba V, Fernandez F, Janciauskiene S, Oikonomopoulou K, de Serres FJ, Fernandez-Bustillo E, Hollenberg MD (2010) Abnormal overexpression of mastocytes in skin biopsies of fibromyalgia patients. Clin Rheumatol 29(12):1403–1412. https://doi.org/10.1007/s10067-010-1474-7
Enestrom S, Bengtson A, Lindstrom F, Johan K (1990) Attachment of IgG to dermal extracellular matrix in patients with fibromyalgia. Clin Exp Rheumatol 8(2):127–135
Enestrom S, Bengtsson A, Frodin T (1997) Dermal IgG deposits and increase of mast cells in patients with fibromyalgia—relevant findings or epiphenomena? Scand J Rheumatol 26(4):308–313
Caro XJ, Wolfe F, Johnston WH, Smith AL (1986) A controlled and blinded study of immunoreactant deposition at the dermal-epidermal junction of patients with primary fibrositis syndrome. J Rheumatol 13(6):1086–1092
Caro XJ (1986) Immunofluorescent studies of skin in primary fibrositis syndrome. Am J Med 81(3A):43–49
Rodriguez-Pinto I, Agmon-Levin N, Howard A, Shoenfeld Y (2014) Fibromyalgia and cytokines. Immunol Lett 161(2):200–203. https://doi.org/10.1016/j.imlet.2014.01.009
Generaal E, Vogelzangs N, Macfarlane GJ, Geenen R, Smit JH, Dekker J, Penninx BW (2014) Basal inflammation and innate immune response in chronic multisite musculoskeletal pain. Pain 155(8):1605–1612. https://doi.org/10.1016/j.pain.2014.05.007
Uceyler N, Hauser W, Sommer C (2011) Systematic review with meta-analysis: cytokines in fibromyalgia syndrome. BMC Musculoskelet Disord 12:245. https://doi.org/10.1186/1471-2474-12-245
Mendieta D, De la Cruz-Aguilera DL, Barrera-Villalpando MI, Becerril-Villanueva E, Arreola R, Hernandez-Ferreira E, Perez-Tapia SM, Perez-Sanchez G, Garces-Alvarez ME, Aguirre-Cruz L, Velasco-Velazquez MA, Pavon L (2016) IL-8 and IL-6 primarily mediate the inflammatory response in fibromyalgia patients. J Neuroimmunol 290:22–25. https://doi.org/10.1016/j.jneuroim.2015.11.011
Gerdle B, Ghafouri B, Ghafouri N, Backryd E, Gordh T (2017) Signs of ongoing inflammation in female patients with chronic widespread pain: a multivariate, explorative, cross-sectional study of blood samples. Medicine 96(9):e6130. https://doi.org/10.1097/MD.0000000000006130
Ranzolin A, Duarte AL, Bredemeier M, da Costa Neto CA, Ascoli BM, Wollenhaupt-Aguiar B, Kapczinski F, Xavier RM (2016) Evaluation of cytokines, oxidative stress markers and brain-derived neurotrophic factor in patients with fibromyalgia—a controlled cross-sectional study. Cytokine 84:25–28. https://doi.org/10.1016/j.cyto.2016.05.011
Littlejohn G (2015) Neurogenic neuroinflammation in fibromyalgia and complex regional pain syndrome. Nat Rev Rheumatol 11(11):639–648. https://doi.org/10.1038/nrrheum.2015.100
Behm FG, Gavin IM, Karpenko O, Lindgren V, Gaitonde S, Gashkoff PA, Gillis BS (2012) Unique immunologic patterns in fibromyalgia. BMC Clin Pathol 12:25. https://doi.org/10.1186/1472-6890-12-25
Christidis N, Ghafouri B, Larsson A, Palstam A, Mannerkorpi K, Bileviciute-Ljungar I, Lofgren M, Bjersing J, Kosek E, Gerdle B, Ernberg M (2015) Comparison of the levels of pro-inflammatory cytokines released in the vastus lateralis muscle of patients with fibromyalgia and healthy controls during contractions of the quadriceps muscle—a microdialysis study. PLoS One 10(12):e0143856. https://doi.org/10.1371/journal.pone.0143856
Heijmans-Antonissen C, Wesseldijk F, Munnikes RJ, Huygen FJ, van der Meijden P, Hop WC, Hooijkaas H, Zijlstra FJ (2006) Multiplex bead array assay for detection of 25 soluble cytokines in blister fluid of patients with complex regional pain syndrome type 1. Mediat Inflamm 2006(1):28398–28398. https://doi.org/10.1155/MI/2006/28398
Huygen FJ, De Bruijn AG, De Bruin MT, Groeneweg JG, Klein J, Zijlstra FJ (2002) Evidence for local inflammation in complex regional pain syndrome type 1. Mediat Inflamm 11(1):47–51. https://doi.org/10.1080/09629350210307
Munnikes RJ, Muis C, Boersma M, Heijmans-Antonissen C, Zijlstra FJ, Huygen FJ (2005) Intermediate stage complex regional pain syndrome type 1 is unrelated to proinflammatory cytokines. Mediat Inflamm 2005(6):366–372. https://doi.org/10.1155/MI.2005.366
Martinez-Martinez LA, Mora T, Vargas A, Fuentes-Iniestra M, Martinez-Lavin M (2014) Sympathetic nervous system dysfunction in fibromyalgia, chronic fatigue syndrome, irritable bowel syndrome, and interstitial cystitis: a review of case-control studies. J Clin Rheumatol 20(3):146–150. https://doi.org/10.1097/RHU.0000000000000089
Lerma C, Martinez A, Ruiz N, Vargas A, Infante O, Martinez-Lavin M (2011) Nocturnal heart rate variability parameters as potential fibromyalgia biomarker: correlation with symptoms severity. Arthritis Res Ther 13(6):R185. https://doi.org/10.1186/ar3513
Lerma C, Martinez-Martinez LA, Ruiz N, Vargas A, Infante O, Martinez-Lavin M (2016) Fibromyalgia beyond reductionism. Heart rhythm fractal analysis to assess autonomic nervous system resilience. Scand J Rheumatol 45(2):151–157. https://doi.org/10.3109/03009742.2015.1055299
Dawson LF, Phillips JK, Finch PM, Inglis JJ, Drummond PD (2011) Expression of alpha1-adrenoceptors on peripheral nociceptive neurons. Neuroscience 175:300–314. https://doi.org/10.1016/j.neuroscience.2010.11.064
Maestroni GJ (2006) Sympathetic nervous system influence on the innate immune response. Ann N Y Acad Sci 1069:195–207. https://doi.org/10.1196/annals.1351.017
Kosek E, Altawil R, Kadetoff D, Finn A, Westman M, Le Maitre E, Andersson M, Jensen-Urstad M, Lampa J (2015) Evidence of different mediators of central inflammation in dysfunctional and inflammatory pain—interleukin-8 in fibromyalgia and interleukin-1 beta in rheumatoid arthritis. J Neuroimmunol 280:49–55. https://doi.org/10.1016/j.jneuroim.2015.02.002
Uceyler N, Zeller D, Kahn AK, Kewenig S, Kittel-Schneider S, Schmid A, Casanova-Molla J, Reiners K, Sommer C (2013) Small fibre pathology in patients with fibromyalgia syndrome. Brain 136(Pt 6):1857–1867. https://doi.org/10.1093/brain/awt053
de Tommaso M, Nolano M, Iannone F, Vecchio E, Ricci K, Lorenzo M, Delussi M, Girolamo F, Lavolpe V, Provitera V, Stancanelli A, Lapadula G, Livrea P (2014) Update on laser-evoked potential findings in fibromyalgia patients in light of clinical and skin biopsy features. J Neurol 261(3):461–472. https://doi.org/10.1007/s00415-013-7211-9
Giannoccaro MP, Donadio V, Incensi A, Avoni P, Liguori R (2014) Small nerve fiber involvement in patients referred for fibromyalgia. Muscle Nerve 49(5):757–759. https://doi.org/10.1002/mus.24156
Kosmidis ML, Koutsogeorgopoulou L, Alexopoulos H, Mamali I, Vlachoyiannopoulos PG, Voulgarelis M, Moutsopoulos HM, Tzioufas AG, Dalakas MC (2014) Reduction of Intraepidermal nerve fiber density (IENFD) in the skin biopsies of patients with fibromyalgia: a controlled study. J Neurol Sci 347(1–2):143–147. https://doi.org/10.1016/j.jns.2014.09.035
Oaklander AL, Herzog ZD, Downs HM, Klein MM (2013) Objective evidence that small-fiber polyneuropathy underlies some illnesses currently labeled as fibromyalgia. Pain 154(11):2310–2316. https://doi.org/10.1016/j.pain.2013.06.001
Caro XJ, Winter EF (2015) The role and importance of small fiber neuropathy in fibromyalgia pain. Curr Pain Headache Rep 19(12):55. https://doi.org/10.1007/s11916-015-0527-7
Doppler K, Rittner HL, Deckart M, Sommer C (2015) Reduced dermal nerve fiber diameter in skin biopsies of patients with fibromyalgia. Pain 156(11):2319–2325. https://doi.org/10.1097/j.pain.0000000000000285
Caro XJ, Winter EF (2014) Evidence of abnormal epidermal nerve fiber density in fibromyalgia: clinical and immunologic implications. Arthritis Rheumatol 66(7):1945–1954. https://doi.org/10.1002/art.38662
Leinders M, Doppler K, Klein T, Deckart M, Rittner H, Sommer C, Uceyler N (2016) Increased cutaneous miR-let-7d expression correlates with small nerve fiber pathology in patients with fibromyalgia syndrome. Pain 157(11):2493–2503. https://doi.org/10.1097/j.pain.0000000000000668
Serra J, Collado A, Sola R, Antonelli F, Torres X, Salgueiro M, Quiles C, Bostock H (2014) Hyperexcitable C nociceptors in fibromyalgia. Ann Neurol 75(2):196–208. https://doi.org/10.1002/ana.24065
Kim SH, Kim DH, Oh DH, Clauw DJ (2008) Characteristic electron microscopic findings in the skin of patients with fibromyalgia: preliminary study. Clin Rheumatol 27(2):219–223. https://doi.org/10.1007/s10067-007-0739-2
Ramirez M, Martinez-Martinez LA, Hernandez-Quintela E, Velazco-Casapia J, Vargas A, Martinez-Lavin M (2015) Small fiber neuropathy in women with fibromyalgia. An in vivo assessment using corneal confocal bio-microscopy. Semin Arthritis Rheum 45(2):214–219. https://doi.org/10.1016/j.semarthrit.2015.03.003
Watson NF, Buchwald D, Goldberg J, Noonan C, Ellenbogen RG (2009) Neurologic signs and symptoms in fibromyalgia. Arthritis Rheum 60(9):2839–2844. https://doi.org/10.1002/art.24772
Harte S, Clauw D, Hayes JM, Feldman EL, St Charles IC, Watson CJ (2017) Reduced intraepidermal nerve fiber density after a sustained increase in insular glutamate: a proof-of-concept study examining the pathogenesis of small fiber pathology in fibromyalgia. Pain Rep 2(e590):1–6
Staud R, Nagel S, Robinson ME, Price DD (2009) Enhanced central pain processing of fibromyalgia patients is maintained by muscle afferent input: a randomized, double-blind, placebo-controlled study. Pain 145(1–2):96–104. https://doi.org/10.1016/j.pain.2009.05.020
Baron R, Hans G, Dickenson AH (2013) Peripheral input and its importance for central sensitization. Ann Neurol 74(5):630–636. https://doi.org/10.1002/ana.24017
Grover S, Srivastava A, Lee R, Tewari AK, Te AE (2011) Role of inflammation in bladder function and interstitial cystitis. Ther Adv Urol 3(1):19–33. https://doi.org/10.1177/1756287211398255
Malhotra R (2016) Understanding migraine: potential role of neurogenic inflammation. Ann Indian Acad Neurol 19(2):175–182. https://doi.org/10.4103/0972-2327.182302
Feng B, La JH, Schwartz ES, Gebhart GF (2012) Irritable bowel syndrome: methods, mechanisms, and pathophysiology. Neural and neuro-immune mechanisms of visceral hypersensitivity in irritable bowel syndrome. Am J Physiol Gastrointest Liver Physiol 302(10):G1085–G1098. https://doi.org/10.1152/ajpgi.00542.2011
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is a contribution to the special issue on Neurogenic Inflammation - Guest Editors: Tony Yaksh and Anna Di Nardo
Rights and permissions
About this article

Cite this article
Littlejohn, G., Guymer, E. Neurogenic inflammation in fibromyalgia. Semin Immunopathol 40, 291–300 (2018). https://doi.org/10.1007/s00281-018-0672-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-018-0672-2