
Abstract
Purpose
Midostaurin, approved for FLT3-mutated acute myeloid leukemia and advanced systemic mastocytosis, is mainly metabolized by cytochrome P450 (CYP) 3A4. Midostaurin exhibited potential inhibitory effects on P-glycoprotein (P-gp), breast cancer resistance protein (BCRP), organic anion-transporting polyprotein 1B1, and CYP2D6 in in vitro studies. This study investigated the pharmacokinetic (PK) effects of midostaurin on P-gp (digoxin), BCRP (rosuvastatin) and CYP2D6 (dextromethorphan) substrates in healthy adults.
Methods
This was an open-label, single-sequence, phase I clinical study evaluating the effect of single-dose midostaurin (100 mg) on the PK of digoxin and rosuvastatin (Arm 1), and dextromethorphan (Arm 2). Participants were followed up for safety 30 days after last dose. In addition, the effect of midostaurin on the PK of dextromethorphan metabolite (dextrorphan) was assessed in participants with functional CYP2D6 genes in Arm 2.
Results
The effect of midostaurin on digoxin was minor and resulted in total exposure (AUC) and peak plasma concentration (Cmax) that were only 20% higher. The effect on rosuvastatin was mild and led to an increase in AUCs of approximately 37-48% and of 100% in Cmax. There was no increase in the primary PK parameters (AUCs and Cmax) of dextromethorphan in the presence of midostaurin. The study treatments were very well tolerated with no occurance of severe adverse events (AEs), AEs of grade ≥ 2, or deaths.
Conclusion
Midostaurin showed only a minor inhibitory effect on P-gp, a mild inhibitory effect on BCRP, and no inhibitory effect on CYP2D6. Study treatments were well tolerated in healthy adults.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Midostaurin, a multi-target tyrosine-kinase inhibitor of class III tyrosine protein kinases, has been developed as an antileukemic agent for patients with acute myeloid leukemia (AML). The FMS-like tyrosine kinase 3 (FLT3) gene is mutated in approximately one-third of patients with AML, either by internal tandem duplications (FLT3-ITD) or by a point mutation, mainly involving the tyrosine kinase domain (FLT3-TKD) [1]. Midostaurin has been shown to inhibit the activity of FLT3 receptor and cell proliferation, thereby inducing apoptosis in leukemic cells expressing FLT3-ITD or FLT3-TKD or overexpressing wild type FLT3, c-kit, src, platelet-derived, and vascular endothelial growth factor receptors [2]. Based on data from the RATIFY trial, midostaurin (Rydapt®) was approved for adults with newly diagnosed FLT3-mutated AML (50 mg orally twice daily with food) in combination with standard cytarabine and daunorubicin induction and cytarabine consolidation in the United States (US), European Union, and other countries [3,4,5]. Midostaurin has also been approved as monotherapy for aggressive systemic mastocytosis (ASM), systemic mastocytosis (SM) with associated hematological neoplasm, and mast cell leukemia [4, 5].
In humans, following an oral administration, the absorption of midostaurin is high (> 90%) and rapid, with a time to reach peak plasma concentration (Tmax) of total radioactivity observed at 1–3 h post-dose. Midostaurin is metabolized mainly by cytochrome P450 (CYP) 3A4 (CYP3A4) via oxidative pathways, and the major plasma components included midostaurin (22.0% ± 4.9%) and its two major active metabolites, CGP62221 and CGP52421, accounting for 27.7% ± 2.7% and 37.97% ± 6.6%, respectively. The recovery of the mass balance was good (81.6%) despite long radioactivity half-lives. The findings from the Human Mass Balance study indicate that fecal excretion is the major route of elimination (77.6% of the dose), mostly as metabolites, whereas unchanged midostaurin accounts for 3.4% of the dose, suggesting predominant hepatic metabolism. The median terminal half-lives (T1/2) of midostaurin, CGP62221, and CGP52421 in plasma are approximately 20.3 h, 33.4 h, and 495 h, respectively [6]. Midostaurin neither inhibits nor induces CYP3A4 and CYP2C8, and weakly induces CYP2B6. Midostaurin at steady state has no clinically relevant PK interaction on hormonal contraceptives [7]. Although concomitantly administered strong CYP3A4 inhibitors increased midostaurin exposure 1.44-fold in patients with FLT3-mutated AML, no clinically relevant differences in safety were noted [8]. In vitro data suggests a potential of midostaurin and its metabolites to inhibit the P-glycoprotein (P-gp), breast cancer resistance protein (BCRP), and organic anion-transporting polyprotein 1B1 (OATP1B1) transporters. Specifically, the half maximal inhibitory concentration (IC50) value for P-gp inhibition by midostaurin was 1.7 ± 0.18 µM (unpublished data). Moreover, midostaurin inhibited BCRP-mediated efflux of fluorescent substrates in cells that over-express the transporter, the IC50 was 0.23 µM. Assessments using a net model based on Food and Drug Administration (FDA) Guidance (2020) on “In vitro drug interaction studies — cytochrome P450 enzyme- and transporter-mediated drug interactions guidance” for industry [9] suggested a risk of possible drug interactions.
Both the metabolites (CGP52421 and CGP62221) have been identified as being pharmacologically active against cells expressing FLT3-ITDs and KIT D8 [10]. Midostaurin (EC50 of cytotoxicity response of 39 and 47 nM for FLT3-ITDs and KIT D816V, respectively) and CGP62221 (EC50 of cytotoxicity response of 30 and 70 nM for FLT3-ITDs and KIT D816V, respectively) showed similar potency, whereas CGP52421 showed an average approximately 10-fold reduced potency (EC50 of cytotoxicity response of 656 and 233 nM for FLT3-ITDs and KIT D816V, respectively).
In vitro experiments also showed that midostaurin and one of its metabolites CGP52421 inhibit CYP2D6 with the respective IC50 of 1 and 5 µM (unpublished data). After correction for microsomal protein binding, the unbound inhibition constant Ki,u values for midostaurin and CGP52421 are 0.25 and 1.5 µM, respectively. However, based on the net effect model described by FDA 2020 recommendation [9], no inhibitory effect was predicted for midostaurin and its metabolites on CYP2D6 activity. The values of unbound Cmax for midostaurin and CGP52421 at 0.32 nM and 0.12 nM were used for the assessment, respectively, where midostaurin total Cmax was 1210 ng/mL (2.1 µM) with corrected unbound fraction of plasma fup of 0.00015 and Cmax of CGP52421 was 328 ng/mL (0.56 µM) corrected for fup of 0.00021 from a single dose of 50 mg in the human ADME study [6].
In absence of supportive in vivo clinical evidence, the present study (CPKC412A2121 study, referred to as A2121 study hereon) was designed to investigate the effect of a single oral dose of 100 mg midostaurin on the activity of P-gp, BCRP and CYP2D6 by evaluating the PK profile of their specific substrates, digoxin (P-gp), rosuvastatin (BCRP and OATP1B1) and dextromethorphan (CYP2D6), respectively, upon co-administration with midostaurin. An absence of effect of the drug substrates on the PK of midostaurin, CGP52421, and CGP62221 was a prerequisite as midostaurin and its metabolites are primarily metabolized via CYP3A4.
Digoxin is a cardiac glycoside, indicated for the treatment of mild to moderate heart failure in adults and for controlling the resting ventricular rate in patients with chronic atrial fibrillation [11]. Following the oral administration of a tablet (0.25 mg), digoxin is rapidly absorbed with a Tmax of 1–3 h. Unlike midostaurin, urinary excretion is the major pathway of elimination for digoxin as the parent drug, and CYP-dependent metabolism plays a minor role in digoxin elimination. The terminal elimination half-life of digoxin in healthy humans with normal renal function is 30–40 h [12]. Digoxin is a substrate of P-gp at the level of intestinal absorption, and renal tubular and biliary-intestinal secretions. Therefore, drugs that induce/inhibit P-gp have the potential to alter the PK profile of digoxin.
Rosuvastatin is a 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG Co-A) reductase inhibitor (statin) used for lowering cholesterol and triglyceride levels [13]. Compared to digoxin, post-dose absorption of rosuvastatin is slightly longer (Tmax = 3–5 h). Similar to midostaurin, fecal excretion is the major elimination route for rosuvastatin and its metabolites. Rosuvastatin is not extensively metabolized with approximately 10% of the administered dose being recovered as metabolite, mainly by CYP2C9. The plasma elimination half-life of rosuvastatin is approximately 19 h. It is a substrate for certain transporter proteins including the hepatic uptake transporter OATP1B1 and efflux transporter BCRP. Thus, midostaurin and its metabolites may influence its PK profile.
Dextromethorphan (a CYP2D6 substrate), a non-opioid, over-the-counter antitussive drug, was also investigated in the present study for a potential drug-drug interaction (DDI) with midostaurin and its metabolites. Dextromethorphan (elimination T1/2: 13 h) is metabolized by O-demethylation to dextrorphan via CYP2D6, comprising of 2% hepatic CYP content, whose genetic expression among individuals is subject to genetic polymorphisms [14]. CYP2D6 is one of the best-known polymorphic drug-metabolizing enzymes with over 75 functionally important allelic variants. Dextromethorphan is recognized as a CYP2D6 substrate for the assessment of CYP2D6 inhibition potential [15, 16]. Individuals with deficient CYP enzyme activity are classified as poor metabolizers (PMs), carrying two non-functional CYP alleles. Individuals with slightly subnormal or normal rates of metabolism are defined as either intermediate metabolizers (IMs), having only one functional allele, or extensive metabolizers (EMs), with two functional alleles. In addition, a subgroup of ultrarapid metabolizers (UMs; multiple copies [3 to 13] of functional alleles) with extremely high enzyme activity has been identified [17,18,19].
The use of digoxin and rosuvastatin as substrates for P-gp and BCRP in a cocktail has been established [9, 20]. The cocktail approach offers advantages such as reduced complexity compared to the administration of individual substrate drugs in separate arms, provided that there is no DDI between substrate drugs, as demonstrated for digoxin and rosuvastatin. Based on the results of a clinical study, rosuvastatin (40 mg once daily to steady state) has no effect on the PK of a single dose of 0.5-mg digoxin, allowing the simultaneous use of both drugs as a cocktail in DDI studies [21]. The present clinical study investigated the impact of a single dose of 100-mg midostaurin on P-gp and BCRP transport with a two-drug cocktail of digoxin (0.25 mg) and rosuvastatin (10 mg), respectively, in Arm 1. In Arm 2, the impact of a single dose of 100-mg midostaurin on the pharmacokinetics (PK) of a single dose of 60-mg dextromethorphan (CYP2D6 substrate) was investigated. As midostaurin and metabolites were not identified as potential inducers or time-dependent inhibitors of CYP2D6, a single-dose study was adequate to assess the potential for CYP2D6 inhibition. Additionally, a single dose of midostaurin was adequate to assess the P-gp and BCRP-mediated absorption-related interaction as recommended by the recent ICH guidance 2024 [22].
This study also sought to evaluate the safety and tolerability of a single oral dose of 100-mg midostaurin when co-administered with digoxin, rosuvastatin, or dextromethorphan in healthy adult participants. Additionally, the study assessed the PK profiles of midostaurin and its metabolites, CGP52421 and CGP62221 (in Period 2 of Arm 1 and Arm 2) with the drug substrates. CYP2D6 genotyping was performed to check the eligibility of the participants in Arm 2.
Methods
Study design
This was an open-label, single-center, single-sequence phase I study conducted in healthy participants with two independent arms based on the substrate used. Each arm included two periods (Period 1 and Period 2). An overview of the study design (including dosing and PK sampling) is schematically depicted in Fig. 1. Study protocol and its amendment were reviewed by the Independent Ethics Committee or Institutional Review Board. Written informed consent was obtained from each participant at screening before any study-specific procedure was performed. The study was conducted in accordance with the International Council for Harmonization E6 guideline for Good Clinical Practice, which has its origin in the Declaration of Helsinki.

Overall study design: Dosing and PK sampling. DIGO, digoxin; DM, dextromethorphan; EoS, end of study; EoT, end of treatment; MIDO, midostaurin; PG, pharmacogenetics; PK, pharmacokinetics; ROSU, rosuvastatin
The participants were screened for eligibility within 21 days of the first baseline assessment. Excluding the screening period, total study treatment duration was 43 days for Arm 1 and 37 days for Arm 2. Safety follow-up was performed for 30 days from the last dose of treatment.
In Arm 1, the effect of a single dose of 100-mg midostaurin on the drug substrates of P-gp (digoxin 0.25 mg) and BCRP (rosuvastatin 10 mg) transporters was investigated using a two-drug cocktail. In Period 1, the participants received a single oral dose of the cocktail substrate drugs, and blood samples were collected up to 192 h post-dose to measure the plasma concentration of digoxin and rosuvastatin. Period 2 was started after a washout of at least 12 days, during which no study treatment was administered. On Day 13, a single oral dose of 100-mg (4 × 25 mg) midostaurin was administered, followed by the two-drug cocktail (within 2 min of midostaurin administration). PK samples were collected for the measurement of the plasma concentration of the drug substrates and midostaurin (and its metabolites).
In Arm 2, the effect of a single dose of 100-mg midostaurin on CYP2D6 substrate (dextromethorphan 0.25 mg) was investigated. In Period 1, participants received a single oral dose of dextromethorphan (2 × 30 mg), and blood samples were collected up to 72 h post-dose for measuring the plasma concentration of dextromethorphan and its metabolite dextrorphan. Period 2 started after a washout period of at least 6 days, during which no study treatment was administered. On Day 7, a single oral dose of 100-mg (4 × 25 mg) midostaurin was co-administered with dextromethorphan. PK samples were collected to measure the plasma concentration of the drug substrates and midostaurin (and its metabolites).
PK sampling
Blood samples were collected at various time points as specified in the assessment schedule (Fig. 1). For both Period 1 and Period 2, PK samples were collected up to 192 h post-dose for digoxin and rosuvastatin (including pre-dose, 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 36, 48, 72, 96, 120, 144, 168, and 192 h) in Arm 1 and up to 72 h post-dose for dextromethorphan and its metabolite dextrorphan (including pre-dose, 0.25, 0.5, 1, 1.5, 2, 4, 6, 8, 12, 24, 36, 48, and 72 h) in Arm 2. In addition, in both study arms, PK samples were collected for midostaurin (and its metabolites CGP62221 and CGP52421) in Period 2 at various timepoints up to 120 h post-dose (including pre-dose, 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 36, 48, 72, 96, and 120 h). Thus, the PK of midostaurin (and of the metabolite CGP62221) could be adequately explored over the evaluation period 3- to 4-fold longer than their elimination half-lives.
Eligibility criteria
All eligible participants were required to be aged 18 to 65 years with a body mass index (BMI) of 18.0 to 29.9 kg/m2 and to be in good health condition, as determined by the absence of clinically significant findings in their medical history, physical examination, vital signs, laboratory parameters (including hematology and standard clinical chemistry parameters), and electrocardiogram (ECG) results; eligible participants also required to provide written informed consent prior to any screening procedures being performed. The key exclusion criteria included a history of cardiac disease, a family history or presence of prolonged QT-interval syndrome, a history of immunodeficiency disease, and contraindication or hypersensitivity to any drug or metabolites from a similar class as the study drug. Genotyping was only performed for the participants in Arm 2 to exclude PMs. Detailed inclusion and exclusion criteria are provided in the supplementary (Table S1).
Study objectives
The primary objective of this study was to evaluate the effect of a single oral dose of 100-mg midostaurin on the PK of digoxin and rosuvastatin in healthy participants (Arm 1) and on the PK of dextromethorphan in healthy participants with functional CYP2D6 genes (EMs, IMs, and UMs) (Arm 2). The secondary objective was to assess the safety and tolerability in healthy participants when receiving digoxin and rosuvastatin or dextromethorphan alone and/or concomitantly with midostaurin. The exploratory objectives were to (i) assess of the effect of a single oral dose of 100-mg midostaurin on the PK of dextrorphan, a metabolite of dextromethorphan, in participants with functional CYP2D6 genes and (ii) evaluate the PK of midostaurin and its metabolites (CGP52421 and CGP62221) following a single oral dose of midostaurin in combination with the substrates (either digoxin and rosuvastatin in Arm 1 or dextromethorphan in Arm 2).
PK assessments
PK parameters were determined using the actual recorded sampling times and a non-compartmental method with Phoenix WinNonlin (Version 8.0). The area under the curve from the time of administration up to the last quantifiable concentration point (AUClast), area under the plasma concentration-time curve from time 0 to time t (AUC0 − t; if needed, e.g., AUC0 − 12 h, AUC0 − 48 h), area under the plasma concentration-time curve extrapolated to infinity (AUCinf), observed maximum plasma concentration (Cmax), time to reach maximum plasma concentration (Tmax), elimination half-life (T1/2), apparent volume of distribution (Vz/F), and clearance (CL/F) were calculated based on the determined plasma levels of the substrate drugs (digoxin, rosuvastatin, and dextromethorphan) and midostaurin. Other PK parameters were calculated as appropriate (e.g., AUC0 − 12 h for midostaurin). The same PK parameters, except CL/F and Vz/F, were calculated for the metabolites of midostaurin and dextromethorphan, when possible. Apparent T1/2 was calculated for the metabolites, when possible. The metabolic molar ratio (MR) of dextromethorphan/dextrorphan was calculated using the following formula:
MR = (AUC0 − 48 h of dextromethorphan/molecular weight of dextromethorphan)/(AUC0 − 48 h of dextrorphan/molecular weight of dextrorphan).
The molecular weights (in g/mol) used for the MR calculation were as follows: for dextromethorphan, 271.40 and for dextrorphan, 257.37. Definitions for the measured PK parameters are provided in Table S2.
Pharmacogenetic assessments
Genotyping of CYP2D6 was performed only in participants in Arm 2 following the CPIC guidelines [23]. After deoxyribonucleic acid (DNA) isolation from ethylenediaminetetraacetic acid blood and polymerase chain reaction identification of the genotype is performed by DNA sequencing. The following allele variants were sequenced: CYP2D6*1,*3,*4,*5,*6,*9,*10,*17,*41, XxN. Detection of the CYP2D6 deletions or amplifications was performed using multiplex ligation-dependent probe amplification technology.
Genotypes were categorized as EMs, IMs, or UMs; PMs were excluded.
Safety assessments
Safety follow-up phone calls were performed 30 days after last dose: Day 43 to 46 for Arm 1 and Day 37 to 40 for Arm 2. Safety assessments consisted of monitoring and recording of all adverse events (AEs), including serious AEs (SAEs) and treatment-related AEs (TRAEs) along with their severity levels. Other safety data collection included the assessment of physical condition, vital signs, height, weight, and BMI, as well as 12-lead ECGs and regular monitoring of various laboratory parameters including hematology, chemistry, coagulation, HIV screening (at screening), hepatitis markers, alcohol test (urine), drug and cotinine screen (urine, breath, or blood) and fertility testing at study visits, as per the protocol.
Sample size calculation
Considering that the two treatment arms were independent of each other, the sample size was calculated separately for each arm under the most conservative assumptions. The intra-subject coefficients of variation (CVs) were 34% for Arm 1 and 40.9% for Arm 2. A total of 16 and 20 evaluable participants were recommended for Arm 1 and Arm 2, respectively. To allow for possible dropouts (20%), a total of 20 participants were to be enrolled to ensure that 16 evaluable participants would complete this study in Arm 1.
For Arm 2, up to 23 participants were to be enrolled to ensure that 20 evaluable participants would complete the study, allowing for a possible dropout rate of 10%.
Bioanalytical method
The plasma concentrations of midostaurin and its metabolites CGP52421 and CGP62221 were determined using a validated liquid chromatography-tandem mass spectrometry (LC-MS/MS) assay with a lower limit of quantitation (LLOQ) of 10 ng/mL (SGS France).
Digoxin was measured by a validated method using solid phase extraction of plasma, followed by evaporation of the extract to dryness, and solid phase extraction of the reconstituted samples by LC-MS/MS assay with a LLOQ of 0.010 ng/mL (Wuxi AppTec, USA).
Rosuvastatin was quantified by a validated method using protein precipitation extraction of the samples, followed by evaporation of the extract to dryness and analysis of the reconstituted samples by LC-MS/MS assay with a LLOQ of 0.050 ng/mL (Wuxi AppTec).
Dextromethorphan and dextrorphan were measured by a validated method using liquid-liquid extraction of the samples, followed by evaporation of the extract to dryness and analysis of the reconstituted samples by LC-MS/MS assay with a LLOQ of 0.1 ng/mL (Wuxi AppTec).
Statistical analysis
Three types of analysis sets were considered: full analysis set (FAS), safety set, and PK analysis set (PAS). The FAS included all participants who received at least one dose of any of the study treatments (digoxin, rosuvastatin, or midostaurin in Arm 1 and dextromethorphan or midostaurin in Arm 2). The FAS and safety sets of each arm were identical. The PK analysis set included all participants who provided an evaluable PK profile for at least one period. Three separate PASs were considered in this study: PAS1 for the substrate drugs digoxin and rosuvastatin in Arm 1, PAS2 for the substrate drug dextromethorphan in Arm 2, and PAS3 for midostaurin. Only PK parameters and concentrations from evaluable periods were included in the PAS.
No formal hypothesis tests were performed. To evaluate the interaction between the substrates and the investigational drug, log-transformed PK parameters (Cmax, AUClast, and AUCinf) in plasma were analyzed separately using linear mixed-effects models. For each comparison, a point estimate and the corresponding two-sided 90% confidence interval (CI) for the difference between the means of the test (substrate plus midostaurin) and reference (substrate) treatment were calculated, and then the point estimate and CI were anti-log transformed to obtain the point estimate and the 90% CI for the geometric mean ratio (GMR) on the original scale. The median and range of differences in the Tmax values were calculated.
For all safety analyses, the respective safety set was used. Safety data were analyzed based on the number and percentage of participants with AEs and tabulated by preferred term (PT).
Results
Disposition of participants
A total of 43 participants were enrolled in the study after screening 90 participants. Of the 43, 20 participants (median age, 59 years [range, 36–64 years]; mean BMI [± SD], 25.3 [± 2.37] kg/m2; male, 70.0%) were enrolled in Arm 1 and 23 participants (median age, 57 years [range, 27–65 years]; mean BMI [± SD], 24.6 [± 2.31] kg/m2; male, 69.6%) were enrolled in Arm 2. Two participants, one each in Arms 1 and 2, discontinued treatment due to vomiting (grade 1 AE) within 4 h of dosing during Period 2 of treatment. Additionally, one participant in Arm 2 discontinued treatment due to a protocol deviation. Detailed disposition and demographics of the participants are available in Table S3 and Table S4.
Pharmacokinetic results
DDI with digoxin, rosuvastatin, and dextromethorphan
A summary of plasma PK parameters of digoxin, rosuvastatin and dextromethorphan when administered alone or concomitantly with midostaurin is shown in Table 1.
The GMR and 90% CIs for the primary PK parameters of digoxin, rosuvastatin and dextromethorphan, when administered with midostaurin (test) or alone (i.e. without midostaurin, reference) were estimated. Details of the statistical analysis of the primary PK parameters for all three drug substrates are given in Table S5. The co-administration of midostaurin with digoxin increased the AUCinf (23%) and AUClast (21%) of digoxin compared to when digoxin was administered alone. The estimated GMRs (90% CI) for AUCinf and AUClast were 1.23 (1.16, 1.31) and 1.21 (1.14, 1.29), respectively. The estimated GMR (90% CI) for Cmax was 1.20 (1.00, 1.43). The effect of concomitant administration of midostaurin was comparatively more pronounced on the substrate of the transporter BCRP (rosuvastatin) with a 37–48% increase in total exposure AUCs (AUCinf: GMR [90% CI] 1.37 [1.18, 1.59]; AUClast: GMR [90% CI] 1.48 [1.33, 1.64]) and a 100% increase in the geometric mean peak plasma concentration (Cmax: GMR [90% CI] 2.01 [1.73, 2.32]). Conversely, when midostaurin was co-administered with a substrate of CYP2D6 enzyme (dextromethorphan), the total exposure AUCs (AUCinf: GMR [90% CI] 0.869 [0.711, 1.06]; AUClast: GMR [90% CI] 0.872 [0.711, 1.07]) was decreased by ∼ 13% and peak plasma concentration (Cmax: GMR [90% CI] 0.716 [0.589, 0.871]) of dextromethorphan was decreased by ∼ 28%.
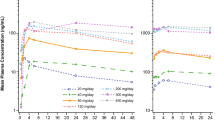
The full plasma concentration-time profiles of each drug substrate alone and in the presence of midostaurin are illustrated in Fig. 2. Digoxin and rosuvastatin were quantifiable in all participants up to 192 h post-dose, whereas dextromethorphan was quantifiable up to 72 h post-dose in all participants. The 24-h profiles for each substrate (digoxin, rosuvastatin, and dextromethorphan) alone and in the presence of midostaurin are provided in Fig. S1.

Plasma concentration-time profile (Arithmetic mean ± SD) of digoxin (a), rosuvastatin (b) dextromethorphan (c)
The effect of midostaurin on the three probe substrates (digoxin, rosuvastatin, and dextromethorphan) as GMR (90% CI) for the primary PK parameters (PAS for the respective probe substrate) is presented in Fig. 3.

The effect of midostaurin as geometric mean ratio (90% CI) on digoxin, rosuvastatin and dextromethorphan. AUCinf, area under the plasma concentration-time curve extrapolated to infinity; AUClast, area under the curve from the time of administration to the last quantifiable concentration point; BCRP, breast cancer resistance protein; CI, confidence interval; Cmax, observed maximum plasma concentration; P-gp, P-glycoprotein
DDI with dextrorphan
The effect of a single oral dose of 100-mg midostaurin on the PK of dextrorphan, a metabolite of dextromethorphan, was assessed in participants with functional CYP2D6 genes (EMs, IMs, and UMs). A summary of PK parameters is listed in Table 2. The AUCs and Cmax for dextrorphan were overall comparable when dextromethorphan was administered alone and in combination with midostaurin. Compared with dextromethorphan alone, the combination of midostaurin with dextromethorphan increased the partial exposure of dextrorphan (AUC0 − 48 h: geometric mean [geo-CV%]) from 88.7 (17.8%) to 98.1 (33.4%) ng·h/mL. The mean Cmax of dextrorphan was slightly decreased in the presence of midostaurin (geometric mean [geo-CV%]: 7.39 ng/mL [91.3%] in dextromethorphan + midostaurin vs. 9.88 ng/mL [48.8%] in dextromethorphan). The Tmax of dextrorphan increased by 2 h in the presence of midostaurin. The metabolic MR of dextromethorphan: dextrorphan for dextromethorphan in combination with midostaurin was slightly lower than that for dextromethorphan alone. The plasma concentration-time profile of dextrorphan alone and in the presence of midostaurin is provided in Fig. S2a.
Impact of probe substrates on midostaurin
The PK parameters of midostaurin and its metabolites CGP52421 and CGP62221 in both study arms were comparable. A summary of PK parameters is presented in Table 2. The midostaurin AUCs were almost identical in both arms and Cmax were very close. The mean T1/2 was 23 h in both arms. The PK of both metabolites CGP52421 and CGP62221 were similar. The plasma concentration-time profiles of midostaurin and its metabolites (pooled for both arms) are provided in Fig. S2b.
Pharmacogenetics data
In Arm 2, 19 EMs, 1 μm, and 3 IMs were identified. The MR values of the 19 EMs in both study arms ranged from approximately 0.1 to 1.4, with the exception of a participant who had MR of 1.68 with dextromethorphan alone and another EM participant who had the maximal MR value of 3.68 with dextromethorphan alone and 2.94 with dextromethorphan plus midostaurin. The three IMs had MR values between 5.35 and 9.23, with no consistent change with or without co-administration of midostaurin. The only UM had MR values of 0.88 with dextromethorphan alone and 0.78 with dextromethorphan plus midostaurin. There were no PMs as PMs were not allowed to participate in Arm 2.
Safety
Overall, a single dose of 100-mg midostaurin, alone or in combination with digoxin, rosuvastatin, and dextromethorphan, was well tolerated. A list of AEs reported in both study arms is presented in Table 3. No deaths or SAEs were reported during the study or follow-up. In addition, none of the AEs were of Grade 3 severity in either study arm. In Arm 1, six participants (30%) reported at least one AE. Four participants (20%) experienced AEs that were suspected to be related to the study treatment. Of the 23 participants in Arm 2, one or more AEs were reported in 18 participants (78.3%). All subjects who reported any AE in Arm 2 (18 participants, 78.3%) had AEs suspected to be related to the study treatment. No clinically significant changes in hematological or clinical chemistry parameters were observed. No clinically significant changes in vital signs from baseline were noted in the participants in either study arm. The mean and median values of ECG parameters and their changes from baseline at different assessments over time were not considered clinically significant by the investigator for participants in either study arm.
Discussion
This phase I, open-label, single-sequence study with two independent arms showed that a single oral dose of 100-mg midostaurin had only minor inhibitory effects on the PK profile of P-gp (digoxin), mild effect on BCRP (rosuvastatin), but no effect on CYP2D6 (dextromethorphan) substrates. The results of this study were in line with the in vitro predictions. The safety assessment showed that midostaurin and the other study treatments were well tolerated in healthy participants with no reported deaths, SAE, or AEs exceeding Grade 2 severity. Moreover, this study analyzed CYP2D6 genotypes among the participants in Arm 2 and correlated their metabolizer status (EM, IM, and UM) to the systemic exposures to dextromethorphan and dextrorphan. No clear conclusion could be inferred based on the observations in the different genotypes due to limited data availability; as next to 19 EMs only 1 μm and 3 IMs were available.
A single oral dose of 100-mg midostaurin was administered to the participants in this study. This dose presents an acceptable safety profile to be administered in healthy participants, as found in the previous single-dose clinical studies [24]. This single dose corresponds to the recommended daily dose of midostaurin in patients with AML (i.e., 50 mg twice daily, orally). The recommended starting dose of midostaurin in patients with ASM is 100 mg twice daily, orally. Thus, in the present study the AUC result would reflect the daily dose in AML and Cmax the single dose in ASM, this is also acceptable from a safety perspective as well (see above). The effects of reversible inhibition of BCRP, P-gp, and CYP2D6 by this dosing regimen could be adequately assessed.
To evaluate the inhibitory effect of midostaurin on P-gp and BCRP enzymes, a cocktail approach was employed, in which midostaurin was co-administered with a combination of digoxin (a P-gp substrate) and rosuvastatin (a BCRP, OATP1B1, OATP1B3 substrate). The results indicated that co-administration of a single dose of midostaurin (100 mg) with digoxin or rosuvastatin in healthy participants increased the AUCs of digoxin by ∼ 20% (i.e., < 1.25-fold) and rosuvastatin by 37–48% (1.37-1.48-fold) compared with those when digoxin or rosuvastatin was administered alone. The Cmax of digoxin and rosuvastatin were also increased by ∼ 20% (< 1.25 fold) and 100% (2.01-fold), respectively, in presence of midostaurin compared to administration of digoxin or rosuvastatin alone. In addition, the secondary PK parameters for both digoxin and rosuvastatin exhibited small differences in presence of midostaurin. As per the FDA and European Medicines Agency (EMA) guidelines [9, 25] regarding clinical drug interaction studies, enzyme inhibitors are classified into three categories: (i) strong inhibitor, which causes a > 5-fold increase in the plasma AUC values or ≥ 80% decrease in oral clearance; (ii) moderate inhibitor, which causes a > 2-fold increase in the plasma AUC or 50% to ≤ 80% inhibition of oral clearance; (iii) mild (as per EMA)/weak (per FDA) inhibitor, which causes 1.25- to 2-fold increase in the plasma AUC or ≤ 50% inhibition of oral clearance.
Thus, a single dose of 100-mg midostaurin has a minor inhibitory effect on digoxin and a mild inhibitory effect on rosuvastatin. Medicinal products with a narrow therapeutic range that are substrates of the transporter BCRP should be used with caution when administered concomitantly with midostaurin and may need dose adjustment to maintain optimal exposure.
Co-administration of a single dose of dextromethorphan (CYP2D6 substrate) with a single dose of midostaurin (100 mg) did not cause any increase in dextromethorphan exposure (approximately 13% decrease in AUCs and approximately 28% decrease in Cmax) compared to administration of dextromethorphan alone. Similar to digoxin and rosuvastatin, the secondary PK parameters for dextromethorphan displayed minimal differences between the two treatments. Therefore, in line with the FDA and EMA classification of enzymes [9, 25], midostaurin has no inhibitory effect on CYP2D6 activity. Thus, clinically relevant DDIs of midostaurin with CYP2D6 substrates are unlikely to occur.
Furthermore, the AUCs of dextrorphan in the two treatment periods were comparable. The MR of dextromethorphan/dextrorphan was slightly lower when dextromethorphan was co-administered with midostaurin than when dextromethorphan was administered alone.
Because of safety concerns, no PK data are available after multiple 100-mg doses of midostaurin on the substrates from the current study. However, a physiologically based pharmacokinetic (PBPK) modeling would enable to bridge a DDI study from a current single dose to, for example, the effect of multiple doses (50 mg and 100 mg twice daily) on rosuvastatin (manuscript under preparation by Gu et al., 2024). The prediction results suggested a mild inhibition of BCRP/OATP1B by midostaurin after multiple doses.
Elderly patients diagnosed with FLT3-mutated AML are typically fragile because of comorbidities and are often polymedicated with treatments such as antifungal, prophylaxis, or immunosuppressants. To allow for a better clinical management of the patients on midostaurin treatment, it is important to understand the potential DDIs of this class of drugs.
Therefore, based on the present results, the labels of midostaurin (Rydapt) (the summary of product characteristics [5] and prescribing information [4]) were adjusted, and the corresponding interaction liabilities of these medications were removed or adapted as appropriate.
The PK parameters of midostaurin and its metabolites CGP52421 and CGP62221 were similar between the two arms, thereby showing the absence of treatment group effect between the arms.
In conclusion, the study results showed that the impact of a single dose of 100-mg midostaurin on digoxin was minor and led to an approximately 20% increase in the digoxin primary PK parameters (AUCs and Cmax). A mild increase of approximately 37–48% in rosuvastatin AUCs was observed. There was no increase in the primary PK parameters of dextromethorphan. Therefore, midostaurin has only a minor inhibitory effect on Pgp, a mild inhibitory effect on BCRP and OATP1B1 / OATP1B3, and no inhibitory effect on CYP2D6. The study treatments were well tolerated by participants with no new safety findings.
Data availability
Novartis is committed to sharing with qualified external researchers, access to patient-level data and supporting clinical documents from eligible studies. These requests are reviewed and approved by an independent review panel on the basis of scientific merit. All data provided are anonymized to respect the privacy of patients who have participated in the trial in line with applicable laws and regulations.
References
Garciaz S, Hospital MA (2023) FMS-like tyrosine kinase 3 inhibitors in the treatment of acute myeloid leukemia: an update on the emerging evidence and safety profile. Onco Targets Ther 16:31–45. https://doi.org/10.2147/OTT.S236740
Gallogly MM, Lazarus HM, Cooper BW (2017) Midostaurin: a novel therapeutic agent for patients with FLT3-mutated acute myeloid leukemia and systemic mastocytosis. Ther Adv Hematol 8(9):245–261. https://doi.org/10.1177/2040620717721459
Stone RM, Manley PW, Larson RA, Capdeville R (2018) Midostaurin: its odyssey from discovery to approval for treating acute myeloid leukemia and advanced systemic mastocytosis. Blood Adv 2(4):444–453. https://doi.org/10.1182/bloodadvances.2017011080
FDA (2023) RYDAPT® (midostaurin) capsules, for oral use initial U.S. approval: 2017 https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/207997s000lbl.pdf
EMA (2023, 26/7/2023). Rydapt Product Information. Retrieved 28 May 2024 from https://www.ema.europa.eu/en/documents/product-information/rydapt-epar-product-information_en.pdf
He H, Tran P, Gu H, Tedesco V, Zhang J, Lin W, Gatlik E, Klein K, Heimbach T (2017) Midostaurin, a novel protein kinase inhibitor for the treatment of acute myelogenous leukemia: insights from human absorption, metabolism, and excretion studies of a BDDCS II drug. Drug Metab Dispos 45(5):540–555. https://doi.org/10.1124/dmd.116.072744
Sechaud R, Gu H, Rahmanzadeh G et al (2024) Midostaurin drug interaction profile: a comprehensive assessment of CYP3A, CYP2B6, and CYP2C8 drug substrates, and oral contraceptives in healthy participants. Cancer Chemother Pharmacol 93(5):439–453. https://doi.org/10.1007/s00280-023-04635-3
Sechaud R, Sinclair K, Grosch K, Ouatas T, Pathak D (2022) Evaluation of drug-drug interactions between midostaurin and strong CYP3A4 inhibitors in patients with FLT-3-mutated acute myeloid leukemia (AML). Cancer Chemother Pharmacol 90(1):19–27. https://doi.org/10.1007/s00280-022-04448-w
FDA (2020) In vitro drug interaction studies — Cytochrome P450 enzyme- and transporter-mediated drug interactions guidance for industry. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/in-vitro-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions Accessed on 28 May 2024
Gu H, Dutreix C, Rebello S, Ouatas T, Wang L, Chun DY, Einolf HJ, He H (2018) Simultaneous physiologically based pharmacokinetic (PBPK) modeling of parent and active metabolites to investigate complex CYP3A4 drug-drug interaction potential: a case example of midostaurin. Drug Metab Dispos 46(2):109–121. https://doi.org/10.1124/dmd.117.078006
FDA (2016) LANOXIN® (digoxin) tablets for oral use initial U.S. approval: 1954. From https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/020405s013lbl.pdf Accessed on 28 May 2024
Digoxin SMPC (2022) https://www.medicines.org.uk/emc/product/5465/smpc/print Accessed on 28 May 2024
FDA (2023) CRESTOR (rosuvastatin) tablets for oral use initial U.S. approval: 2003 https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/021366s043s044lbl.pdf Accessed on 28 May 2024
Taylor CP, Traynelis SF, Siffert J, Pope LE, Matsumoto RR (2016) Pharmacology of dextromethorphan: relevance to dextromethorphan/quinidine (Nuedexta®) clinical use. Pharmacol Ther 164:170–182. https://doi.org/10.1016/j.pharmthera.2016.04.010
FDA (2023) Drug development and drug interactions | table of substrates, inhibitors and inducers. https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers
Tornio A, Filppula AM, Niemi M, Backman JT (2019) Clinical studies on drug–drug interactions involving metabolism and transport: methodology, pitfalls, and interpretation. Clin Pharmacol Ther 105:1345–1361. https://doi.org/10.1002/cpt.1435
Sheffield LJ, Phillimore HE (2009) Clinical use of pharmacogenomic tests in 2009. Clin Biochem Rev 30(2):55–65
Mas S, Gassò P, Álvarez S et al (2012) Intuitive pharmacogenetics: spontaneous risperidone dosage is related to CYP2D6, CYP3A5 and ABCB1 genotypes. Pharmacogenomics J 12:255–259. https://doi.org/10.1038/tpj.2010.91
Storelli F, Matthey A, Lenglet S, Thomas A, Desmeules J, Daali Y (2018) Impact of CYP2D6 functional allelic variations on phenoconversion and drug-drug interactions. Clin Pharmacol Ther 104(1):148–157. https://doi.org/10.1002/cpt.889
Stopfer P, Giessmann T, Hohl K et al (2016) Pharmacokinetic evaluation of a drug transporter cocktail consisting of digoxin, furosemide, metformin, and rosuvastatin. Clin Pharmacol Ther 100(3):259–267. https://doi.org/10.1002/cpt.406
Martin PD, Kemp J, Dane AL, Warwick MJ, Schneck DW (2002) No effect of rosuvastatin on the pharmacokinetics of digoxin in healthy volunteers. J Clin Pharmacol 42(12):1352–1357. https://doi.org/10.1177/0091270002042012008
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (2024) ICH Harmonised guideline. M12 Drug Interaction Studies ICH_M12_Step4_Guideline_2024_0521.pdf Adopted 21 May 2024
Clinical Pharmacogenetics Implementation Consortium (CPIC) (2015) Guideline for CYP2D6 and CYP2C19 genotypes and dosing of selective serotonin reuptake inhibitors https://cpicpgx.org/guidelines/
Dutreix C, Munarini F, Lorenzo S, Roesel J, Wang Y (2013) Investigation into CYP3A4-mediated drug-drug interactions on midostaurin in healthy volunteers. Cancer Chemother Pharmacol 72(6):1223–1234. https://doi.org/10.1007/s00280-013-2287-6
EMA (2012) Guideline on the investigation of drug interactions. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions-revision-1_en.pdf
Acknowledgements
The authors thank all the investigators, trial site staff, and volunteers who participated in the trials. The authors thank Twarita Chakraborty, PhD (Novartis Healthcare Pvt. Ltd. India) for providing medical writing support in accordance with Good Publication Practice (GPP4) guidelines.
Funding
The study was funded by Novartis.
Author information
Authors and Affiliations
Contributions
RS, HG, GR, OC, AB, and HM performed conception and design, data review and interpretation, drafting the manuscript, critical revision of the manuscript for important intellectual content, and final approval of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
R Sechaud, G Rahmanzadeh, and H Menssen are employees of Novartis Pharma AG, Switzerland. H Gu and O Chiparus, are employees of Novartis Pharmaceuticals Corporation, USA. A Breitschaft is an employee of Parexel International GmbH, Germany.
Ethics approval and informed consent
The study was performed in accordance with the ethical principles, which have their origin in the Declaration of Helsinki and are consistent with Good Clinical Practice and applicable regulatory requirements. All participants of the study provided written informed consent according to international standards. The study conducted at the Early Phase Clinical Unit of PAREXEL International GmbH in Berlin, Germany. The study protocol was reviewed and approved by the State Office of Health and Social Affairs Ethics Committee of Berlin (Landesamt für Gesundheit und Soziales Ethik-Kommission des Landes Berlin) for PAREXEL International GmbH. The A2121 study was registered under EudraCT number: EudraCT number: 2018-000971-34, and it obtained a favorable opinion from the IEC. The study participants were informed about the study procedures, risks, and benefits of their participation. Informed consent was documented by the investigator.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sechaud, R., Gu, H., Rahmanzadeh, G. et al. Effect of midostaurin on the pharmacokinetics of P-gp, BCRP, and CYP2D6 substrates: assessing potential drug-drug interactions in healthy participants. Cancer Chemother Pharmacol (2024). https://doi.org/10.1007/s00280-024-04683-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00280-024-04683-3