
Abstract
Qinghaosu, known as artemisinin (ARS), has been for over two millennia, one of the most common herbs prescribed in traditional Chinese medicine (TCM). ARS was developed as an antimalarial drug and currently belongs to the established standard treatments of malaria as a combination therapy worldwide. In addition to the antimalarial bioactivity of ARS, anticancer activities have been shown both in vitro and in vivo. Like other natural products, ARS acts in a multi-specific manner also against hematological malignancies. The chemical structure of ARS is a sesquiterpene lactone, which contains an endoperoxide bridge essential for activity. The main mechanism of action of ARS and its derivatives (artesunate, dihydroartemisinin, artemether) toward leukemia, multiple myeloma, and lymphoma cells comprises oxidative stress response, inhibition of proliferation, induction of various types of cell death as apoptosis, autophagy, ferroptosis, inhibition of angiogenesis, and signal transducers, as NF-κB, MYC, amongst others. Therefore, new pharmaceutically active compounds, dimers, trimers, and hybrid molecules, could enhance the existing therapeutic alternatives in combating hematologic malignancies. Owing to the high potency and good tolerance without side effects of ARS-type drugs, combination therapies with standard chemotherapies could be applied in the future after further clinical trials in hematological malignancies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
For over two millennia Qinghaosu, known as artemisinin (ARS) in Western cultures, has been one of the most common herbs prescribed to treat febrile symptoms in traditional Chinese medicine (TCM). The earliest mention occurs in the Hou Bei Ji Fang (A Handbook of Prescriptions for Emergency Treatment) by Ge Hong (317–420 A.D.) as a remedy with anti-pyretic activity. Subsequently, qinghaosu soup, pills, and powders have been described for relieving malaria symptoms. Later, the herbalist Li Shi Zhen published in Compendium of Materia Medica (1596) that “fever and colds” could be treated with qinghaosu preparations [1].
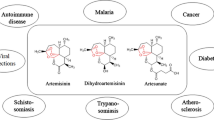
Artemisia annua L. came under the spotlight during the Vietnam War when the Vietnamese government asked China for help to manage the effects of malaria that infected nearly half of the militaries. A program for the discovery of new anti-malarial drugs, known as National Project 523 was launched in 1967 by the Chinese government that established a screening program of traditional Chinese plants. A modified extraction procedure was later able to isolate ARS, the active principle of Artemisia annua L. (sweet wormwood) [2]. The structure of Artemisia annua L. was elucidated in 1974, showing a sesquiterpene lactone, with an endoperoxide bridge essential for activity. Together with the corresponding derivatives, ARS attracted worldwide attention, and ARS-based combination therapies nowadays belong to the globally established standard treatments for malaria [1, 3]. Following the elucidation of this structure, many derivatives were synthesized with substitutions at the lactone carbonyl group to improve solubility in both water and oil. Water-soluble derivative such as artesunate (ART) and dihydroartemisinin (DHA), the latter considered to be the main active metabolite of ARS, and oil-soluble derivatives such as artemether (ARM) and arteether (Fig. 1) have been synthesized [1, 4].

Chemical structure of compounds derived from traditional Chinese medicine (TCM)
In the 1980s, the World Health Organization (WHO) officially recommended ARS-type derivatives for malaria treatment, particularly as a part of combination therapies with other antimalarial drugs for their impressive activity against multidrug-resistant forms of Plasmodium falciparum both in vitro and in vivo [1]. Tu Youyou was honored with the 2015 Nobel Prize for Physiology or Medicine [3] for having isolated and studied ARS which saved the lives of millions of malaria patients.
Numerous natural products originated from Chinese medicine exhibit anti-cancer activities which include antiproliferative, pro-apoptotic, anti-angiogenic effects, as well as regulating autophagy, reversing multidrug resistance, balancing immunity, and enhancing chemotherapy. ARS-like compounds also exert profound activity against tumor cells in vitro and in vivo [5,6,7,8] suggesting that ARS-type drugs could be applied in clinical oncology, probably as part of combination therapy [9,10,11,12].
Our group has been working over the past 2 decades with ARS-type compounds and has demonstrated the antiulcerogenic effects of two isolates from A. annua [13], activity against parasite [14] and the toxicity effects in pregnant mice [15]. Recently, the development of a transdermal bioadhesive treatment for children’s treatment has been demonstrated [16]. Driven by the demand for alternative drugs to improve the survival and decrease the relapse rate of acute myeloid leukemia (AML), our group has also demonstrated the antileukemic effects of natural products such as catechins and quercetin over the last decade [17, 18].
In addition to acute leukemias, other hematological malignancies can also benefit from natural products. Hematologic malignancies are cancers that begin in the cells of blood-forming tissue such as the bone marrow, or in the cells of the immune system and may be considered liquid tumors. A single germline or somatic mutation in the lymphohematopoietic stem cell may be prone to clonal expansion depending on new mutations acquired [19].
Multiple myeloma (MM), for example, is a malignancy of terminally differentiated plasma cells that primarily reside in the bone marrow (BM); however, in a later stage of the disease, they can also be detected in peripheral blood and extramedullary sites [20]. Lymphomas arise from lymphocytes that are at different stages of development and the subtype characteristics, B-cell and T-cell neoplasms reflect the cell from which they originated [21]. Leukemias are, in general, a heterogeneous group of disorders characterized by clonal expansion, abnormal proliferation of undifferentiated myeloid or lymphoid progenitors, and variable response to therapy [22].
Currently, chemotherapy, immunotherapy, and hematopoietic stem cell transplantation are the main therapeutic approaches against these diseases. Despite the progress in treatment, the outcome of AML in adult patients remains dismal [22]. Approximately 35%–40% of patients under the age of 60 years old are cured. The prognosis for the elderly is improving but remains grim. Furthermore, older patients have unfavorable cytogenetic features probably reflecting antecedent myeloid disorders which underwent clonal evolution. Relapse and/or disease refractoriness and drug resistance to standard chemotherapies are the major causes of treatment failure [23].
One current challenge in the treatment of cancer is to overcome cancer drug resistance. Combination therapies were applied to prevent resistance by combining drugs with different targets, modes of action and distribution of the side effects in the body to reduce toxicities. Natural products act in a multi-specific manner, exhibiting several modes of action simultaneously [11, 12]. For example, ARS-type drugs induce oxidation, and can lead to DNA damage and breaking of double-strands. Thus, many studies have shown the additive or synergistic effects of ARS-type drugs with standard chemotherapies. Doxorubicin, frequently used for chemotherapy of hematological cancers, exhibited profound synergism with ART in various MM cell lines [24]. Moreover, a synergistic interaction was observed with cytarabine plus ART or DHA in AML [25], as well as in other tumor cell lines. Interestingly, the spectrum of drugs that can be combined with ARS and its derivatives is remarkably broad and comprises natural products, radiotherapy, and photodynamic therapy, and also antibodies or recombinant proteins [11, 12].
A PubMed search using the keywords leukemia, lymphoma, multiple myeloma, artemisinin, artesunate, artemether, and dihydroartemisinin yielded a result of 82 papers published over the period of 25 years.
This review supplies an update on the mechanism of action, synergistic effects, and new hybrid compounds of ARS-type drugs, as well as a possible role in the treatment of hematological malignancies.
Cytotoxicity activity of ARS and derivatives
In the 1990s, many researchers described the cytotoxicity of ARS and its derivatives in leukemia cell lines (Table 1). Nine compounds isolated from Artemisia annua L., including terpenoids and flavonoids, were tested in vitro on a P388 murine lymphocytic leukemia cell line and ARS showed the highest cytotoxicity [26]. Furthermore, a panel of 55 tumor cell lines showed that ART, a semi-synthetic derivative, was most active against leukemia and colon cancer cell lines [5].
Efflux pumps of ATP-binding cassette (ABC) transporter family extrudes drug molecules that passively diffuse into cancer cells in an active ATP-consuming manner out of the cells. These pumps reduce the intracellular accumulation of many anticancer drugs to sub-therapeutic levels leading to the survival of cancer cells and consequently failure of chemotherapy. ARS-type drugs exhibit multi-specific interaction with P-gp. The derivatives can act on both sides, as a substrate or an inhibitor of P-gp. The currently ARS derivatives, ART, DHA, and ARM, are not transported by P-gp. In additional, they can act as an inhibitor of P-gp, which exhibits the potential to reverse MDR [27]. Efferth and collaborators tested 22 TCM derived compounds in multidrug-resistant acute lymphoblastic leukemia cell lines (CCRF-CEM), such as doxorubicin-selected P-glycoprotein (P-gp)/MDR1-expressing CEM/ADR5000, vinblastine-selected P-gp/MDR1-expressing CEM/VLB100, and epirubicin-selected multidrug resistance-related protein 1 (MRP1)-expressing CEM/E1000 sublines. ART, homoharringtonine, and bufalin were the most active and potent compounds and had the lowest IC50 in wild-type CCRF-CEM cells. ART modulated multidrug resistance and increased daunorubicin uptake in CEM/E1000 cells [28].
Ethanolic leaf extracts of A. annua from Brazilian (hybrid CPQBA 2/39 × PL5) and Chinese origins were tested on leukocytes and Molt-4 (human acute lymphoblastic leukemia) cell line for comparison with DHA. An increased biological activity was expected since A. annua leaves have flavonoids that may synergize with ARS. Both extracts had high antioxidant capacity and toxicity toward leukemia cells; however, whereas ethanolic extracts were more potent in killing Molt-4 cells at 24 h, DHA was significantly more potent than ethanolic extracts in killing Molt-4 cells at 48 and 72 h. Furthermore, DHA presented less toxicity in leukocytes than the Brazilian and Chinese extracts with the Brazilian extract revealing a better safety index (LD50 value 28.23 μg/ml) compared to Chinese extract at 24 h [29].
The combination of DHA and sodium salicylate (SS) significantly reduced cancer cell proliferation [30]; however, no interaction between DHA and SS was indicated. Nutritional supplements that affect the oxidative status of cells such as vitamin C, with antioxidant proprieties, and vitamin D3, and hydrogen peroxide (H2O2) with pro-oxidant properties, cause significant Molt-4 cell death when combined with DHA [31]. Furthermore, the interaction between H2O2 and DHA was found to be additive, which may be due to mechanisms similar to apoptosis mediation by reactive oxygen species (ROS).
Moreover, ART showed high cytotoxicity toward MV4-11 and MOLM-13 cell lines with the downregulation of SRC, a key protein in cell proliferation and survival. The antileukemic activity of ART was confirmed in various xenograft models resulting in significant survival prolongation [32]. Xenograft mice models transplanted with MOLM14 cells treated with ART-838, a semi-synthetic ARS-derived trioxane diphenylphosphate dimer, presented repression of tumor growth (83% less growth than in controls). Indeed, a B-acute lymphoblastic primagraft leukemia model treated with ART-838 had an extended mean survival of 20 days (55% prolongation) compared to controls [6]. Drenberg and coauthors also showed lower leukemic infiltration in a xenograft mice leukemia (ML-2) model treated with ART twice daily, in another xenograft model (MOLM-13), ART treatment added no survival benefit [25].
Interestingly, ARS suppressed proliferation via the downregulation of NOTCH1 signaling which may be up-regulated in several cancers [33]. In addition, ARS and derivatives also inhibited cell growth in MM and lymphoma cells. Holien and coauthors observed that ART decreased cell growth in nine MM and five lymphoma cell lines [34].
Role of iron
Since the onset of studies with ARS derivatives and cancer, many studies have shown a crucial role of iron in the anticancer activity of ARS-type drugs. Lai and Singh showed, for the first time in leukemia cell lines, that combined incubation of holotransferrin and DHA can selectively destroy cancer cells, whereas the effect was significantly less on normal lymphocytes [50]. The same group also showed that the addition of sodium butyrate (1 mM) to the culture medium together with DHA (20 μM) and holotransferrin (12 μM) acted synergistically in Molt-4 cell lines [51]. Moreover, Efferth and collaborators showed that iron (II)-glycine sulfate (Ferrosanol®) and transferrin enhanced the cytotoxicity of ART, maltosyl-β-cyclodextrin-encapsulated-ART (ART-MCD) and ARS against leukemia cell lines compared to each derivative without iron. Furthermore, expression levels of mitochondrial aconitase (ACO2) and ceruloplasmin (CP) correlated with the IC50 values of several artemisinin derivatives when administered with ferrous iron [52]. ART treatment also regulated the expression of drug efflux pumps involved in iron homeostasis as observed after ART incubation with the CCRF-CEM cell line which increased the expression of the ATP-binding cassette (ABC) transporter ABCB6 and reduced ABCB7 expression [53].
In fact, tumor cells express significantly more transferrin receptor on their cell surface than normal cells and transferrin endocytosis is higher in tumor cells compared to normal cells. Tagging an ARS analog to transferrin, both iron and ARS were transported into cancer cells and the “tagged-compound” was very potent and selective in killing cancer cells [54]. To improve the cytotoxicity of ARS, the same group of researchers covalently tagged ARS to a peptide that binds to a cavity on the surface of the transferrin receptor (ARS-TfR). After endocytosis, iron released from TfR reacted with the ARS moiety and formed free radicals, leading to cell death. They also demonstrated that ARS-TfR was more potent and an extremely selective anti-tumor agent compared to ARS itself [55]. Two synthetic ARS compounds, ARS dimer-alcohol (dimer-OH) and ARS-tagged holotransferrin (ART-TF), were more potent and showed no significant cross-resistance toward a DHA-resistant Molt-4 (RTN) cell line [56]. Indeed, combined ARS, pretreated with holotransferrin, and hyperbaric oxygen, resulted in an additional 22% decrease in growth of Molt-4 cell line compared with ARS treated alone [57].
Moreover, DHA, 10β-(p-bromophenoxy) DHA (PBrDHA), and 10β-(p-fluorophenoxy) DHA (PFDHA) demonstrated selective cytotoxicity activity of the endoperoxide group toward leukemia cells over normal peripheral blood mononuclear cells (PBMC). In HL-60 cells, the compounds with endoperoxide induced caspase-dependent apoptotic cell death characterized by mitochondrial membrane depolarization, caspases-3 and -7 activation, and sub-G0/G1 DNA formation, concentration- and time-dependent. Deoxy-10β-(p-fluorophenoxy) DHA (dPFDHA), which lacks the endoperoxide bridge, was less active, confirming the importance of this functional group [35]. In addition, ARS-type drugs exerted effects in lymphoma cells. Wang and coauthors demonstrated enhanced cytotoxicity effects of co-treatment of DHA plus holotransferrin in T-cell lymphoma cells with inhibition of TfR mRNA expression [58] (Table 2).
Oxidative stress response
Efferth showed that ART induced apoptosis of leukemia cells mainly through the mitochondrial pathway via generation of reactive oxygen species (ROS) [32, 63]. Furthermore, the induction of ROS by ART was accompanied by increased phosphorylation of histone H2AX (γ-H2AX), a marker for double-strand DNA damage, and activation of c-Jun N-terminal kinase (JNK), a mitogen-activated protein kinases (MAPK) family member [32], suggesting a strong pro-oxidant effect of ART on leukemic cells.
Moreover, Zhang and collaborators showed that the cytotoxicity induced by DHA correlated with superoxide (O2−) levels, measured by dihydroethidine (HET) fluorescence, in a concentration-dependent way. Furthermore, the co-incubation with the superoxide scavenger TEMPOL dramatically reduced the HET fluorescence. Indeed, an increase in protein levels of four antioxidant enzymes, catalase, copper/zinc-superoxide dismutase (CuZnSOD), manganese-superoxide dismutase (MnSOD), and glutathione peroxidases 1 and 2 (GPX 1/2) were observed in Molt-4 cell lines [64]. Another evidence of the antileukemic mechanism of ARS derivatives through ROS generation was obtained using N-tert-butyl-alpha-phenylnitrone (PBN), a compound that effectively sequesters free radicals, or deferoxamine (DX), an iron chelating agent, which attenuated the cytotoxicity of DHA. Altogether, these results suggest that DHA induced the formation of toxic-free radicals via an iron-mediated process [65] (Table 3).
Cell cycle arrest
Two novel derivatives, containing cyano and aryl groups at C-10 carbon of the ARS structure, showed a potent antiproliferative in vitro effect, leading to cell cycle arrest (Table 4) in G0/G1 phase [38]. The active metabolite of ARS, DHA, also arrested the cell cycle at G0/G1 phase with downregulation of cyclin D, CDK2, and CDK4 [36]. Furthermore, ARS-type drugs induced cell cycle arrest in MM and lymphoma cells. DHA induced cell cycle arrest at sub-G0/G1 phase in U266 MM cells [40]. Moreover, ARM induced cell cycle arrest at G0/G1 phase in diffuse large B-cell lymphoma (DLBCL) cells with a decrease of cyclin D1, CDK2, and CDK4 [44]. In addition, the hydrophilic ARS-derivative, ART, caused cell cycle arrest in both G0/G1 and G2/M phase in adult T-cell leukemia/lymphoma cells with decreased levels of activator protein-1 (AP-1) and NF-κB signaling [49].
Programmed cell death
Apoptosis
Numerous studies have demonstrated the role of ARS and derivatives in the induction of apoptosis (Table 5). Singh and Lai demonstrated that DHA induces apoptosis, but not necrosis in Molt-4 cell line [61]. Increased phosphorylation of p38 mitogen-activated protein kinases (MAPK), but not JNK, or extracellular signal-regulated kinase (ERK) is required for DHA-induced apoptosis through both intrinsic and extrinsic pathways in HL-60 cells [59] while ROS is dispensable. Furthermore, DHA induced apoptosis of U937, Jurkat, and HL-60 cell lines, primary human AML and acute lymphoma leukemia (ALL) cells in vitro accompanied by inactivation of MEK/ERK, MCL-1 down-regulation, caspase activation and finally apoptosis. Moreover, DHA-mediated inhibition of xenograft tumor growth associated with apoptosis induction, MCL-1 down-regulation, and ERK inactivation [67].
The active metabolite, DHA and its derivative X-11, acted through a NOXA-mediated pathway and downregulated MCL-1 through a new cascade of O2−/FOXO3a/NOXA [7]. The endoperoxide moiety of DHA or X-11 interacted with iron to form carbon-center radicals. Thereby, increased levels of O2− through the endoperoxide moiety contributed to NOXA induction and finally apoptosis. NOXA protein bound to MCL-1 leads to Bak activation, and finally apoptosis [7].
The Bcr/Abl fusion gene is the pathogenic factor for chronic myeloid leukemia development. Lee and colleagues showed that DHA blocked BCR/ABL tyrosine phosphorylation and suppressed, in a concentration-dependent manner, downstream signaling pathways such as AKT and ERK, and also suppressed NF-κB protein expression, leading to the release of cytochrome c from mitochondria and activation of caspase cascade [68]. Furthermore, DHA suppresses Bcr/Abl mRNA amplification in imatinib-resistant cell lines [69].
DHA further downregulated MCL-1 expression, though not mRNA expression, suggesting a MCL-1 turnover by the proteasome. Ddit3, which encodes CHOP protein, a key regulator of the endoplasmic reticulum (ER) stress pathway contributed to repression of MCL-1 protein by DHA in B-acute lymphoblastic leukemia (B-ALL) mice cells containing the BCR-ABL protein (BCR-ABL+). Interestingly, DHA synergized with ABT-263, a BH3-mimetic, inducing apoptosis, supporting the hypothesis that DHA can repress MCL-1 protein expression. Combined treatment of DHA and ABT-263 extended survival and showed repression in circulating leukemia cells of mice transplanted with BCR-ABL+B-ALL cells. Furthermore, a decrease of MCL-1 protein was observed in ex vivo analysis of splenic blast cell of BCR-ABL+B-ALL mouse after in vivo DHA treatment [70]. Indeed, in HL-60 cells, DHA treatment decreased TfR expression at the mRNA and protein level, upregulated the proapoptotic protein Bax and downregulated the antiapoptotic protein BCL-2, resulting in the activation of caspase-3 and apoptosis [60].
Combined treatment of ARS derivatives with other compounds has also shown effects on the apoptosis of leukemic cells. For example, DHA and an inhibitor of 6-phosphogluconate dehydrogenase (6PGD) induced synergistic apoptosis of the chronic myeloid leukemia K562 cell line through AMPK (AMP-activated protein kinase) signaling pathway. Moreover, combined treatment significantly decreased tumor growth in a xenograft model beyond the increased levels of phospho-AMPK [71]. ART and arsenic trioxide (ATO) also increased K562 cell line apoptosis and necrosis [72].
ART induced apoptosis of leukemic T cells through the intrinsic pathway with cytochrome c release and caspase-9 activation [63]. In another study, ART treatment of KBM-5 (chronic myeloid leukemia) cell line exerted apoptosis effects through suppression of multiple signaling pathways including suppression of p38/ERK/STAT5/CREB phosphorylation [8]. Moreover, ART showed high cytotoxicity toward MV4-11 and MOLM-13 cells, Bcl-2 reduction, loss of mitochondrial membrane potential (MMP) and induction of the intrinsic mitochondrial pathway [32]. In vitro studies also showed that, in the acute monocytic leukemia cell line THP-1, ART decreased STAT3 protein levels and activated caspase-3 and -8. These results were reproduced in a xenograft model [73]. Furthermore, Cao and collaborators described DHA-induced apoptosis of THP-1 cell line through AKT and ERK downregulation at mRNA and protein levels and activation of caspase-3 [74].
Drenberg and collaborators showed potent activity of ART and DHA in AML cell lines (ML-2, CMS, MV4-11, U937, M07e, MOLM-13) representing subtypes and genetic lesions associated with high risk and poor prognosis. MV4-11 cells were the most sensitive with IC50 value of 0.092 μM and 0.24 μM for ART and DHA, respectively. Both derivatives induced apoptosis, caspase-3 and -7 activation, in addiction, ROS and lysosomal induction. ARS derivatives also showed synergistic interaction with cytarabine (Ara-C) when drugs were administered sequentially, pre-treatment with DHA followed by Ara-C (CI range 0.37–0.72) and pre-treatment with Ara-C followed by ART (CI range 0.49–0.9). ART produced modest inhibition in patient samples with the same mutations of the cell lines and the combination therapy with Ara-C demonstrated synergistic effects [25]. The semi-synthetic ARS-derived trioxane diphenylphosphate dimer 838 (ART-838) was more potent than ART in 23 leukemia cell lines and caspase-dependent apoptosis was observed [6].
ART also induced apoptosis in MM and lymphoma cell lines, with downregulation of MYC and anti-apoptotic proteins of BCL-2 family beyond caspase-3 activation [34]. Furthermore, ART overcame drug resistance in MM lineage and induced apoptosis predominantly through the non-caspase-mediated pathway by increasing ROS levels and leading to a loss of mitochondrial membrane permeabilization early in time. Thereafter, ART translocated AIF and EndoG, factors of this type of apoptosis, from the mitochondria to the cytoplasm and subsequently to the nucleus inducing apoptosis together with increased levels of superoxide [24]. Wang and the authors observed an apoptosis induction through JNK signaling pathway activation in MM cell line by DHA [40]. Li and collaborators showed apoptosis induction and proliferation inhibition of SP2/0 (murine myeloma) cells together with decreased NF-κB protein and transcriptional activity in addition to increased IκBα, which inactivated NF-κB when they were combined, by ART [43].
The c-Myc transcriptional factor regulated cell proliferation, differentiation, and apoptosis. The combined use of DHA and siNotch1 therapy induced the reduction of Notch1 and c-Myc levels, the last downstream target of Notch1, at mRNA and protein levels, and also increased caspase-3 mRNA and protein levels, subsequently inducing apoptosis and suppressed cell proliferation in T-cell lymphoma cells [46]. Furthermore, DHA reduced c-Myc protein expression at the transcriptional level and could exert antitumor effect by inhibiting the AKT/GSK3β pathway in T-cell lymphoma cells. DHA also induced apoptosis with increased Bax/BCL-2 ratio [47] and through Bak-dependent intrinsic pathway [75].
ART further demonstrated a potent anti-tumor effect against B-cell lymphoma cells. ART induced ER stress and unfolded protein response (UPR) through increased levels of ATF-4, ATF-6, and CHOP at mRNA and protein levels in BL-41 (Burkitt lymphoma) and SU-DHL-6 (large cell lymphocyte) cells, leading to apoptosis. In addition, ART suppressed the overall metabolism, affecting both respiration and glycolysis [45]. Moreover, Sieber and collaborators showed a potent cytotoxic effect of combination therapy using mAb rituximab and ART in B-cell lymphoma cells in which upstream transcriptional factors of apoptosis process, YY1 and Sp1, regulated Fas/CD95 expression leading to the intrinsic apoptosis pathway. Combination therapy also induced downregulation of antioxidant proteins [48]. Indeed, ART induced apoptosis through caspase-dependent and -independent pathway together with a suppression of NF-κB and activator protein-1 (AP-1) signaling in adult T-cell leukemia/lymphoma [49] cells. Furthermore, the lipophilic ARS-derivative, ARM, induced apoptosis in DLBCL cell lines with caspase-3 induction [44].
Autophagy
Regulation of cancer cell autophagy by ARS-type drugs involves decreased phosphorylation of proteins of the PI3K/AKT/mTOR pathway and increased levels of Beclin1 mediated by the JNK pathway. ARS and derivatives also inhibit NF-κB activity through blocking Rel/p65 translocation to the nucleus and activate ER stress through the stress-regulated protein p8 [76]. In leukemia, Wang and collaborators reported that DHA induced autophagy of K562 cells, pretreated with holotransferrin, through ROS generation followed by LC3-II expression and caspase-3 activation [62].
A new ARS-derivative, SM1044, induced autophagy mediated by the CaMKK2/AMPK/ULK1 pathway through promoting de novo synthesis of ceramide in lymphoma cells. Furthermore, the new derivative also induced autophagy-dependent apoptosis through acetylation of Survivin protein and enhanced interaction of Survivin and LC3-II [77]. Moreover, DHA treatment suppressed NF-κB activity by preventing the translocation of the Rel/p65 subunit to the nucleus consequently contributing to autophagy in MM and leukemia cell lines [78].
Ferroptosis
Ferroptosis is an iron-dependent programmed cell death pathway dependent on the lipid peroxidation process. In contrast with other programmed cell deaths, reduction of glutathione peroxidase 4 (GPX4) repairs enzymes, accumulation of lipid peroxidation products and ROS derived from iron metabolism characterizes ferroptosis. ARS-type drugs are capable of reversing ferroptosis resistance of head and neck cancer cells, through inhibition of the Nrf2-ARE antioxidant signaling [79]. Moreover, in pancreatic cells, the induction of ferroptosis by ART is enhanced by GRP78 knockdown, chaperone of ER [80]. Recently, Du and collaborators described that DHA treatment of the HL-60 leukemic cell line induced ferroptosis through degradation of the heavy ferritin chain by autophagy [36]. Indeed, ART activated ATF4/CHOP/CHAC1 pathway, an ER stress response, and enhanced ferroptosis [66] in Burkitt’s lymphoma cell lines.
Anti-angiogenic effects
Angiogenesis plays a fundamental role in the neoplastic process. Effects of ARS-type drugs on cancer cells rely on perturbations of the MAPK pathway, which comprise ERK1/2 reduction or JNK and p38 MAPK activation, NF-κB inhibition and also AKT and mTOR inhibition promoting induction of proliferation and apoptosis of endothelial cell and reduction on the vascular endothelial growth factor (VEGF) production [81]. VEGF is the most potent angiogenic factor and plays an important role in the development and progression of leukemias (Table 6) and MM. Interestingly, ART and DHA reduced the expression and secretion of VEGF in RPMI8226 myeloma and K562 leukemia cell lines, respectively [37, 41] and inhibited the formation of new microvessels in an in vivo model of chick chorioallantoic membrane (CAM) loaded with RPMI8226 conditioned medium (CM) under hypoxic conditions [42]. Furthermore, ART, the other water soluble ARS-derivative compound, inhibited the production of new microvessel on aortic sprouting, reduced CML angiogenesis in an in vivo model of CAM [82], inhibited vascular endothelial cell (HUVEC) migration and reduced expression of VEGF and angiopoietin-1 (Ang-1) proteins [41].
Differentiation induced by ARS and derivatives
ARS potentiated 1α,25-dihydoxyvitamin D3 and all-trans retinoic acid induced differentiation in HL-60 promyelocytic leukemia cells. ARS in combination with low concentration of 1,25-(OH)2D3 increased HL-60 differentiation into monocytes through ERK and PKC pathways with an increased PKCβ1 isoform. Indeed, ARS plus all-trans retinoic acid induced cell differentiation into granulocytes through ERK pathway [83]. ARS has a synergistically effect with interferon-α (IFN-α) in enhancing HL-60 cell differentiation through PKCα/ERK signaling pathway [84]. On the other hand, DHA inhibited erythroid cell differentiation with a decreased expression of glycophorin A (GpA) surface receptor and γ-globin synthesis [85] (Table 7).
New hybrid compounds
Hybridization is a new approach used to improve the activity of chemical compounds [86] (Table 8). Homodimers of two artesunic acid molecules and heterohybrids of artesunic acid and betulin, a natural product that exhibited cytotoxicity activity toward human lung cancer, were tested in human sensitive and multidrug-resistant cells. CEM/ADR5000, multidrug-resistant leukemia cells, were not cross-resistant to the novel compounds and sensitivity was also observed for artesunic acid homodimer in CCRF-CEM cell line. Furthermore, artesunic acid and artesunic acid homodimer increased ROS formation and induced apoptosis and G0/G1 cell cycle arrest [87].
As structure modification may improve ARS anti-cancer activity, a series of DHA chalcone hybrids and derivatives were synthetized [88] and demonstrated a high antiproliferative and cytotoxicity effect in HL-60 cell lines [89]. Additionally, new ARS–spermidine conjugates were designed to upregulate polyamine transporter. Amine-linked conjugates were approximately 1.5–2 times more active than amide-linked conjugates in HL-60 leukemia cells and all of them were higher than DHA [90]. Furthermore, new dimers phosphate ester, screening against human leukemia and normal cell lines, exhibited very high potency against cancer cells with no toxicity to normal cells [91]. Dimers and trimers were more active against CCRF-CEM cells [92]. Interestingly, molecular docking showed that most derivatives revealed similar binding sites at the transmembrane region of the multidrug P-glycoprotein [93] transporter.
Three hybrid molecules having aliphatic, aromatic, or alcoholic linkers were analyzed for their activity against human multidrug-resistant leukemia cells. The multidrug-resistant cells were not cross-resistant to any of the dimers [94]. Therefore, new ARS-derived hybrids incorporating cholic acid moieties were efficient against the CCRF-CEM and multidrug-resistant CEM/ADR5000 cells. The majority of the compounds proved to be more active than ARS and ART alone [95]. Furthermore, a series of novel 1,2,4-trioxane-based hybrids incorporating egonol and/or ferrocene fragments were synthesized and showed remarkable cytotoxicity toward CCRF-CEM cells or against multidrug-resistant leukemia cells [96, 97].
ARS-based hydroxamic acids were synthesized with a possible dual mechanism of both endoperoxide bridge and hydroxamic acid moiety. All the compounds exhibited moderate inhibition against HL-60 cells and, interestingly, docking studies of two very active compounds showed that they were capable of binding to HDAC2 with high affinity, even higher than the HDAC inhibitor, suberoylanilide hydroxamic acid (SAHA) [98].
Another modification of ARS derivative that has been demonstrated to improve their activity was a sugar attachment which enhanced specificity of drug delivery, polarity, and solubility, to attenuate toxicity. Thus, N-glycosylated DHA-piperazine, glucose, maltose, and ribose were most active and specific against leukemic cells than DHA and artemisone [99].
A novel synthesis of quinazoline–ARS [100] and thymoquinone–ARS [101] hybrids were investigated and showed promising results for multidrug-resistant cell line [100, 101]. Furthermore, a hybrid containing egonol, isolated from Styrax officinalis L. and homoegonol, with anti-inflammatory, antioxidant and anticancer activity, combined with thymoquinone and ARS were synthetized and demonstrated high activity [102].
The antimalarial and anticancer mechanism of ARS-type drugs
This class of compounds acts in a multi-specific manner, exhibiting several modes of action simultaneously. Interestingly, mechanisms of induction of malaria parasite death can be transposable to cancer cells. During the erythrocyte infection, plasmodia consume hemoglobin as a source of amino acids, leading to the generation of ROS by heme–iron which cleaves the endoperoxide moiety of the ARS-type drugs by a Fe+2 Fenton-type reaction, with more free radical intermediate formation [103]. This can lead to macromolecular damage and, consequently, death to the parasite.
Another mechanism of action is based on its structural similarity with thapsigargin, which is a highly specific inhibitor of sarcoendoplasmic reticulum Ca+2-ATPase (SERCA). Thus, ARS has been shown to inhibit the SERCA orthologue (PfATP6) of the P. falciparum [104] and that DHA, the active metabolite of ARS-type drugs, damages proteins and inhibits the proteasome, causing ER stress response [105].
Cancer cells have more intracellular iron than normal cells, thus ARS-like drugs can react with intracellular free iron to form cytotoxic free radicals that promote the death of cancer cells. A plethora of articles has revealed the importance of endoperoxide moiety for anticancer activity. Thus, the cleavage of the endoperoxide bridge leads to reactive oxygen species (ROS) formation and consequently, oxidative stress that induces cancer cell death. Moreover, the endoperoxide-reduced form of ARS, deoxyartemisinin, which lacks the peroxide, is inactive [7] against leukemic cells. Furthermore, similar to the mechanism proposed for antimalarial activity, the anticancer activity of ARS-type drug can be targeting the unfolded protein response (UPR) and endoplasmic reticulum (ER) response of the cancer cell leading to apoptosis or ferroptosis [66].
Most of the changes in the ARS structure that improve the pharmacological properties and pharmacokinetics of ARS compounds occur at C-10 carbon of DHA, enabling derivation to drugs with increased biological efficacy, reduced undesired side effects, selected profile (e.g., lower toxicity), and better bioavailability.
Conclusions
In conclusion, ARS and its derivatives are active against hematological malignancy cells in vitro and in vivo [6, 7, 25]. Hybrids, dimers, and trimers improved cytotoxicity against leukemia cells, proposing high potency even in MDR cells [87, 92]. It is speculated that the main mechanism of action is the bioactivation of the endoperoxide pharmacophore group by iron that leads ROS formation and free radical intermediates formation [50, 54, 63], inducing cell death as occurs with the malaria parasite. ARS-type drugs also enhance chemotherapeutic anticancer activity contributing mechanistically to additive or synergistic effects [25]. Collectively, this review summarizes the main studies that indicated the role of ARS-type drugs in the treatment of leukemia, lymphoma, and multiple myeloma through apoptotic and non-apoptotic cell death (downstream mechanism), mainly by inducing cell cycle arrest at G0/G1 phase, inhibition of proliferation, inhibition of angiogenesis with downregulation of VEGF, and signal transduction modulation (Fig. 2). Importantly, combination therapies with standard chemotherapy drugs enhanced the potential of ARS-type drugs in adjuvant therapy in clinical oncology. No clinical trial for hematological malignancies has been conducted yet. Thus, clinical trials should be encouraged to provide more compelling evidence regarding the use of ARS-derivatives in hematological cancer treatment.

Summary of the mechanisms of action of ARS-type drugs against hematological malignancies
Abbreviations
- AML:
-
Acute myeloid leukemia
- AMoL:
-
Acute monocytic leukemia
- AMPK:
-
AMP-activated protein kinase
- AP-1:
-
Activator protein-1
- Ara-C:
-
Cytarabine
- ARM:
-
Artemether
- ARS:
-
Artemisinin
- ART:
-
Artesunate
- ATO:
-
Arsenic trioxide
- B-ALL:
-
B-acute lymphoblastic leukemia
- BM:
-
Bone marrow
- CML:
-
Chronic myeloid leukemia
- CuZnSOD:
-
Copper, zinc-superoxide dismutase
- DA:
-
Decitabine
- DHA:
-
Dihydroartemisinin
- DLBCL:
-
Diffuse large B-cell lymphoma
- DM:
-
Dexamethasone
- DX:
-
Deferoxamine
- ER:
-
Endoplasmic reticulum
- ERK:
-
Extracellular signal-regulated kinase
- GEM:
-
Genetically-engineered mouse
- GpA:
-
Glycophorin A receptor
- GPX1/2:
-
Glutathione peroxidases 1 and 2
- HBO2 :
-
Hyperbaric oxygen
- HET:
-
Dihydroethidine
- H2O2 :
-
Hydrogen peroxide
- IFN-α:
-
Interferon-α
- JNK:
-
C-Jun-N-terminal kinase
- MAPK:
-
Mitogen-activated protein kinases
- MM:
-
Multiple myeloma
- MMP:
-
Mitochondrial membrane potential
- MnSOD:
-
Manganese-superoxide dismutase
- MTT:
-
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- O2 − :
-
Superoxide
- PBMC:
-
Peripheral blood mononuclear cells
- PBN:
-
N-Tert-butyl-alpha-phenylnitrone
- P-gp:
-
P-Glycoprotein
- PPP:
-
Pentose phosphate pathway
- ROS:
-
Reactive oxygen species
- SS:
-
Sodium salicylate
- STAT3:
-
Signal transducer and activator of transcription-3
- TCM:
-
Traditional Chinese medicine
- TfR:
-
Transferrin receptor
- VEGF:
-
Vascular endothelial growth factor
- WHO:
-
World Health Organization
References
Klayman DL (1985) Qinghaosu (artemisinin): an antimalarial drug from China. Science 228:1049–1055
Miller LH, Su X (2011) Artemisinin: discovery from the Chinese herbal garden. Cell 146:855–858
Tu Y (2016) Artemisinin—a gift from traditional chinese medicine to the world (nobel lecture). Angew Chem. https://doi.org/10.1002/anie.201601967
Hien TT, White NJ (1993) Qinghaosu. Lancet 341:603–608
Efferth T, Dunstan F, Sauerbrey A, Miyachi H, Chitambar CR (2001) The anti-malarial artesunate is also active against cancer. Int J Oncol 18:767–773
Fox JM, Moynihan JR, Mott BT, Mazzone JR, Anders NM, Brown PA, Rudek MA, Liu JO, Arav-Borger R, Posner GH, Civin CI, Chen X (2016) Artemisinin-derived dimer ART-838 potently inhibited human acute leukemias, persisted in vivo, and synergized with antileukemic drugs. Oncotarget 7:7268–7279
Zhao X, Zhong H, Wang R, Liu D, Waxman S, Zhao L, Jing Y (2015) Dihydroartemisinin and its derivative induce apoptosis in acute myeloid leukemia through Noxa-mediated pathway requiring iron and endoperoxide moiety. Oncotarget. https://doi.org/10.18632/oncotarget.3336
Kim C, Lee JH, Kim S-H, Sethi G, Ahn KS (2015) Artesunate suppresses tumor growth and induces apoptosis through the modulation of multiple oncogenic cascades in a chronic myeloid leukemia xenograft mouse model. Oncotarget. https://doi.org/10.18632/oncotarget.3004.
Lam NS, Long X, Wong JW, Griffin RC, Doery JCG (2019) Artemisinin and its derivatives: a potential treatment for leukemia. Anticancer Drugs 30:1–18
Efferth T (2017a) From ancient herb to modern drug: artemisia annua and artemisinin for cancer therapy. Semin Cancer Biol. https://doi.org/10.1016/j.semcancer.2017.02.009
Efferth T (2017b) Cancer combination therapies with artemisinin-type drugs. Biochem Pharmacol. https://doi.org/10.1016/j.bcp.2017.03.019
Kumar MS, Yadav TT, Khair RR, Peters GJ, Yergeri MC (2019) Combination therapies of artemisinin and its derivatives as a viable approach for future cancer treatment. Curr Pharm Des 25:3323–3338
Foglio MA, Dias PC, Antônio MA, Possenti A, Ferreira Rodrigues RA, Ferreira da Silva E, Rehder VL, de Carvalho JE (2002) Antiulcerogenic activity of some sesquiterpene lactones isolated from artemisia annua. Planta Med 68:515–518
Ives CS, Pedro MM, Ilza MOS, Mary AF, Eduardo L, Sérgio NE, Chagas ACS (2017) Anthelmintic activity of Artemisia annua in sheep-model. J Med Plants Res 11:137–143
Boareto AC, Muller JC, Bufalo AC, Botelho GGK, de Araujo SL, Foglio MA, de Morais RN, Dalsenter PR (2008) Toxicity of artemisinin [Artemisia annua L.] in two different periods of pregnancy in Wistar rats. Reprod Toxicol 25:239–246
Volpe Zanutto F, McAlister E, Marucci Pereira Tangerina M, Fonseca-Santos B, Costa Salles TH, Oliveira Souza IM, Brisibe A, Vilegas W, Chorilli M, d’Ávila MA, Donnelly RF, Foglio MA (2019) Semisynthetic derivative of Artemisia annua-Loaded transdermal bioadhesive for the treatment of uncomplicated malaria caused by Plasmodium falciparum in children. J Pharm Sci 108:1177–1188
Torello CO, Shiraishi RN, Della Via FI, de Castro TCL, Longhini AL, Santos I, Bombeiro AL, Araujo Silva CL, Queiroz ML, Rego EM, Saad STO (2018) Reactive oxygen species production triggers green tea-induced anti-leukaemic effects on acute promyelocytic leukaemia model. Cancer Lett 414:116–126
Alvarez MC, Maso V, Torello CO, Ferro KP, Saad STO (2018) The polyphenol quercetin induces cell death in leukemia by targeting epigenetic regulators of pro-apoptotic genes. Clin Epigenetics. https://doi.org/10.1186/s13148-018-0563-3
Bowman RL, Busque L, Levine RL (2018) Clonal Hematopoiesis and Evolution to Hematopoietic Malignancies. Cell Stem Cell 22:157–170
Kumar SK, Rajkumar V, Kyle RA, Van Duin M, Sonneveld P, Mateos MV, Gay F, Anderson KC (2017) Multiple myeloma. Nat Rev Dis Prim. https://doi.org/10.1038/nrdp.2017.46
Armitage JO, Gascoyne RD, Lunning MA, Cavalli F (2017) Non-Hodgkin lymphoma. Lancet 390:298–310
Gurnari C, Voso MT, Maciejewski JP, Visconte V (2020) From bench to bedside and beyond: therapeutic scenario in acute myeloid leukemia. Cancers (Basel) 12:1–20
De Kouchkovsky I, Abdul-Hay M (2016) Acute myeloid leukemia: a comprehensive review and 2016 update. Blood Cancer J. https://doi.org/10.1038/bcj.2016.50
Papanikolaou X, Johnson S, Garg T, Tian E, Tytarenko R, Zhang Q, Stein J, Barlogie B, Epstein J, Heuck C (2014) Artesunate overcomes drug resistance in multiple myeloma by inducing mitochondrial stress and non-caspase apoptosis. Oncotarget 5:4118–4128
Drenberg CD, Buaboonnam J, Orwick SJ, Hu S, Li L, Fan Y, Shelat AA, Guy RK, Rubnitz J, Baker SD (2016) Evaluation of artemisinins for the treatment of acute myeloid leukemia. Cancer Chemother Pharmacol 77:1231–1243
Zheng G-Q (1993) Cytotoxic terpenoids and flavonoids from Artemisia annua. Planta Med. https://doi.org/10.1055/s-2006-959408
Wang Y, Li Y, Shang D, Efferth T (2019) Interactions between artemisinin derivatives and P-glycoprotein. Phytomedicine. https://doi.org/10.1016/j.phymed.2019.152998
Efferth T, Davey M, Olbrich A, Rücker G, Gebhart E, Davey R (2002) Activity of drugs from traditional Chinese medicine toward sensitive and MDR1- or MRP1-overexpressing multidrug-resistant human CCRF-CEM leukemia cells. Blood Cells, Mol Dis 28:160–168
Singh NP, Ferreira JFS, Park JS, Lai HC (2011) Cytotoxicity of ethanolic extracts of Artemisia annua to molt-4 human leukemia cells. Planta Med 77:1788–1793
Wickerath M, Singh NP (2014) Additive cytotoxic effects of dihydroartemisinin and sodium salicylate on cancer cells. Anticancer Res 34:3399–3401
Gerhardt T, Jones R, Park J, Lu R, Chan HW, Fang Q, Singh N, Lai H (2015) Effects of antioxidants and pro-oxidants on cytotoxicity of dihydroartemisinin to Molt-4 human leukemia cells. Anticancer Res 35:1867–1872
Kumar B, Kalvala A, Chu S, Rosen S, Forman SJ, Marcucci G, Chen CC, Pullarkat V (2017) Antileukemic activity and cellular effects of the antimalarial agent artesunate in acute myeloid leukemia. Leuk Res 59:124–135
Ohtaka M, Itoh M, Tohda S (2017) BMI1 inhibitors down-regulate NOTCH signaling and suppress proliferation of acute leukemia cells. Anticancer Res 37:6047–6053
Holien T, Olsen OE, Misund K, Hella H, Waage A, Rø TB, Sudan A (2013) Lymphoma and myeloma cells are highly sensitive to growth arrest and apoptosis induced by artesunate. Eur J Haematol 91:339–346
Mercer AE, Maggs JL, Sun XM, Cohen GM, Chadwick J, O’Neill PM, Park BK (2007) Evidence for the involvement of carbon-centered radicals in the induction of apoptotic cell death by artemisinin compounds. J Biol Chem 282:9372–9382
Du J, Wang T, Li Y, Zhou Y, Wang X, Yu X, Ren X, An Y, Wu Y, Sun W, Fan W, Zhu Q, Wang Y, Tong X (2019) DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic Biol Med 131:356–369
Lee J, Zhou HJ, Wu XH (2006) Dihydroartemisinin downregulates vascular endothelial growth factor expression and induces apoptosis in chronic myeloid leukemia K562 cells. Cancer Chemother Pharmacol 57:213–230
Li Y, Shan F, Wu JM, Wu GS, Ding J, Xiao D, Yang WY, Atassi G, Léonce S, Caignard DH, Renard P (2001) Novel antitumor artemisinin derivatives targeting G1 phase of the cell cycle. Bioorg Med Chen Lett 11:5–8
Li Y, Wu JM, Shan F, Wu GS, Ding J, Xiao D, Han JX, Atassi G, Leonce S, Caignard DH, Renard P (2003) Synthesis and cytotoxicity of dihydroartemisinin ethers containing cyanoarylmethyl group. Bioorg Med Chem 11:977–984
Wang Y, Xu X, Wu X, Chen W, Huang F, Gui X (2018) Dihydroartemisinin treatment of multiple myeloma cells causes activation of c-jun leading to cell apoptosis. Oncol Lett 15:2562–2566
Chen H, Shi L, Yang X, Li S, Guo X, Pan L (2010) Artesunate inhibiting angiogenesis induced by human myeloma RPMI8226 cells. Int J Hematol 92:587–597
Wu XH, Zhou HJ, Lee J (2006) Dihydroartemisinin inhibits angiogenesis induced by multiple myeloma RPMI8226 cells under hypoxic conditions via downregulation of vascular endothelial growth factor expression and suppression of vascular endothelial growth factor secretion. Anticancer Drugs 17:839–848
Li S, Xue F, Cheng Z, Yang X, Wang S, Geng F, Pan L (2009) Effect of artesunate on inhibiting proliferation and inducing apoptosis of SP2/0 myeloma cells through affecting NFκB p65. Int J Hematol 90:513–521
Zhao X, Guo X, Yue W, Wang J, Yang J, Chen J (2017) Artemether suppresses cell proliferation and induces apoptosis in diffuse large B cell lymphoma cells. Exp Ther Med 14:4083–4090
Våtsveen TK, Myhre MR, Steen CB, Wälchli S, Lingjærde OC, Bai B, Dillard P, Theodossiou TD, Holien T, Sudan A, Inderberg EM, Smeland EB, Myklebust JH, Oksvold MP (2018) Artesunate shows potent anti-tumor activity in B-cell lymphoma. J Hematol Oncol. https://doi.org/10.1186/s13045-018-0561-0
Huo L, Wei W, Wu S, Zhao X, Zhao C, Zhao H, Sun L (2018) Effect of dihydroarteminin combined with siRNA targetingnotch1 on Notch1/c-Myc signaling in T-cell lymphoma cells. Exp Ther Med 15:3059–3065
Wei W, Zhao X, Wu S, Zhao C, Zhao H, Sun L, Cui Y (2018) Dihydroartemisinin triggers c-Myc proteolysis and inhibits protein kinase B/glycogen synthase kinase 3β pathway in T-cell lymphoma cells. Oncol Lett 16:6838–6846
Sieber S, Gdynia G, Roth W, Bona Vida B, Efferth T (2009) Combination treatment of malignant B cells using the anti-CD20 antibody rituximab and the anti-malarial artesuante. Int J Oncol 35:149–158
Ishikawa C, Senba M, Mori N (2020) Evaluation of artesunate for the treatment of adult T-cell leukemia/lymphoma. Eur J Pharmacol. https://doi.org/10.1016/j.ejphar.2020.172953
Lai H, Singh NP (1995) Selective cancer cell cytotoxicity from exposure to dihydroartemisinin and holotransferrin. Cancer Lett 91:41–46
Singh NP, Lai HC (2005) Synergistic cytotoxicity of artemisinin and sodium butyrate on human cancer cells. Anticancer Res 25:4325–4331
Efferth T, Benakis A, Romero MR, Tomicic M, Rauh R, Steinbach D, Hafer R, Stamminger T, Oesch F, Kaina B, Marschall M (2004) Enhancement of cytotoxicity of artemisinins toward cancer cells by ferrous iron. Free Radic Biol Med 37:998–1009
Kelter G, Steinbach D, Konkimalla VB, Tahara T, Taketani S, Fiebig HH, Efferth T (2007) Role of transferrin receptor and the ABC transporters ABCB6 and ABCB7 for resistance and differentiation of tumor cells towards artesunate. PLoS ONE. https://doi.org/10.1371/journal.pone.0000798
Lai H, Sasaki T, Singh NP, Messay A (2005) Effects of artemisinin-tagged holotransferrin on cancer cells. Life Sci 76:1267–1279
Oh S, Kim BJ, Singh NP, Lai H, Sasaki T (2009) Synthesis and anti-cancer activity of covalent conjugates of artemisinin and a transferrin-receptor targeting peptide. Cancer Lett 274:33–39
Park J, Lai HC, Singh M, Sasaki T, Singh NP (2014) Development of a dihydroartemisinin-resistant molt-4 leukemia cell line. Anticancer Res 34:2807–2810
Ohgami Y, Elstad CA, Chung E, Shirachi DY, Quock RM, Lai HC (2010) Effect of hyperbaric oxygen on the anticancer effect of artemisinin on molt-4 human leukemia cells. Anticancer Res 30:4467–4470
Wang Q, Wu S, Zhao X, Zhao C, Zhao H, Huo L (2015) Mechanisms of dihydroartemisinin and dihydroartemisinin/holotransferrin cytotoxicity in T-cell lymphoma cells. PLoS ONE 10:1–12
Lu JJ, Meng LH, Cai YJ, Chen Q, Tong LJ, Lin LP, Ding J (2008) Dihydroartemisinin induces apoptosis in HL-60 leukemia cells dependent of iron and p38 mitogen-activated protein kinase activation but independent of reactive oxygen species. Cancer Biol Ther 7:1017–1023
Zhou H-J, Wang Z, Li A (2007) Dihydroartemisinin induces apoptosis in human leukemia cells HL60 via downregulation of transferrin receptor expression. Anti Cancer Drugs. https://doi.org/10.1097/CAD.0b013e3282f3f152.
Singh NP, Lai HC (2004) Artemisinin induces apoptosis in human cancer cells. Anticancer Res 24:2277–2280
Wang Z, Hu W, Zhang JL, Wu XH, Zhou HJ (2012) Dihydroartemisinin induces autophagy and inhibits the growth of iron-loaded human myeloid leukemia K562 cells via ROS toxicity. FEBS Open Biol 2:103–112
Efferth T, Glaisi M, Merling A, Krammer PH, Li-Weber M (2007) Artesunate induces ROS-mediated apoptosis in Doxorubicin-resistant T leukemia cells. PLoS ONE. https://doi.org/10.1371/journal.pone.0000693
Zhang S, Chen H, Gerhard GS (2010) Heme synthesis increases artemisinin-induced radical formation and cytotoxicity that can be suppressed by superoxide scavengers. Chem Biol Interact 186:30–35
Chan HW, Singh NP, Lai HC (2013) Cytotoxicity of dihydroartemisinin toward Molt-4 cells attenuated by N-tert-butyl-alpha-phenylnitrone and deferoxamine. Anticancer Res 33:4389–4394
Wang N, Zeng GZ, Yin JL, Bian ZX (2019) Artesunate activates the ATF4-CHOP-CHAC1 pathway and affects ferroptosis in Burkitt’s lymphoma. Biochem Biophys Res Commun 519:533–539
Gao N, Budhraja A, Cheng S, Liu EH, Huang C, Chen J, Yang Z, Chen D, Zhang Z, Shi X (2011) Interruption of the MEK/ERK signaling cascade promotes dihydroartemisinin-induced apoptosis in vitro and in vivo. Apoptosis 16:511–523
Lee J, Zhang G, Wu X, Xu F, Zhou J, Zhang X (2012) Growth inhibitory effect of dihydroartemisinin on Bcr/Abl+chronic myeloid leukemia K562 cells involve AKT, ERK and NF-κB modulation. J Cancer Res Clin Oncol 138:2095–2102
Lee J, Shen P, Zhang G, Wu X, Zhang X (2013) Dihydroartemisinin inhibits the Bcr/Abl oncogene at the mRNA level in chronic myeloid leukemia sensitive or resistant to imatinib. Biomed Pharmacother 67:157–163
Budhraja A, Turnis ME, Churchman ML, Kothari A, Yang X, Xu H, Kaminska E, Panetta JC, Finkelstein D, Mullighan CG, Opferman JT (2017) Modulation of navitoclax sensitivity by dihydroartemisinin-mediated MCL-1 repression in BCR-ABL+ B-lineage acute lymphoblastic leukemia. Clin Cancer Res 23:7558–7568
Elf S, Lin R, Xia S, Pan Y, Shan C, Wu S, Lonial S, Gaddh M, Arellano ML, Khoury HJ, Khuri FR, Lee BH, Boggon TJ, Fan J, Chen J (2017) Targeting 6-phosphogluconate dehydrogenase in the oxidative PPP sensitizes leukemia cells to antimalarial agent dihydroartemisinin. Oncogene 36:254–262
Li Y, Feng L, Jiang W, Shan N, Wang X (2014) Artesunate possesses anti-leukemia properties that can be enhanced by arsenic trioxide. Leuk Lymphoma 55:1366–1372
Tan M, Rong Y, Su Q, Chen Y (2017) Artesunate induces apoptosis via inhibition of STAT3 in THP-1 cells. Leuk Res 62:98–103
Cao JT, Mo HM, Wang Y, Zhao K, Zhang TT, Wang CQ, Xu KL, Han ZH (2018) Dihydroartemisinin-induced apoptosis in human acute monocytic leukemia cells. Oncol Lett 15:3178–3184
Handrick R, Ontikatze T, Bauer KD, Freier F, Rübel A, Dürig J, Belka C, Jendrossek V (2010) Dihydroartemisinin induces apoptosis by a bak-dependent intrinsic pathway. Mol Cancer Ther 9:2497–2510
Sun X, Yan P, Zou C, Wong YK, Shu Y, Lee YM, Zhang C, Yang ND, Wang J, Zhang J (2019) Targeting autophagy enhances the anticancer effect of artemisinin and its derivatives. Med Res Rev 39:2172–2193
Cheng C, Wang T, Song Z, Peng L, Gao M, Hermine O, Rousseaux S, Khochbin S, Mi JQ, Wang J (2018) Induction of autophagy and autophagy-dependent apoptosis in diffuse large B-cell lymphoma by a new antimalarial artemisinin derivative, SM1044. Cancer Med 7:380–396
Hu W, Chen SS, Zhang JL, Lou XE, Zhou HJ (2014) Dihydroartemisinin induces autophagy by suppressing NF-κB activation. Cancer Lett 343:239–248
Roh JL, Kim EH, Jang H, Shin D (2017) Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol 11:254–262
Wang K, Zhang Z, Wang M, Cao X, Qi J, Wang D, Gong A, Zhu H (2019) Role of GRP78 inhibiting artesunate-induced ferroptosis in KRAS mutant pancreatic cancer cells. Drug Des Dev Ther 13:2135–2144
Wei T, Liu J (2017) Anti-angiogenic properties of artemisinin derivatives. Int J Mol Med 40:972–978
Zhou HJ, Wang WQ, Wu GD, Lee J, Li A (2007) Artesunate inhibits angiogenesis and downregulates vascular endothelial growth factor expression in chronic myeloid leukemia K562 cells. Vascul Pharmacol 47:131–138
Kim SH, Kim HJ, Kim TS (2003) Differential involvement of protein kinase C in human promyelocytic leukemia cell differentiation enhanced by artemisinin. Eur J Pharmacol 482:67–76
Kim SH, Chun SY, Kim TS (2008) Interferon-α enhances artemisinin-induced differentiation of HL-60 leukemia cells via a PKCα/ERK pathway. Eur J Pharmacol 587:65–72
Finaurini S, Basilico N, Corbett Y, D’Alessandro S, Parapini S, Olliaro P, Haynes RK, Taramelli D (2012) Dihydroartemisinin inhibits the human erythroid cell differentiation by altering the cell cycle. Toxicology 300:57–66
Lu X, Efferth T (2020) Repurposing of artemisinin-type drugs for the treatment of acute leukemia. Semin Cancer Biol. https://doi.org/10.1016/j.semcancer.2020.05.016
Horwedel C, Tsogoeva SB, Wei S, Efferth T (2010) Cytotoxicity of artesunic acid homo- and heterodimer molecules toward sensitive and multidrug-resistant CCRF-CEM leukemia cells. J Med Chem 53:4842–4848
Yang X, Wang W, Tan J, Song D, Li M, Liu D, Jing Y, Zhao L (2009) Synthesis of a series of novel dihydroartemisinin derivatives containing a substituted chalcone with greater cytotoxic effects in leukemia cells. Bioorg Med Chem Lett 19:4385–4388
Gaur R, Pathania AS, Malik FA, Bhakuni RS, Verma RK (2016) Synthesis of a series of novel dihydroartemisinin monomers and dimers containing chalcone as a linker and their anticancer activity. Eur J Med Chem 122:232–246
Chadwick J, Jones M, Mercer AE, Stocks PA, Ward SA, Park BK, O’Neill PM (2010) Design, synthesis and antimalarial/anticancer evaluation of spermidine linked artemisinin conjugates designed to exploit polyamine transporters in Plasmodium falciparum and HL-60 cancer cell lines. Bioorg Med Chem 18:2586–2597
Mott BT, He R, Chen X, Fox JM, Civin CI, Arav-Boger R, Posner GH (2013) Artemisinin-derived dimer phosphate esters as potent anti-cytomegalovirus (anti-CMV) and anti-cancer agents: A structure-activity study. Bioorg Med Chem 21:3702–3707
Reiter C, Fröhlich T, Gruber L, Hutterer C, Marschall M, Voigtländer C, Friedrich O, Kappes O, Efferth T, Tsogoeva, (2015) Highly potent artemisinin-derived dimers and trimers: Synthesis and evaluation of their antimalarial, antileukemia and antiviral activities. Bioorg Med Chem 23:5452–5458
Gruber L, Abdelfatah S, Frohlich T, Reiter C, Klein V, Tsogoeva SB, Efferth T (2018) Treatment of multidrug-resistant leukemia cells by novel artemisinin-, egonol-, and thymoquinone-derived hybrid compounds. Molecules. https://doi.org/10.3390/molecules23040841
Reiter C, Herrmann A, Çapci A, Efferth T, Tsogoeva SB (2012) New artesunic acid homodimers: Potent reversal agents of multidrug resistance in leukemia cells. Bioorg Med Chem 20:5637–5641
Letis AS, Seo EJ, Nikolaropoulos SS, Efferth T, Giannis A, Fousteris MA (2017) Synthesis and cytotoxic activity of new artemisinin hybrid molecules against human leukemia cells. Bioorg Med Chem 25:3357–3367
Reiter C, Çapci Karagöz A, Fröhlich T, Klein V, Zeino M, Viertel K, Held J, Mordmuller OSE, Anil H, Efferth T, Tsoegoeva SB (2014) Synthesis and study of cytotoxic activity of 1,2,4-trioxane- and egonol-derived hybrid molecules against Plasmodium falciparum and multidrug-resistant human leukemia cells. Eur J Med Chem 75:403–412
Reiter C, Fröhlich T, Zeino M, Marschall M, Bahsi H, Leidenberger M, Friedrich O, Kappes B, Hampel F, Efferth T, Tsogoeva SB (2015) New efficient artemisinin derived agents against human leukemia cells, human cytomegalovirus and Plasmodium falciparum: 2nd generation 1,2,4-trioxane-ferrocene hybrids. Eur J Med Chem 97:164–172
Ha VT, Kien VT, Binh LH, Tien VD, My NTT, Nam NH, Baltas M, Hahn H, Han BW, Thao DT, Vu TK (2016) Design, synthesis and biological evaluation of novel hydroxamic acids bearing artemisinin skeleton. Bioorg Chem 1(66):63–71. https://doi.org/10.1016/j.bioorg.2016.03.008
Wu Y, Parapini S, Williams ID, Misiano P, Wong HN, Taramelli D, Basilico N, Haynes RK (2018) Facile preparation of N-glycosylated 10-piperazinyl artemisinin derivatives and evaluation of their antimalarial and cytotoxic Activities. Molecules. https://doi.org/10.3390/molecules23040841
Fröhlich T, Reiter C, Ibrahim MM, Beutel J, Hutterer C, Zeitträger I, Bahsi H, Leidenberger M, Friedrich O, Kappes B, Efferth T, Marschall M, Tsogoeva SB (2017) Synthesis of novel hybrids of quinazoline and artemisinin with high activities against plasmodium falciparum, human cytomegalovirus, and leukemia cells. ACS Omega 2:2422–2431
Fröhlich T, Reiter C, Saeed MEM, Hutterer C, Hahn F, Leidenberger M, Friedrich O, Kappes B, Marschall M, Efferth T, Tsogoeva SB (2018) Synthesis of thymoquinone-artemisinin hybrids: new potent antileukemia, antiviral, and antimalarial agents. ACS Med Chem Lett 9:534–539
Çapcı Karagöz A, Reiter C, Seo EJ, Gruber L, Hahn F, Leidenberger M, Klein V, Hampel F, Friedrich O, Marschall M, Kappes B, Efferth T, Tsogoeva SB (2018) Access to new highly potent antileukemia, antiviral and antimalarial agents via hybridization of natural products (homo)egonol, thymoquinone and artemisinin. Bioorg Med Chem 26:3610–3618
Posner GH, Cumming JN, Ploypradith P, Oh CH (1995) Evidence for Fe(IV)=O in the molecular mechanism of action of the trioxane antimalarial artemisinin. J Am Chem Soc 117:5885–5886
Eckstein-Ludwig U, Webb RJ, van Goethem DA, East JM, Lee AG, Kimura M, O’Neill PM, Bray PG, Ward SA, Krishna S (2003) Artemisinin target the SERCA of Plasmodium falciparum. Nature 424:957–961
Bridgford JL, Xie SC, Cobbold SA, Pasaje CFA, Herrmann S, Yang T, Gillett DL, Dick LR, Ralph SA, Dogovski C, Spillman NJ, Tilley L (2018) Artemisinin kills malaria parasites by damaging proteins and inhibiting the proteasome. Nat Commun. https://doi.org/10.1038/s41467-018-06221-1
Acknowledgements
The authors would like to thank São Paulo Research Foundation (FAPESP) and Ministry of Science, Technology, Innovations and Communications (CNPq) for financial support and Raquel S. Foglio for the writing assistance and English revision.
Funding
This study was funded by grant # 2017/21801-2 (STOS), 2014/16008-3 (MAF), 2016/18384-8 (MAF), São Paulo Research Foundation (FAPESP) and Ministry of Science, Technology, Innovations and Communications (CNPq) grant # 301676/2013-5 (STOS) CNPq #142286/2017 (RIM) and 401904/2012-1 (MAF).
Author information
Authors and Affiliations
Contributions
Design of the Review: RIM and STOS; Bibliographical review: RIM, STOS, MAF; Manuscript preparation: all authors. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mancuso, R.I., Foglio, M.A. & Olalla Saad, S.T. Artemisinin-type drugs for the treatment of hematological malignancies. Cancer Chemother Pharmacol 87, 1–22 (2021). https://doi.org/10.1007/s00280-020-04170-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-020-04170-5