
Abstract
Deficits in immunological tolerance against self-antigens and antigens provided by the diet and commensal microbiota can result in the development of inflammatory and autoimmune disorders. Dendritic cells (DCs) are pivotal regulators of the immune response, specialized in antigen presentation to drive T cell priming and differentiation. DCs also have a tolerogenic function, participating in the enforcement of central and peripheral tolerance and the resolution of ongoing immune responses. Thus, DCs control effector and regulatory mechanisms relevant to the pathology of autoimmune disorders. In this review, we discuss recent findings regarding the control of the adaptive immune response by tolerogenic DCs. A thorough understanding of the mechanisms that control the tolerogenic DC phenotype will guide the development of novel strategies for the treatment of autoimmunity.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Tolerance is a dynamic and active process through which innate and adaptive immune cells limit immune system activation and tissue damage. Regulatory T cells (Tregs) are crucial in the maintenance of peripheral tolerance in mice and humans [1]. Several Treg populations have been described; the best characterized are those defined by the expression of the transcription factor FoxP3, which can be classified in natural and induced Treg cells [2–4]. Natural Tregs (nTregs) develop in the thymus, but Tregs are also differentiated in the periphery (pTregs) in response to specific factors such as transforming growth factor beta (TGFβ), kynurenine, and retinoic acid (RA) [2, 4–6]. Additional regulatory T cell populations have been described as well, for example, FoxP3-negative interleukin (IL)-10-producing type 1 regulatory T cells (Tr1 cells) [7–12].
Dendritic cells (DCs) promote immune tolerance by participating in the negative selection of autoreactive T cells in the thymus during central tolerance [13–15]. DCs also limit effector T cells and promote Treg differentiation in the periphery through various mechanisms including the production of cytokines such as IL-10, IL-27, and TGFβ; the expression of indoleamine 2,3-dioxygenase (IDO); and the control of the extracellular levels of adenosine triphosphate (ATP) and adenosine [16–20]. In this review, we discuss tolerogenic DC phenotypes and the molecular pathways that control their development and maintenance.
Overview on DCs
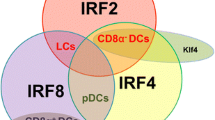
DCs constitute a heterogeneous cell population that plays a pivotal role in linking the innate and adaptive immune response. Based on their morphological features and function, DCs can be classified in classical DCs (cDCs) and plasmocytoid DCs (pDCs). Both cDCs and pDCs are derived from the common DC progenitors (CDPs) in an Flt3-dependent manner [21, 22], but the differentiation of each DC subset is driven by different transcription factors (Fig. 1 ).

Dendritic cell subsets in lymphatic organs. Two major classes of DCs have been identified based on their morphological and functional features: classical DCs (cDCs) and plasmacytoid DCs (pDCs). cDCs are also classified in two types based on the expression of CD8 and CD11b. All subsets of DCs are derived from a common dendritic cell precursor (CDP) in a process controlled by lineage-specific transcriptional programs
pDCs are a small subset of DCs characterized by a spherical shape that resembles plasma cells and are present mainly in the blood and lymphoid tissues. In the steady state, pDCs express low levels of major histocompatibility complex class II (MHC-II) and co-stimulatory molecules and low levels of the integrin CD11c. pDCs can efficiently prime CD4+ T cell responses and cross-prime CD8+ T cell responses and are characterized by the production of high amounts of type I interferon (IFN) upon recognition of foreign nucleic acids [23].
cDCs are highly phagocytic cells with a dendritic morphology that express high levels of MHC-II molecules and are endowed with potent antigen-presenting cell (APC) function. So far, cDCs have been classified in at least two major subsets characterized by the expression of either CD8α and CD103 or CD11b [24]. CD8α+ cDCs efficiently present exogenous antigens to CD8+ T cells; these cells also require Id2 and the basic leucine zipper transcription factor, ATF-like 3 (Batf3) for their differentiation [25, 26], but a Batf3-independent pathway has also been described for their generation [27]. CD11b+ cDCs preferentially activate CD4+ T cells and require Kruppel-like factor 4 (KLF4) for their differentiation [28]. Moreover, DCs are also present in nonlymphoid tissues, where cDCs represent 1–5 % of tissue cells depending on the organ and consist of two major subsets, CD103+ CD11b− and CD11b+ cDCs, in which CD103+ CD11b− cDCs share their origin and function with lymphoid tissue CD8+ cDCs, and CD11b+ cDCs consist of a mixture of tissue cDCs and macrophages [24].
DCs sense environmental signals through specific surface and intracellular pattern recognition receptors (PRRs). Many classes of PRRs have been identified, including Toll-like receptors (TLRs), NOD-like receptors (NLRs), and the RNA helicase RIG-I-like receptor (RIG), which trigger different signaling cascades [29]. PRRs recognize molecules produced by microorganisms. However, endogenous molecules released from damaged tissues or dead cells can also activate specific PRRs [30], suggesting that DC activation in the context of chronic autoimmune disorders can be initiated by infections and perpetuated by the release of self-molecules from damaged tissue even after the initial infection has been cleared.
During inflammation and infection, DCs are activated by PRR signaling, and then they migrate into T cell areas in lymphoid organs to present pathogen-derived antigens to antigen-specific T cells. In addition to antigen presentation via MHC molecules and the provision of co-stimulatory molecules, DCs produce cytokines that polarize T cells into effector and regulatory subtypes. Different cytokines are produced by DCs in response to diverse stimuli [31, 32], resulting in the generation of varied effector and regulatory T cell responses against self- and nonself-antigens.
Tolerogenic DCs: cell lineage or maturation status?
Pioneering studies on the role of DCs in peripheral tolerance suggested that the regulatory function of DCs is determined by their activation status. For example, the delivery of the antigen HEL to DCs in vivo using an antibody reactive against the DC surface antigen DEC-205 results induces transient antigen-specific T cell activation followed by T cell deletion and unresponsiveness [33]. Similar effects were observed when the same experiment was performed using fusions of autoantigens and DEC-205 reactive antibodies to ameliorate experimental autoimmunity [34–38]. However, these antigen-specific tolerogenic effects of DC-targeted antigen administration were abrogated when the mice were co-treated with an anti-CD40 antibody that induces DC activation [33]. Thus, the induction of self-tolerance or immunity is controlled by the activation status of the DCs.
As discussed below, increasing evidence suggests that mature DCs can limit effector T cell responses and promote immune tolerance in response to signaling triggered by IL-27, IL-10, vitamin A, or ligands of the aryl hydrocarbon receptor (AhR) [19, 39, 40]. Indeed, some DC subpopulations limit the development of experimental autoimmunity [41] and metabolic disorders [42]. It is still unclear, however, whether specific tolerogenic DC lineages exist in vivo or whether the tolerogenic phenotype of DCs reflects their activation status.
Perforin-expressing DCs
In vitro experiments identified a subset of conventional CD11c+ DCs expressing perforin (perf-DCs) that enforces peripheral tolerance by deleting T cells [43]. The role of perf-DCs in limiting inflammation in vivo was recently demonstrated using chimeric mice in which perforin expression was selectively impaired in CD11c+ DCs. Chimeric mice lacking perf-DCs show increased weight gain and exhibit features of metabolic syndrome, concomitantly with an oligoclonal expansion of T cells in adipose tissue. Perforin deficiency in DCs also enhances susceptibility to experimental autoimmune encephalomyelitis (EAE) and is associated with higher frequencies of Th1 and Th17 effector cells in the central nervous system [42]. Collectively, these data identify perf-DCs as an important population of tolerogenic DCs that limit autoreactive T cells in vivo through a perforin-mediated mechanism. However, several questions remain with regards to perf-DC biology in health and disease [44]. What is the role of perf-DCs in human inflammatory disorders and tumors? Do perf-DCs represent a specific lineage associated with a unique transcriptional program, or do they simply represent an activation stage of DCs?
CD103+ DCs
Another subset of cDCs with steady-state tolerogenic properties is the population of CD103+ DCs present into the intestinal mucosa, where they play a central role in enforcing tolerance to commensal bacteria and food antigens [45]. The CD103+ DC subset is derived from the common DC precursor, and its development depends on the growth factor Flt3 ligand [46]. Lamina propria CD103+ DCs induce the expression of gut-homing receptors CCR9 and α4β7 on T cells and induce FoxP3+ pTreg cell differentiation through a mechanism mediated by RA, a metabolite of vitamin A [5, 47–49]. In vitro and in vivo studies demonstrated that TGFβ and RA produced by CD103+ DCs in the lamina propria, but not in the spleen, mediate the differentiation of FoxP3 pTreg cells, which is abrogated by treatment with a retinoic acid receptor (RAR) antagonist [5, 49].
The mucosal administration of antigens has been shown to induce the differentiation of FoxP3+ pTreg cells and suppress pathological inflammatory responses [50]. Mucida and co-workers recently showed that monocyte-derived APCs are dispensable, while cDCs are critical, for the induction of peripheral FoxP3+ CD4+ Treg cells and tolerance by oral antigen administration [51]. Indeed, although the establishment of oral tolerance involves several subsets of cDCs in the mesenteric lymph nodes, there is a hierarchy of cDC subsets that drive the differentiation of FoxP3+ Treg cells, which is associated at least in part with the expression of genes related to the TGFβ and RA pathways. In particular, the mesenteric lymph node CD11b− CD103+ DCs were the most efficient inducers of FoxP3+ Tregs, followed by CD8α+ DCs, CD11b+ CD103+ DCs, and CD11b+ CD103− DCs [51].
CD103+ DCs in the gut also participate in intestinal homeostasis in a T cell-independent manner. Using a conditional knockout approach that allowed the deletion of specific subsets of CD103+ DC in a model of experimental colitis, Muzaki et al. demonstrated that intestinal CD103+ CD11b− DCs, the migratory subset of DC, are the major regulators of intestinal homeostasis by controlling the production of IFN-γ-induced anti-inflammatory proteins in intestinal epithelial cells (IECs) such as IDO1 and IL-18bp [52].
CD103+ DCs are crucial for gut homeostasis, but the mechanisms involved in their acquisition of a tolerogenic phenotype are not yet fully understood. Some studies have shown that factors present in the lamina propria contribute to the establishment of the regulatory function in CD103+ DCs, including luminal bacteria, dietary constituents, IECs, other leukocytes, stromal cells, and neuroendocrine factors. Among the most studied of these factors are vitamin A and its metabolite RA, which participates in the regulation of the Th17/FoxP3 Treg cell ratio by CD103+ lamina propria DCs by boosting FoxP3+ Treg cell differentiation [5] and inducing the expression of gut-homing receptors on T cells [48]. RA also controls the homeostasis of the migratory CD11b+ CD103+ DC subset within the gut [53].
Another nutrient provided by diet that determines the differentiation of tolerogenic DCs is the essential amino acid tryptophan (trp). Dietary trp is metabolized into agonists for the AhR through a series of cooperative biochemical reactions catalyzed by enzymes provided by gut commensal bacteria and the host [40, 54]. Tryptophan-derived AhR ligands induce the production of anti-inflammatory IL-10 and IL-27 by DCs, cytokines that favor the generation of FoxP3+ and Tr1 regulatory cells [55].
Finally, the gut mucosa is densely innervated and therefore exposed to the local release of neurotransmitters. For example, norepinephrine regulates some aspects of DC function, mainly inhibiting the production of pro-inflammatory cytokines and boosting IL-10 production [56–58]. Collectively, these data suggest that metabolites provided by the diet and the gut flora act in concert with endogenous signals to regulate the ability of DCs to control T cell responses and tissue homeostasis.
Induction of tolerogenic DCs by cytokines
Both in mice and humans, IL-10 and IL-27 are linked to the induction of CD4+ T cells with regulatory function [7, 9, 10]. However, accumulating evidence shows that these cytokines also have a role in promoting DC tolerogenic function [19, 59].
We showed that IL-27 signaling in DCs limits the differentiation of effector T cells and the development of EAE, while expanding FoxP3+ Tregs and Tr1 cells [19]. Through detailed transcriptional and epigenetic analyses, we found that IL-27 promotes the expression of the ectonucleotidase CD39 in DCs by a mechanism mediated by STAT3 [19]. CD39 catalyzes the degradation of immunostimulatory extracellular ATP released in the context of immune activation [60]. Indeed, we found that IL-27-induced CD39 in DCs limits the differentiation of Th1 and Th17 cells by depleting extracellular ATP, consequently reducing P2rX7-dependent NLRP3 activation and the production of IL-1β and IL-18 (19). Interestingly, we recently found that CD39 expression is also induced by IL-27 in Tr1 cells where it participates in their suppressive function [11], suggesting that the IL-27/CD39 axis is co-opted by several immune cells to regulate ongoing immune responses.
IL-10 is an anti-inflammatory cytokine that impairs the activation of DCs and the production of pro-inflammatory cytokines by immune cells [39]. Indeed, IL-10 inhibits the production of IL-12 and the expression of co-stimulatory molecules by various types of DC, which correlates with its ability to inhibit primary alloantigen-specific T cell responses [61, 62]. IL-10-treated DCs drive the induction of anergy in alloantigen- and peptide-activated T cells [59, 63, 64].
Interestingly, both IL-10 and IL-27 exert their immunosuppressive functions through signaling mediated by STAT3. Indeed, STAT3 deficiency restricted to DCs results in the spontaneous development of inflammation, suggesting that cytokines that activate STAT3 signaling induce a tolerogenic phenotype in DCs under homeostatic conditions and that STAT3-controlled DCs have an important role in enforcing immune tolerance in health and disease [65]. Altogether, these data suggest that cytokines signaling via STAT3 induce a tolerogenic phenotype in DCs.
Induction of tolerogenic DCs by AhR activation
AhR is a ligand-activated transcription factor activated by a broad array of synthetic and natural agonists [66]. The inactive AhR is located in the cytoplasm as part of a protein complex that includes the 90-kDa heat shock protein (HSP90), the proto-oncogene tyrosine-protein kinase SRC (c-SRC) protein kinase, and the AhR-interacting protein Ara9 [67]. After activation by its agonists, AhR forms a complex with the AhR nuclear translocator protein (ARNT) and translocates to the nucleus, where the AhR/ARNT complex establish protein–protein interactions with additional co-activators and transcription factors [67]. These AhR-containing protein complexes interact with specific sequences in target genes to control their transcriptional activity in what is known as the “genomic pathway of AhR signaling” [68]. Of note, several AhR interactions with other proteins are only triggered by specific AhR ligands, suggesting that some transcriptional partners of AhR are recruited in a ligand-specific manner [69–71]. Thus, it is possible that different AhR ligands trigger the formation of specific protein complexes that differ in their composition, leading to the recognition of noncanonical DNA sequences [72].
In addition, AhR also regulates cellular responses through nongenomic mechanisms. When AhR is activated by its ligands, c-SRC is released from the AhR/HSP90/c-SRC complex and binds to multiple cellular targets to control their activity through phosphorylation-dependent mechanisms [73]. Moreover, it has been reported that AhR is a ligand-dependent E3 ubiquitin ligase, involved in the degradation of AhR-interacting proteins through the proteasome [74]. However, the relevance of these pathways for the immunoregulatory roles of AhR is still unknown.
AhR was initially characterized as the receptor for dioxins, but the immune and liver defects observed in AhR-deficient mice suggest that natural AhR ligands play a role in normal physiology [75, 76]. As we already mentioned, the diet is an important source of AhR ligands, and flavonoids represent the largest group of naturally occurring dietary AhR ligands [77, 78]. However, the AhR ligands that have been more deeply studied with regards to their effects on the immune response are those derived from tryptophan, such as 6-formylindolo[3,2-b]carbazole (FICZ), 2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE), and l-kynurenine [79–81]. l-Kynurenine, for example, is generated by the enzymatic degradation of tryptophan through a pathway controlled by the rate-limiting enzymes IDO or tryptophan 2,3-dioxygenase (TDO) [81]. IDO expression is induced in various cell types, mainly in DCs under different stimuli as IFN-γ, TNF-α, IL-1β, and IL-6 [82].
AhR has been shown to regulate DC activity in experimental models, but the molecular mechanisms involved are mostly unknown [11, 83–85]. AhR is known to interact with and regulate the activity and degradation of transcription factors important for the control of DC function, such as nuclear factor κB (NF-κB) and AP-1 [86, 87]. Several mechanisms mediate the control of NF-κB activation by AhR, including the regulation of NF-κB expression and direct NF-κB/AhR protein interactions [86, 88–91]. We recently showed that AhR induces Socs2 expression in DCs, interfering with NF-κB activation by TRAF6 and therefore limiting the production of pro-inflammatory IL-6 and IL-12 by DCs [85]. Thus, through its effects on NF-κB and other pathways, AhR activation inhibits DC activation, decreasing the expression of MHC-II and co-stimulatory molecules and the production of pro-inflammatory cytokines that drive Th1 and Th17 polarization [55, 83, 92–94].
Concomitantly, AhR activation boosts the ability of DCs to promote the differentiation and expansion of FoxP3+ Tregs through the production of kynurenins and RA [55, 94, 95]. Indeed, AhR activation upregulates the expression of IDO in DCs, which catalyzes the production of kynurenine. Strikingly, kynurenine activates AhR in T cells, promoting FoxP3+ Treg differentiation through different molecular mechanisms [94–97]. In addition, AhR signaling in DCs upregulates the expression of the enzymatic machinery that controls the production of RA, a metabolite that promotes the differentiation of FoxP3+ Treg cells [10]. Indeed, the differentiation of FoxP3+ Tregs by DCs pretreated with the AhR ligand ITE can be significantly inhibited with inhibitors of RA signaling [10].
Altogether, these data demonstrate that AhR activation induces tolerogenic properties in DCs, which limit effector responses and promote the generation and expansion of Tregs. These findings suggest that targeting AhR in DCs may provide a therapeutic avenue to induce antigen-specific tolerance in immune-mediated disorders. We have recently tested this approach using two models of T cell-dependent autoimmunity.
We first used gold nanoparticles (NPs) to co-deliver the tolerogenic AhR agonist ITE and myelin autoantigen to DCs. We found that these NPs induce a tolerogenic phenotype in DCs that limits effector T cell differentiation and promotes the differentiation of Tregs in vitro and in vivo (Fig. 2). Indeed, these tolerogenic NPs suppress EAE development in preventive and therapeutic paradigms [83]. More recently, we constructed NPs loaded with ITE and β cell antigens (recombinant pro-insulin or relevant peptides) to modulate the T cell response that drives the immune destruction of insulin-producing β cells in type 1 diabetes (T1D). When evaluated in spontaneous and cyclophosphamide-accelerated T1D models in NOD mice, NPs arrested T1D development even when administered after the onset of insulitis [85]. The anti-diabetogenic effects of NPs were linked to the induction of tolerogenic DCs through a mechanism mediated by Socs2. Interestingly, these tolerogenic NPs were also able to induce a tolerogenic phenotype in human DCs, limiting their ability to activate β cell-reactive T cells from T1D patients [85]. Collectively, these data suggest that NPs engineered to activate AhR and induce tolerogenic DCs while delivering relevant immune targets may provide a new method for the re-establishment of antigen-specific tolerance in human autoimmune disorders.

AhR induces tolerogenic phenotype in DCs. Nanoparticles (NPs) engineered to co-deliver the non-toxic tolerogenic AHR ligand ITE and a specific antigen to DCs. Activation of AHR in DCs induces a tolerogenic phenotype that promotes the generation of antigen-specific FoxP3+ Treg cells (Treg) and type-1 regulatory T cells (Tr1) that suppress Th1 and Th17 effector T cells. Moreover, AHR-targeting NPs interfere with the differentiation of effector Th1 and Th17 cells by DCs by modulating the production of polarizing cytokines
Conclusions
DCs with a tolerogenic phenotype are an important component of the immune system and regulate the immune response in health and disease. It is yet unclear, however, whether tolerogenic DCs constitute a specific lineage or simply reflect a particular activation status of DCs. Indeed, these options are not mutually exclusive and this issue should be the focus of future investigations. Nevertheless, it is expectable that the tolerogenic phenotype is controlled by specific signaling and transcriptional programs, such as those controlled by STAT3, AhR, and Socs2. The identification of these molecular programs will delineate the cytokines and metabolites that control the induction and stability of the tolerogenic DC phenotype in health and disease, as well as their modulation by environmental factors. In addition, the identification of the mechanisms responsible for the tolerogenic DC phenotype will guide new therapeutic strategies for immune-mediated diseases.
References
Josefowicz SZ, Lu LF, Rudensky AY (2012) Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol 30:531–564
Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M (2011) Pillars article: immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995. J Immunol 186:3808–3821
Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY, Regulatory T (2005) Cell lineage specification by the forkhead transcription factor foxp3. Immunity 22:329–341
Mucida D, Pino-Lagos K, Kim G, Nowak E, Benson MJ, Kronenberg M, Noelle RJ, Cheroutre H (2009) Retinoic acid can directly promote TGF-beta-mediated Foxp3(+) Treg cell conversion of naive T cells. Immunity 30:471–472 author reply 472-473
Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, Cheroutre H (2007) Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 317:256–260
Curti A, Pandolfi S, Valzasina B, Aluigi M, Isidori A, Ferri E, Salvestrini V, Bonanno G, Rutella S, Durelli I, Horenstein AL, Fiore F, Massaia M, Colombo MP, Baccarani M, Lemoli RM (2007) Modulation of tryptophan catabolism by human leukemic cells results in the conversion of CD25- into CD25+ T regulatory cells. Blood 109:2871–2877
Roncarolo MG, Bacchetta R, Bordignon C, Narula S, Levings MK (2001) Type 1 T regulatory cells. Immunol Rev 182:68–79
Groux H, O’Garra A, Bigler M, Rouleau M, Antonenko S, de Vries JE, Roncarolo MG (1997) A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature 389:737–742
Apetoh L, Quintana FJ, Pot C, Joller N, Xiao S, Kumar D, Burns EJ, Sherr DH, Weiner HL, Kuchroo VK (2010) The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat Immunol 11:854–861
Gandhi R, Kumar D, Burns EJ, Nadeau M, Dake B, Laroni A, Kozoriz D, Weiner HL, Quintana FJ (2010) Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3(+) regulatory T cells. Nat Immunol 11:846–853
Mascanfroni ID, Takenaka MC, Yeste A, Patel B, Wu Y, Kenison JE, Siddiqui S, Basso AS, Otterbein LE, Pardoll DM, Pan F, Priel A, Clish CB, Robson SC, Quintana FJ (2015) Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-alpha. Nat Med 21:638–646
Farez MF, Mascanfroni ID, Mendez-Huergo SP, Yeste A, Murugaiyan G, Garo LP, Aguirre MEB, Patel B, Ysrraelit MC, Zhu C, Kuchroo VK, Rabinovich GA, Quintana FJ, Correale J (2015) Melatonin contributes to the seasonality of multiple sclerosis relapses. Cell 162:1338–1352
Bonasio R, Scimone ML, Schaerli P, Grabie N, Lichtman AH, von Andrian UH (2006) Clonal deletion of thymocytes by circulating dendritic cells homing to the thymus. Nat Immunol 7:1092–1100
Ohnmacht C, Pullner A, King SB, Drexler I, Meier S, Brocker T, Voehringer D (2009) Constitutive ablation of dendritic cells breaks self-tolerance of CD4 T cells and results in spontaneous fatal autoimmunity. J Exp Med 206:549–559
Quintana FJ, Yeste A, Mascanfroni ID (2015) Role and therapeutic value of dendritic cells in central nervous system autoimmunity. Cell Death Differ 22:215–224
Akbari O, DeKruyff RH, Umetsu DT (2001) Pulmonary dendritic cells producing IL-10 mediate tolerance induced by respiratory exposure to antigen. Nat Immunol 2:725–731
Munn DH, Sharma MD, Lee JR, Jhaver KG, Johnson TS, Keskin DB, Marshall B, Chandler P, Antonia SJ, Burgess R, Slingluff CL Jr, Mellor AL (2002) Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science 297:1867–1870
Travis MA, Reizis B, Melton AC, Masteller E, Tang Q, Proctor JM, Wang Y, Bernstein X, Huang X, Reichardt LF, Bluestone JA, Sheppard D (2007) Loss of integrin alpha(v)beta8 on dendritic cells causes autoimmunity and colitis in mice. Nature 449:361–365
Mascanfroni ID, Yeste A, Vieira SM, Burns EJ, Patel B, Sloma I, Wu Y, Mayo L, Ben-Hamo R, Efroni S, Kuchroo VK, Robson SC, Quintana FJ (2013) IL-27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nat Immunol 14:1054–1063
Mahnke K, Qian Y, Knop J, Enk AH (2003) Induction of CD4+/CD25+ regulatory T cells by targeting of antigens to immature dendritic cells. Blood 101:4862–4869
Onai N, Obata-Onai A, Schmid MA, Ohteki T, Jarrossay D, Manz MG (2007) Identification of clonogenic common Flt3+M-CSFR+ plasmacytoid and conventional dendritic cell progenitors in mouse bone marrow. Nat Immunol 8:1207–1216
Naik SH, Sathe P, Park HY, Metcalf D, Proietto AI, Dakic A, Carotta S, O’Keeffe M, Bahlo M, Papenfuss A, Kwak JY, Wu L, Shortman K (2007) Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat Immunol 8:1217–1226
Reizis B, Bunin A, Ghosh HS, Lewis KL, Sisirak V (2011) Plasmacytoid dendritic cells: recent progress and open questions. Annu Rev Immunol 29:163–183
Merad M, Sathe P, Helft J, Miller J, Mortha A (2013) The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol 31:563–604
Hacker C, Kirsch RD, Ju XS, Hieronymus T, Gust TC, Kuhl C, Jorgas T, Kurz SM, Rose-John S, Yokota Y, Zenke M (2003) Transcriptional profiling identifies Id2 function in dendritic cell development. Nat Immunol 4:380–386
Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, Schreiber RD, Murphy TL, Murphy KM (2008) Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322:1097–1100
Tussiwand R, Lee WL, Murphy TL, Mashayekhi M, KC W, Albring JC, Satpathy AT, Rotondo JA, Edelson BT, Kretzer NM, Wu X, Weiss LA, Glasmacher E, Li P, Liao W, Behnke M, Lam SS, Aurthur CT, Leonard WJ, Singh H, Stallings CL, Sibley LD, Schreiber RD, Murphy KM (2012) Compensatory dendritic cell development mediated by BATF-IRF interactions. Nature 490:502–507
Park CS, Lee PH, Yamada T, Burns A, Shen Y, Puppi M, Lacorazza HD (2012) Kruppel-like factor 4 (KLF4) promotes the survival of natural killer cells and maintains the number of conventional dendritic cells in the spleen. J Leukoc Biol 91:739–750
Akira S, Uematsu S, Takeuchi O (2006) Pathogen recognition and innate immunity. Cell 124:783–801
Yu L, Wang L, Chen S (2010) Endogenous toll-like receptor ligands and their biological significance. J Cell Mol Med 14:2592–2603
Negishi H, Yanai H, Nakajima A, Koshiba R, Atarashi K, Matsuda A, Matsuki K, Miki S, Doi T, Aderem A, Nishio J, Smale ST, Honda K, Taniguchi T (2012) Cross-interference of RLR and TLR signaling pathways modulates antibacterial T cell responses. Nat Immunol 13:659–666
Roses RE, Xu S, Xu M, Koldovsky U, Koski G, Czerniecki BJ (2008) Differential production of IL-23 and IL-12 by myeloid-derived dendritic cells in response to TLR agonists. J Immunol 181:5120–5127
Hawiger D, Inaba K, Dorsett Y, Guo M, Mahnke K, Rivera M, Ravetch JV, Steinman RM, Nussenzweig MC (2001) Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J Exp Med 194:769–779
Hawiger D, Masilamani RF, Bettelli E, Kuchroo VK, Nussenzweig MC (2004) Immunological unresponsiveness characterized by increased expression of CD5 on peripheral T cells induced by dendritic cells in vivo. Immunity 20:695–705
Idoyaga J, Fiorese C, Zbytnuik L, Lubkin A, Miller J, Malissen B, Mucida D, Merad M, Steinman RM (2013) Specialized role of migratory dendritic cells in peripheral tolerance induction. J Clin Invest 123:844–854
Ring S, Maas M, Nettelbeck DM, Enk AH, Mahnke K (2013) Targeting of autoantigens to DEC205(+) dendritic cells in vivo suppresses experimental allergic encephalomyelitis in mice. J Immunol 191:2938–2947
Spiering R, Margry B, Keijzer C, Petzold C, Hoek A, Wagenaar-Hilbers J, van der Zee R, van Eden W, Kretschmer K, Broere F (2015) DEC205+ dendritic cell-targeted tolerogenic vaccination promotes immune tolerance in experimental autoimmune arthritis. J Immunol 194:4804–4813
Stern JN, Keskin DB, Kato Z, Waldner H, Schallenberg S, Anderson A, Boehmer H v, Kretschmer K, Strominger JL (2010) Promoting tolerance to proteolipid protein-induced experimental autoimmune encephalomyelitis through targeting dendritic cells. Proc Natl Acad Sci U S A 107:17280–17285
Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A (2001) Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol 19:683–765
Quintana FJ, Sherr DH (2013) Aryl hydrocarbon receptor control of adaptive immunity. Pharmacol Rev 65:1148–1161
Yogev N, Frommer F, Lukas D, Kautz-Neu K, Karram K, Ielo D, Stebut E v, Probst HC, van den Broek M, Riethmacher D, Birnberg T, Blank T, Reizis B, Korn T, Wiendl H, Jung S, Prinz M, Kurschus FC, Waisman A (2012) Dendritic cells ameliorate autoimmunity in the CNS by controlling the homeostasis of PD-1 receptor(+) regulatory T cells. Immunity 37:264–275
Zlotnikov-Klionsky Y, Nathansohn-Levi B, Shezen E, Rosen C, Kagan S, Bar-On L, Jung S, Shifrut E, Reich-Zeliger S, Friedman N, Aharoni R, Arnon R, Yifa O, Aronovich A, Reisner Y (2015) , Perforin-positive dendritic cells exhibit an immuno-regulatory role in metabolic syndrome and autoimmunity. Immunity 43:776–787
Zangi L, Klionsky YZ, Yarimi L, Bachar-Lustig E, Eidelstein Y, Shezen E, Hagin D, Ito Y, Takai T, Reich-Zeliger S, Lask A, Milstein O, Jung S, Shinder V, Reisner Y (2012) Deletion of cognate CD8 T cells by immature dendritic cells: a novel role for perforin, granzyme A, TREM-1, and TLR7. Blood 120:1647–1657
Takenaka MC, Quintana FJ (2016) Achieving tolerance with perforin-secreting dendritic cells. Trends Mol Med 22:3–4
Scott CL, Aumeunier AM, Mowat AM (2011) Intestinal CD103+ dendritic cells: master regulators of tolerance? Trends Immunol 32:412–419
Bogunovic M, Ginhoux F, Helft J, Shang L, Hashimoto D, Greter M, Liu K, Jakubzick C, Ingersoll MA, Leboeuf M, Stanley ER, Nussenzweig M, Lira SA, Randolph GJ, Merad M (2009) Origin of the lamina propria dendritic cell network. Immunity 31:513–525
Johansson-Lindbom B, Svensson M, Pabst O, Palmqvist C, Marquez G, Forster R, Agace WW (2005) Functional specialization of gut CD103+ dendritic cells in the regulation of tissue-selective T cell homing. J Exp Med 202:1063–1073
Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY (2004) Retinoic acid imprints gut-homing specificity on T cells. Immunity 21:527–538
Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, Belkaid Y (2007) Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med 204:1775–1785
Weiner HL, da Cunha AP, Quintana F, Wu H (2011) Oral tolerance. Immunol Rev 241:241–259
Esterházy D, Loschko J, London M, Jove V, Oliveira TY, Mucida D (2016) Classical dendritic cells are required for dietary antigen-mediated induction of peripheral Treg cells and tolerance. Nat Immunol 17:545–555
Muzaki AR, Tetlak P, Sheng J, Loh SC, Setiagani YA, Poidinger M, Zolezzi F, Karjalainen K, Ruedl C (2016) Intestinal CD103(+)CD11b(−) dendritic cells restrain colitis via IFN-γ-induced anti-inflammatory response in epithelial cells. Mucosal Immunol 9:336–351
Klebanoff CA, Spencer SP, Torabi-Parizi P, Grainger JR, Roychoudhuri R, Ji Y, Sukumar M, Muranski P, Scott CD, Hall JA, Ferreyra GA, Leonardi AJ, Borman ZA, Wang J, Palmer DC, Wilhelm C, Cai R, Sun J, Napoli JL, Danner RL, Gattinoni L, Belkaid Y, Restifo NP (2013) Retinoic acid controls the homeostasis of pre-cDC-derived splenic and intestinal dendritic cells. J Exp Med 210:1961–1976
Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, Pieraccini G, Zecchi R, D’Angelo C, Massi-Benedetti C, Fallarino F, Carvalho A, Puccetti P, Romani L (2013) Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 39:372–385
Quintana FJ, Murugaiyan G, Farez MF, Mitsdoerffer M, Tukpah AM, Burns EJ, Weiner HL An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A 107:20768–20773
Takenaka MC, Araujo LP, Maricato JT, Nascimento VM, Guereschi MG, Rezende RM, Quintana FJ, Basso AS (2016) Norepinephrine controls effector T cell differentiation through β2-adrenergic receptor-mediated inhibition of NF-κB and AP-1 in dendritic cells. J Immunol 196:637–644
Maestroni GJ, Mazzola P (2003) Langerhans cells beta 2-adrenoceptors: role in migration, cytokine production, Th priming and contact hypersensitivity. J Neuroimmunol 144:91–99
Hervé J, Dubreil L, Tardif V, Terme M, Pogu S, Anegon I, Rozec B, Gauthier C, Bach JM, Blancou P (2013) β2-Adrenoreceptor agonist inhibits antigen cross-presentation by dendritic cells. J Immunol 190:3163–3171
Steinbrink K, Wolfl M, Jonuleit H, Knop J, Enk AH (1997) Induction of tolerance by IL-10-treated dendritic cells. J Immunol 159:4772–4780
Takenaka MC, Robson S, Quintana FJ (2016) Regulation of the T Cell Response by CD39. Trends Immunol 37(7):427–439
Macatonia SE, Doherty TM, Knight SC, O’Garra A (1993) Differential effect of IL-10 on dendritic cell-induced T cell proliferation and IFN-gamma production. J Immunol 150:3755–3765
Mitra RS, Judge TA, Nestle FO, Turka LA, Nickoloff BJ (1995) Psoriatic skin-derived dendritic cell function is inhibited by exogenous IL-10. Differential modulation of B7-1 (CD80) and B7-2 (CD86) expression. J Immunol 154:2668–2677
Steinbrink K, Jonuleit H, Müller G, Schuler G, Knop J, Enk AH (1999) Interleukin-10-treated human dendritic cells induce a melanoma-antigen-specific anergy in CD8(+) T cells resulting in a failure to lyse tumor cells. Blood 93:1634–1642
Enk AH, Jonuleit H, Saloga J, Knop J (1997) Dendritic cells as mediators of tumor-induced tolerance in metastatic melanoma. Int J Cancer 73:309–316
Melillo JA, Song L, Bhagat G, Blazquez AB, Plumlee CR, Lee C, Berin C, Reizis B, Schindler C (2010) Dendritic cell (DC)-specific targeting reveals Stat3 as a negative regulator of DC function. J Immunol 184:2638–2645
Nguyen LP, Bradfield CA (2008) The search for endogenous activators of the aryl hydrocarbon receptor. Chem Res Toxicol 21:102–116
McIntosh BE, Hogenesch JB, Bradfield CA (2010) Mammalian Per-Arnt-Sim proteins in environmental adaptation. Annu Rev Physiol 72:625–645
Lo R, Matthews J (2012) High-resolution genome-wide mapping of AHR and ARNT binding sites by ChIP-Seq. Toxicol Sci 130:349–361
Zhang S, Rowlands C, Safe S (2008) Ligand-dependent interactions of the Ah receptor with coactivators in a mammalian two-hybrid assay. Toxicol Appl Pharmacol 227:196–206
Boronat S, Casado S, Navas JM, Piña B (2007) Modulation of aryl hydrocarbon receptor transactivation by carbaryl, a nonconventional ligand. FEBS J 274:3327–3339
Murray IA, Morales JL, Flaveny CA, Dinatale BC, Chiaro C, Gowdahalli K, Amin S, Perdew GH (2010) Evidence for ligand-mediated selective modulation of aryl hydrocarbon receptor activity. Mol Pharmacol 77:247–254
Matikainen T, Perez GI, Jurisicova A, Pru JK, Schlezinger JJ, Ryu HY, Laine J, Sakai T, Korsmeyer SJ, Casper RF, Sherr DH, Tilly JL (2001) Aromatic hydrocarbon receptor-driven Bax gene expression is required for premature ovarian failure caused by biohazardous environmental chemicals. Nat Genet 28:355–360
Matsumura F (2009) The significance of the nongenomic pathway in mediating inflammatory signaling of the dioxin-activated Ah receptor to cause toxic effects. Biochem Pharmacol 77:608–626
Ohtake F, Baba A, Takada I, Okada M, Iwasaki K, Miki H, Takahashi S, Kouzmenko A, Nohara K, Chiba T, Fujii-Kuriyama Y, Kato S (2007) Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature 446:562–566
Temchura VV, Frericks M, Nacken W, Esser C (2005) Role of the aryl hydrocarbon receptor in thymocyte emigration in vivo. Eur J Immunol 35:2738–2747
Fernandez-Salguero P, Pineau T, Hilbert DM, McPhail T, Lee SS, Kimura S, Nebert DW, Rudikoff S, Ward JM, Gonzalez FJ (1995) Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science 268:722–726
Denison MS, Nagy SR (2003) Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol 43:309–334
Denison MS, Pandini A, Nagy SR, Baldwin EP, Bonati L (2002) Ligand binding and activation of the Ah receptor. Chem Biol Interact 141:3–24
Song J, Clagett-Dame M, Peterson RE, Hahn ME, Westler WM, Sicinski RR, DeLuca HF A ligand for the aryl hydrocarbon receptor isolated from lung. Proc Natl Acad Sci U S A 99:14694–14699
Wei YD, Helleberg H, Rannug U, Rannug A (1998) Rapid and transient induction of CYP1A1 gene expression in human cells by the tryptophan photoproduct 6-formylindolo[3,2-b]carbazole. Chem Biol Interact 110:39–55
Moffett JR, Namboodiri MA (2003) Tryptophan and the immune response. Immunol Cell Biol 81:247–265
Nguyen NT, Nakahama T, Le DH, Van Son L, Chu HH, Kishimoto T (2014) Aryl hydrocarbon receptor and kynurenine: recent advances in autoimmune disease research. Front Immunol 5:551
Yeste A, Nadeau M, Burns EJ, Weiner HL, Quintana FJ Nanoparticle-mediated codelivery of myelin antigen and a tolerogenic small molecule suppresses experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A 109:11270–11275
Wu HY, Quintana FJ, da Cunha AP, Dake BT, Koeglsperger T, Starossom SC, Weiner HL (2011) In vivo induction of Tr1 cells via mucosal dendritic cells and AHR signaling. PLoS One 6:e23618
Yeste A, Takenaka MC, Mascanfroni ID, Nadeau M, Kenison JE, Patel B, Tukpah AM, Babon JA, DeNicola M, Kent SC, Pozo D, Quintana FJ (2016) Tolerogenic nanoparticles inhibit T cell-mediated autoimmunity through SOCS2. Sci Signal 9:ra61
Tian Y, Ke S, Denison MS, Rabson AB, Gallo MA (1999) Ah receptor and NF-kappa B interactions, a potential mechanism for dioxin toxicity. J Biol Chem 274:510–515
Gillesby BE, Stanostefano M, Porter W, Safe S, Wu ZF, Zacharewski TR (1997) Identification of a motif within the 5’ regulatory region of pS2 which is responsible for AP-1 binding and TCDD-mediated suppression. Biochemistry 36:6080–6089
Vogel CF, Wu D, Goth SR, Baek J, Lollies A, Domhardt R, Grindel A, Pessah IN (2013) Aryl hydrocarbon receptor signaling regulates NF-kappa B RelB activation during dendritic-cell differentiation. Immunol Cell Biol 91:568–575
Vogel CF, Matsumura F (2009) A new cross-talk between the aryl hydrocarbon receptor and RelB, a member of the NF-kappa B family. Biochem Pharmacol 77:734–745
Lee JA, Hwang JA, Sung HN, Jeon CH, Gill BC, Youn HJ, Park JH (2007) 2,3,7,8-Tetrachlorodibenzo-p-dioxin modulates functional differentiation of mouse bone marrow-derived dendritic cells Downregulation of RelB by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Lett 173:31–40
Ruby CE, Leid M, Kerkvliet NI (2002) 2,3,7,8-Tetrachlorodibenzo-p-dioxin suppresses tumor necrosis factor-alpha and anti-CD40-induced activation of NF-kappaB/Rel in dendritic cells: p 50 homodimer activation is not affected. Mol Pharmacol 62:722–728
Benson JM, Shepherd DM (2011) Dietary ligands of the aryl hydrocarbon receptor induce anti-inflammatory and immunoregulatory effects on murine dendritic cells. Toxicol Sci 124:327–338
Hauben E, Gregori S, Draghici E, Migliavacca B, Olivieri S, Woisetschlager M, Roncarolo MG (2008) Activation of the aryl hydrocarbon receptor promotes allograft-specific tolerance through direct and dendritic cell-mediated effects on regulatory T cells. Blood 112:1214–1222
Nguyen NT, Kimura A, Nakahama T, Chinen I, Masuda K, Nohara K, Fujii-Kuriyama Y, Kishimoto T (2010) Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc Natl Acad Sci U S A 107:19961–19966
Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA (2010) An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol 185:3190–3198
Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL (2008) Control of T (reg) and T (H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 453:65–71
Jaronen M, Quintana FJ (2014) Immunological relevance of the coevolution of IDO1 and AHR. Front Immunol 5:521
Acknowledgments
Research in the Quintana laboratory is supported by the National Institutes of Health, the National Multiple Sclerosis Society, the International Progressive MS Alliance, and the American Cancer Society.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is a contribution to the special issue on Dendritic Cell Subsets and Immune-mediated Diseases - Guest Editor: Francisco Quintana
Rights and permissions
About this article

Cite this article
Takenaka, M.C., Quintana, F.J. Tolerogenic dendritic cells. Semin Immunopathol 39, 113–120 (2017). https://doi.org/10.1007/s00281-016-0587-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-016-0587-8