
Abstract
Purpose
Plitidepsin absorption, distribution, metabolism and excretion characteristics were investigated in a mass balance study, in which six patients received a 3-h intravenous infusion containing 7 mg 14C-plitidepsin with a maximum radioactivity of 100 µCi.
Methods
Blood samples were drawn and excreta were collected until less than 1% of the administered radioactivity was excreted per matrix for two consecutive days. Samples were pooled within-patients and between-patients and samples were screened for metabolites. Afterwards, metabolites were identified and quantified. Analysis was done using Liquid Chromatography linked to an Ion Trap Mass Spectrometer and offline Liquid Scintillation Counting (LC-Ion Trap MS-LSC).
Results
On average 4.5 and 62.4% of the administered dose was excreted via urine over the first 24 h and in faeces over 240 h, respectively. Most metabolites were found in faeces.
Conclusion
Plitidepsin is extensively metabolised and it undergoes dealkylation (demethylation), oxidation, carbonyl reduction, and (internal) hydrolysis. The chemical formula of several metabolites was confirmed using high resolution mass data.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
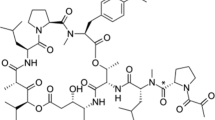
Plitidepsin (Aplidin®; Fig. 1.) is a novel anti-cancer drug currently investigated in late phases of clinical development. The compound was originally isolated from the Mediterranean tunicate Aplidium albicans in 1988, but it is now chemically synthesised [1, 2]. It has been shown to have antitumour activity, mainly in patients with multiple myeloma, peripheral T-cell lymphoma, and melanoma [3]. Plitidepsin’s antitumour activity is induced by targeting eEF1A2, one of the isoforms of the alpha subunit of the eukaryotic Elongation Factor 1A2. This eEF1A2 protein is overexpressed in human tumours and is known to possess oncogenic properties, favouring tumour cell proliferation while at the same time inhibiting apoptosis [4]. Numerous clinical trials are underway investigating plitidepsin’s use in combination therapy [3, 5,6,7,8,9]. As part of the drug development program, regulatory authorities have specified the need for a mass balance and metabolite profiling study, in which excretory pathways and circulating metabolites are investigated. This information is, in fact, part of regulatory filing of the new drug [10, 11].

Molecular structure of plitidepsin. The brackets show the different parts of this molecule susceptible for MS fragmentation. The names of these parts are used throughout the manuscript to describe where biotransformations have taken place. Di-methyl Tyr Di-methyl tyrosine, Hip Hydroxyisovalerylpropionyl, Leu Leucine, Pro Proline, Thr Threonine
A mass balance study was conducted to characterise the absorption, distribution, metabolism and excretion (ADME) of 14C-plitidepsin [12]. Evaluable patients received a 3 h intravenous (i.v.) infusion containing a flat dose of 7 mg (100 µCi) radiolabelled plitidepsin. Plasma, whole blood, urine and faecal samples were collected and analysed by Liquid scintillation counting (LSC) to determine the total radioactivity (TRA). High performance liquid chromatography–tandem mass spectrometry (HPLC-MS/MS) was used to quantify plitidepsin in plasma, whole blood and urine. TRA measurements from this mass balance study have been reported before [12]. After the complete collection period, a mean of 6.1% of the administered radioactive dose was recovered in urine, whereas 71.3% was excreted via faeces.
Previous research by Brandon et al. [13, 14] has shown the formation of four metabolites in incubated pooled human microsomes and incubated human plasma, liver preparations, cytochrome P450 (CYP) and uridine diphosphoglucuronosyl transferase (UGT) supersomes. These metabolites include plitidepsin hydroxylated at the isopropyl group (Apli-h), plitidepsin C-dealkylated at the N(Me)-leucine group (Apli-da), plitidepsin with both these converted amino acid residues (Apli-da/h), and plitidepsin C-demethylated at the threonine group (Apli-dm) [13]. It was also shown that plitidepsin was metabolised by carboxylesterases in human plasma. In particular, CYP2A6 was responsible for the conversion of plitidepsin into C-demethylated plitidepsin, whereas CYP3A4 caused the formation of the other three metabolites [14]. CYP2E1 and CYP4A11 were shown to be involved in plitidepsin metabolism as well, but the identity of the metabolites generated by these experiments remained unknown. Moreover, it was discovered that plitidepsin metabolites formed by CYP could be further conjugated by phase II enzymes UGT, glutathione S-transferase (GST) and sulfotransferase (SULT).
The aim of the present work was to search for in vivo plitidepsin metabolites in human plasma, urine and faeces as part of a mass balance study with the 14C-radioactivity-labelled drug.
Materials and methods
Chemicals
Plitidepsin reference standard was provided by Pharma Mar S.A. (Colmenar Viejo, Madrid, Spain). Acetonitrile, methanol and water (Supra-Gradient grade) were obtained from Biosolve Ltd (Valkenswaard, The Netherlands). Formic acid (≥ 98%; analytical grade), 2-propanol (> 99.8%) and 25% ammonia were purchased from Merck (Amsterdam, the Netherlands). Ammonium acetate (LC-MS grade) was supplied by Sigma-Aldrich (Zwijndrecht, the Netherlands). Ultima Gold™ was obtained from PerkinElmer (Groningen, the Netherlands). Water (non-HPLC grade) was supplied by B. Braun (Oss, the Netherlands).
Sample collection and storage
Six evaluable patients were enrolled in the phase I mass balance clinical study. On Day 1, patients received a 3 h i.v. infusion containing a flat dose of 7 mg of radiolabelled plitidepsin with a radioactivity of 100 µCi (PRA Health Sciences, Zuidlaren, the Netherlands). More information about the enrolment specifics can be found elsewhere [12]. Blood sampling was done at the following time points: pre-infusion, halfway through infusion, just before the end of infusion (EOI) and 15 min, 30 min, 1, 1.5, 2, 4, 9, 24, 48, 72, 96, 120 and 192 h after EOI. Blood samples were drawn using a peripheral line and plasma was obtained by centrifugation (3000 rpm, 10 min, 4 °C). Urine and faeces were collected for the duration of the study period, which ended when less than 1% was recovered in the matrix for two consecutive days. Urine was collected over 2-h periods for the first 12 h, and over the next 12 h afterwards. Additionally, urine was collected over 8-h periods for the next day and from Day 3 onwards urine was collected over 24 h. After each collection period, portions were transferred to smaller tubes and stored at − 80 °C. Faeces were collected per stool. Each stool was diluted with water (3:1, w/w) and homogenised using a T25 basic Ultra Turrax (IKA Works, Staufen, Germany). Afterwards, aliquots were stored at − 80 °C.
Selection of samples for screening and quantification
Metabolite profiling was performed on a representative selection of samples. Selection of samples was based on the amount of radioactivity recovered in individual samples, as well as on the difference between TRA levels and plitidepsin concentrations measured in plasma and urine by LC-MS/MS, the results of which were published before [12]. It was important to select samples in which the amount of radioactivity was sufficient for analysis by LSC, but it was also vital to choose time points which had shown a difference between TRA and plitidepsin levels since this is indicative of circulating metabolites. Radioactivity as well as plitidepsin levels were rather low in plasma. Therefore, besides a pre-dose, a sample around the maximum concentration was selected for metabolite screening to ensure sufficient sensitivity during analysis.
Similar problems were encountered in urine samples. Again, radioactivity levels were low, as well as plitidepsin concentrations. A mean of 6.1% of the administered dose of plitidepsin was recovered in urine over 240 h and 4.6% accounted for unchanged parent drug [12]. Only 1.5% was attributed to plitidepsin metabolites. The following intra-patient pools were prepared first: pre-dose, 0–24 h and 48–72 h. Afterwards, these samples were pooled between patients. Inter-patient pools were used for screening and intra-patient pools for quantification.
For faeces, the following pools were made: pre-dose, 0–72, 72–144 and 144–240 h. Again, inter-patient pools were analysed during screening and the intra-patient pools were analysed afterwards for metabolite quantification.
Preparation of biological samples
Plasma samples (500 µL) were extracted once with 1000 µL acetonitrile–methanol (50:50, v/v). The obtained supernatant was evaporated to dryness under a gentle stream of nitrogen. The dried supernatant was reconstituted in 75 µL 0.1% formic acid in acetonitrile and 75 µL 0.1% formic acid in water.
Faeces homogenate samples (300 µL) were extracted once with 900 µL acetonitrile–methanol (50:50, v/v). The obtained supernatant was evaporated to dryness and the dried supernatant was reconstituted in 300 µL 1 M aqueous ammonia in water–acetonitrile (50:50, v/v). An aliquot of the plasma and faeces extracts was analysed for TRA to calculate pre-treatment recovery. The remaining supernatant was used for LC-Ion Trap MS-LSC analysis. Urine samples were injected onto the LC system without sample pre-treatment.
LC-MS/MSn systems
Two chromatographic systems were developed for metabolite screening and identification. For both systems, a LC-20AD pump and a SIL-HTc autosampler (Shimadzu, Kyoto, Japan) were used. For metabolite separation in plasma and urine, the LC system (referred to as method I) was supplied with a Symmetry C18 column (150 × 4.6 mm, 3.5 µm, Waters). The oven was set at 30 °C and a flow rate of 1 mL/min was applied. Separation was achieved with the following gradient: 10%B, to 90%B in 31.1 min, held for 5 min, before returning to start conditions (36.1–50 min). For faeces, the chromatographic system did not achieve sufficient separation between the numerous metabolites. Therefore, a different column was used (Synergi Hydro-RP 250 × 4.6 mm, 4 µm, Phenomenex) and a longer gradient was applied (method II): from 10 to 47%B in 8.3 min, held until 28.3 min, to 90%B in 50 min, held for 8.3 min, after which the gradient returned to starting conditions (total run time of 78 min). Moreover, the column temperature was set at 80 °C as opposed to 30 °C for method I. Mobile phase A consisted of 0.1% formic acid in water and 0.04% formic acid in water for methods I and II, respectively. Mobile phase B was 0.1% formic acid in acetonitrile and 0.04% formic acid in acetonitrile for method, I and II, respectively. Detection was performed on a LTQ XL mass spectrometer (Thermo Electron, Waltham, MA, USA). Structural information on plitidepsin metabolites was obtained using data-dependent acquisition, causing further fragmentation of the most abundant ions to obtain MS2 and MS3 spectra.
One fourth of the flow was directed into the MS and the remainder was directed to a fraction collector (Fraction Collector III, Waters). The collector was set to collect one fraction per minute and for each fraction radioactivity was measured by LSC (Tri-Carb 4910TR, PerkinElmer). Since one fraction per minute was collected, this meant that the fraction number corresponded directly to the retention time in the chromatogram, i.e. a peak eluting between 4 and 5 min would be found in fraction 5.
Accurate mass measurements were performed on an LTQ Orbitrap Discovery (Thermo Scientific) to confirm the chemical structures of the metabolites. For accurate high resolution analysis, two additional systems were developed and were made compatible with micro LC. These were referred to as method III (for plasma and urine) and method IV (for faeces). Both systems made use of an Ultimate 3000 RLSC NanoSystem LC (Dionex, Amsterdam, the Netherlands). Settings for all four systems are summarised in Table 1.
Metabolite screening and identification
Data-dependent acquisition using a predefined parent list was used to collect the MS data. Online, total ion current (TIC) chromatograms were obtained and from each m/z signal above a threshold of 750 cps, MS/MS spectra were generated when a match was found with a (predicted) parent list. Spectra of unpredicted metabolites were obtained by comparing TIC chromatograms of pre-dose samples with those of post-dose samples. Potential plitidepsin-related ions were selected and added to the parent list. Samples were re-injected and the MS/MS spectra of the unpredicted metabolites were obtained. The nomenclature used throughout the manuscript to identify parts of plitidepsin’s structure is derived from Ngoka et al. (1999) and Brandon et al. (2005) [13, 15].
Results
The search for metabolites was initially based on predicted metabolites using low resolution ion trap data (LTQ MS ion trap data), after which fractions containing high radioactive levels were further investigated for the presence of plitidepsin-related compounds. For this purpose, the search for specific isotopic patterns (12C/14C ratio) in the [M + H]+, [M + Na]+ and [M + K]+ ion clusters was started, and furthermore interpretation of MS2 and MS3 spectra was performed. Finally, high resolution data were obtained to confirm the atomic composition of the suggested molecular structures.
Screening and identification
Metabolites in plasma
Radioactivity levels in plasma were low and plasma samples were not available in excessive amounts. Straightforward extraction procedures did not achieve high recoveries (± 50%), and several approaches were tried to improve the extraction efficiency. This resulted in an improved extraction recovery of 66% using acetonitrile–methanol (50:50, v/v), after which it was decided to not further develop the pre-treatment method due to limited sample availability. Ultimately, screening of metabolites was done using samples that contained relatively higher radioactivity levels (> 10 Bq/g). Mean Cmax of plitidepsin and TRA in these samples was 17.6 and 24.5 ng/mL, respectively, and the volume of distribution of plitidepsin was 2180 L [12]. Plitidepsin is highly distributed to peripheral tissue and the difference between the Cmax of plitidepsin and TRA is small. Hence, low concentrations of metabolites are expected. The radiochromatograms of the samples are shown in Fig. 2. Even in these samples, only small traces of radioactivity were detected. The majority of the radioactivity found in these samples was attributed to plitidepsin, but radioactivity levels did not exceed 16 DPM, while the LOD was 4 DPM. Amounts were so low that it was impossible to obtain MS2 and MS3 spectra, which made structural characterisation impossible. In the recently published mass balance and pharmacokinetic study, the area under the plasma concentration–time curve from zero to last measured time point (AUC0–last) was 81.1 ng h/mL for plitidepsin compared to a TRA AUC0–last of 281.7 ng h/mL. A large amount of the AUC should therefore be explained by circulating metabolites, but they could not be elucidated. The metabolite screening could only detect unchanged plitidepsin. This could mean that the metabolites are present in even smaller amounts than plitidepsin. Nevertheless, it is highly unlikely that individual metabolites account for more than 10% of the AUC of all drug-related material.

Radiochromatograms of two 15 min after EOI plasma samples collected after administration of ~ 7 mg 14C-plitidepsin (~ 80 µCi) over a 3-h infusion to two cancer patients
Metabolites in urine
Figure 3 shows the radiochromatograms of the two urine screening samples. It was apparent from LC-MS/MS and TRA data that only 6.1% of the total administered dose was excreted via urine, of which the large majority was accounted for by the parent compound [12]. Moreover, already 4.5% was excreted over the first 24 h. After screening for metabolites in three inter-patient pooled samples, only low metabolite levels were detected. Besides a peak corresponding to plitidepsin, identified by comparing the MS spectra with the authentic standard, only two peaks were observed in urine (U), one eluted between 2 and 5 min, which was given the name Pliti-M1 and one eluted in fraction U25. No structural information could be obtained for Pliti-M1, and it was confirmed that the metabolite in fraction U25 was identical to metabolite Pliti-M19 found in faeces.

Radiochromatograms of a 0–24 h (A) urine screening sample and a 24–48 h (B) urine screening sample collected after a 3-h i.v. infusion of ~ 7 mg 14C-plitidepsin (~ 80 µCi) to cancer patients. Fraction U26-27 contains plitidepsin. Please note that the scale of the y-axes is different
Metabolites in faeces
Figure 4 shows the radiochromatograms from faeces samples during screening. During the development of a chromatographic method it became evident that many metabolites were formed and excreted via the biliary route, or directly excreted directly into the intestines. Efforts were made to achieve sufficient separation between these metabolites, which resulted in a different elution gradient (method II) as opposed to the one for plasma and urine (method I). Finally, separation was improved, however, no baseline separation was achieved for all metabolites on the Symmetry C18 column.

Radiochromatograms of the 0–72 h A faeces screening sample, the 72–144 h B faeces screening sample and the 144–240 h C faeces screening sample collected after a 3-h i.v. infusion of ~ 7 mg 14C-plitidepsin (~ 80 µCi) to cancer patients. Please note that the scale of the y-axes is different
A few fractions contained single chromatographic peaks; these included F34, F41–42, F43 and F44–45, where F refers to the faeces origin and the number on the fraction in the LC analysis. Other fractions contained multiple peaks which could not always be separated from each other, nor could they always be identified. Quantification was therefore based on (combined) fractions.
Metabolite names were given as follows: “Pliti”, followed by the letter M (metabolite) and a number. The numbers in the names were given in chronological order. A few metabolites were not structurally identified. These included Pliti-M2, Pliti-M4, Pliti-M5, Pliti-M9 and Pliti-M13.
Below is an overview of all other fractions analysed with MS and structural elucidation of metabolites.
F44–45
Between 43 and 45 min a peak with [M + H]+ m/z 1111 was found but the [M + Na]+ ion (m/z 1133) was more abundant and was therefore selected for MS2 analysis. It was identified as plitidepsin and this was confirmed by comparing the spectra to the authentic reference standard (see Fig. 5). This spectrum showed the distinctive fragmentation pattern reported previously: the loss of the di-methyl tyrosine (Tyr) moiety (resulting in a fragment with m/z 941), and the subsequent losses of proline (Pro; m/z 844), leucine (Leu) and hydroxyisovalerylpropionyl (Hip) (m/z 575) groups (The ester bond between Leu and Hip does not break) [13, 15]. Plitidepsin was further fragmented by loss of isostatin, resulting in fragment m/z 418.

Mass spectra and fragmentation pattern of plitidepsin
F17–18
Fraction F17–18 showed a chromatographic peak between 16 and 18 min with an [M + H]+ of 1057. No MS2 and MS3 spectra were obtained, but it was confirmed that the metabolite in this fraction resulted from a loss of 54 Da, which was supported by accurate mass data to be a net loss of C4H6. The metabolite in this fraction was given the name Pliti-M3.
F23–26
Between 22 and 26 min multiple peaks were observed: two with m/z values of 1071 (at 23.2 and 24.5 min), and one with m/z 1041. For the two peaks with m/z 1071, the MS2 and MS3 data indicated a loss of 40 Da in the side chain or in the threonine (Thr) group, since all fragments observed were 40 Da lower than those for plitidepsin, meaning that the loss must have taken place in the smallest plitidepsin fragment. From the accurate mass data, it could be concluded that this loss of 40 Da was the result of the net loss of C3H4. The metabolites were referred to as Pliti-M6 and Pliti-M7.
The MS2 and MS3 data corresponding to m/z 1041 indicated a loss of 14 Da and a loss of 56 Da. The 56 Da was lost from the Hip or Leu group, because the smallest fragment similar to plitidepsin’s fragmentation pattern is 575, which is the part of the molecule without the Hip/Leu group. All other fragments were similar, meaning that the 14 Da change must have taken place in the di-methyl Tyr group. From the accurate mass data, it could be concluded that the loss of 14 Da and the loss of 56 Da were the results of a methyl dealkylation (–CH2) and the net loss of C3H4O, respectively. The metabolite was referred to as Pliti-M8.
F27–33
Two peaks were found in F27–33: m/z 1071, similar to the one in F23–26, and also m/z 1113. The peak with m/z 1071 was identified in the same way as mentioned above and was named Pliti-M10.
The other peak observed in these fractions, m/z 1113, was 2 Da higher than the parent mass. The MS2 and MS3 data revealed an addition of 16 Da in the 941-fragment (the Pro, Leu or the Hip group). The loss of 14 Da most likely occurred in the di-methyl Tyr group, since all smaller fragments are similar to plitidepsin’s fragments. Together with the accurate mass data, it could be concluded that the loss of 14 Da and an addition of 16 Da were the results of a methyl dealkylation (–CH2) and the addition of an O, respectively. The proposed biotransformation was a methyl dealkylation (–CH2) in the di-methyl Tyr group and an oxidation/hydroxylation (+ O) in the Pro, Leu or Hip group. This metabolite was identified as oxidated demethylated plitidepsin and was named Pliti-M11.
F34
F34 contained a chromatographic peak with a [M + H]+ of 1071, similar to the ones found in F23–26 and F27–33, except in this case no MS3 could be obtained due to sensitivity issues. The only peak visible was m/z 901, which means that the loss of 40 could have taken place anywhere in the molecule, except in the di-methyl Tyr group. Accurate mass data confirmed the conclusion that this loss of 40 Da was the result of the net loss of C3H4. The exact identity of this metabolite could not be elucidated. The metabolite is therefore referred to as Pliti-M12.
F35–38
Two masses were observed in F35–38: [M + H]+ of 1099 and [M + Na]+ of 1151. The first showed a loss of 12 Da compared to the parent mass. The MS2 and MS3 data indicated an addition of 2 Da in the side chain or Thr group, since all fragments had a 2 Da increase. The 14 Da loss must therefore have occurred in the largest fragment, so in the di-methyl Tyr group. Together with the accurate mass data, it could be concluded that the loss of 14 Da and the addition of 2 Da were the results of a methyl dealkylation (–CH2) and a reduction, respectively. The proposed biotransformation is a methyl dealkylation in the di-methyl Tyr group and the transformation from a ketone to alcohol (+ H2) in the side chain or Thr group, and the metabolite was, therefore, referred to as carbonyl-reduced demethylated plitidepsin and was named Pliti-M14.
[M + Na]+ m/z of 1151 showed an addition of 18 Da compared to the parent mass. No additional MS2 and MS3 data were available, but from the accurate mass data, it could be concluded that this addition of 18 Da is due to the addition of H2O. Internal hydrolysis in the ring structure is therefore the proposed biotransformation and this hydrolysed plitidepsin metabolite was named Pliti-M15.
F39–40
Two masses were observed in F39–40: [M + Na]+ of m/z 1119 and 1077. 1119 was the result from a loss of 14 Da compared to plitidepsin. The MS2 and MS3 data showed identical fragments as compared to plitidepsin. The change must therefore have taken place in the largest fragment, the di-methyl Tyr group. The demethylated plitidepsin metabolite was given the name Pliti-M16.
MS2 and MS3 data revealed that the loss of 56 Da, resulting in m/z 1077, must have occurred in the side chain or Thr group, since all fragments showed the 56 Da loss. From the accurate mass data, it could be concluded that this loss of 56 Da was caused by the net loss of C3H4O. The metabolite was named Pliti-M17.
F41–42
[M + Na]+ ion of 1149 was found in F41–42, which meant an addition of 16 Da compared to the parent mass. MS2 data indicated a 16 Da increase of the 941-fragment. All other fragments were unchanged, which means that the addition of 16 Da must have occurred in the Pro, Leu or Hip group. MS3 data were not available, but from the accurate mass data, it could be concluded that this addition of 16 Da was due to the addition of O. The suggested biotransformation is an oxidation/hydroxylation (+ O) in the Pro, Leu or Hip group, and this oxidated metabolite was given the name Pliti-M18.
F43
A chromatographic peak with similar m/z values and similar MS2 and MS3 data was found in both urine and faeces samples. This metabolite (m/z 1113) had an addition of 2 Da compared to the parent compound. Another fraction also contained a mass of 1113 (in F27–33), but had different fragmentation patterns. Here, the MS2 and MS3 data revealed an addition of 2 Da in all fragments, meaning that the addition occurred in the smallest fragment: the side chain or Thr group. From the accurate mass data, it could be concluded that this addition of 2 Da was the result of a reduction. The suggested biotransformation is the reduction from a ketone to alcohol (+ H2) in the side chain or Thr group and the metabolite was named Pliti-M19.
The proposed metabolic pathway involves plitidepsin being hydrolysed, oxidated, dealkylated, (demethylated) and carbonyl reduced. Both the oxidated and carbonyl-reduced plitidepsin are demethylated as well, the demethylated plitidepsin is in turn oxidated or carbonyl reduced. An overview of the proposed metabolic pathway can be found in Fig. 6.

Proposed metabolic pathway of plitidepsin. U urine metabolite (analysed using system I), F faeces metabolite (analysed using system II)
Quantification of metabolites
Using LC system I, plitidepsin was detected around 25–27 min and accounted for 2.8% of the amount of radioactivity in urine samples from 0 to 24 h. U3-5 accounted for 0.8% and U25 for less than 0.1% of the administered radioactivity and are therefore considered minor metabolites.
In total, eleven (combined) metabolite fractions were distinguished (Fig. 4) in three inter-patient pooled faeces samples, of which one, F43, corresponded to metabolite U25 in urine and was named Pliti-M19. The largest group of metabolite fractions was combined F1–16 (from 0 to 16 min), accounting for 11.3% of the administered dose. When the MS spectra obtained in fractions F1–16 were compared to the blank samples, however, no significant differences were detected and therefore no MS spectra of the metabolites could be obtained. Other metabolites detected in metabolite fractions included F17–18 (accounting for 3.0% of the administered radioactive dose), F19–22 (2.7%), F23–26 (5.2%), F27–33 (4.0%), F34 (3.1%), F35-38 (4.5%), F39–40 (8.8%), F41–42 (4.2%) and F43 (1.5%). Unchanged parent drug (F44–45) was responsible for 2.3% of the administered dose. This means that a total of 50.6% of the 62.4% excreted via faeces up to 240 h could be attributed to parent drug and metabolites. Only 0.1% was not identified in the radiochromatograms and 12.7% was lost during sample pre-treatment. Pooling accuracy was 6.6% and 7.7% of the radioactivity was lost during sample storage, probably due to the hydrophobic nature of the compounds in the aqueous faeces homogenate. A summary of these data is shown in Fig. 7.

Summary of plitidepsin metabolite excretion up to 240 h after a single i.v. dose of 14C-plitidepsin. Quantification is based on combined fractions, rather than individual metabolites since they could not always be separated from each other
Discussion
Discussion of the results
This clinical trial investigated the metabolic profile of the anti-cancer compound plitidepsin. The study aimed to identify and quantify metabolites in human plasma, urine and faeces after i.v. administration of a flat dose of 7 mg of 14C-plitidepsin containing a radioactivity of 100 µCi.
The TRA curve in whole blood is comparable with the plitidepsin curve from 0 to 24 h after dosing, indicating that the presence of plitidepsin metabolites in this compartment is limited during the first 24 h after dosing [12]. From 24 h, the TRA concentrations expressed in ng.eq/mL are higher than the plitidepsin concentrations (in ng/mL) due to the formation of low abundant metabolites. When, on the other hand, the TRA curve in plasma is compared with the plitidepsin plasma curve, it can be concluded that the drug is metabolised and the absolute differences in concentrations are maximal at the end of infusion. However, at these time points, total radioactivity levels are still low (around 150 DPM) and after fractionation only plitidepsin could be detected in the plasma radiochromatograms. Plitidepsin concentrations were already quantified using LC-MS/MS [12], and therefore, no further metabolite profiling work was performed in plasma.
Urinary excretion is a minor elimination route. The average recovery after 240 h was 6.1 ± 1.5% and the majority was excreted as unchanged drug (4.6 ± 0.5%). Metabolites in urine accounted for approximately 1.5% of the administered dose. These metabolites, Pliti-M1 and Pliti-M19, eluted in the beginning of LC-gradient system I and just before the parent peak (U26-27). The total radioactivity in these fractions was quantified and accounted for 0.8% (U3-5) and less than 0.1% (U25) of the administered dose. The chemical structures of the metabolites in U3-5 could not be elucidated and in U25 carbonyl-reduced plitidepsin was found.
The majority of the radioactivity was recovered in faeces. After a delay of 24 h, plitidepsin and plitidepsin-related compounds were excreted in faeces and after 20 days (480 h after the start of the infusion). 71.3 ± 2.6% of the administered radioactivity was recovered. In total, 77.4% of the initial dose was recovered in excreta after 480 h. The mean faecal sample pre-treatment recovery was 87.3% and showed no time-related trend in the recovery. Using LC-gradient system II, inter-patient pooled faecal samples were analysed and the radiochromatograms revealed that plitidepsin is highly metabolised.
In vitro studies with human liver microsomes, aimed at describing the plitidepsin biotransformation pathways, identified four plitidepsin-related metabolites [14]. In the formation of three of them (Apli-da, Apli-h and Apli-da/h), CYP3A4 was the involved cytochrome, while Apli-dm metabolite was produced only by CYP2A6. In the human mass balance study, two of these metabolites were found in faeces: Pliti-M18 (Apli-h) and Pliti-M16 (Apli-dm). Hence, total correlation between in vitro and in vivo results could not be achieved, which may be partially explained as follows. First of all, in vitro plitidepsin concentrations were far higher (1–100 µg/mL) than those found in patients (25.4 ng.eq/mL plitidepsin). And second, the very low plasma concentration of most plitidepsin-related metabolites seriously hampered their accurate identification. On the other hand, some of the metabolites identified in faeces may be products of chemical degradation of either plitidepsin or/and plitidepsin-related metabolites.
Brandon et al. (2006) [14] already concluded that the metabolites formed by CYP enzymes are less toxic than plitidepsin [14] and that the liver is responsible for its detoxification. One of the metabolites that correlates with the in vitro data (Pliti-M18) is generated by CYP3A4, hence this enzyme is at least moderately involved in plitidepsin metabolism. Therefore, care should be taken when plitidepsin is administered in combination with other CYP3A4 substrates.
An overview of structural information of the detected metabolites obtained by low and high resolution MS is presented in Table 2. In faeces, the majority of the metabolites was identified (confirmed by high-resolution MS data), however, the chemical structures of a few metabolites could not be unambiguously elucidated. Some of these, including peaks in F1–16, F17–18, F19–22, F27–33 and F35–38, did not contain individual fractions accounting for more than 10% of 14C-plitidepsin-related radioactivity, and thus, further nonclinical characterisation is considered unnecessary, since metabolites are considered to be major metabolites if they account for more than 10% of the parent drug exposure [16].
Occasionally, metabolite identification was hampered for various reasons, which are discussed below.
Discussion of the limitations
Essentially, the limitations seen in this study were very similar to those in the trabectedin metabolite-profiling study, another marine-derived compound [17]. Plitidepsin is also given as an i.v. infusion containing only a few milligrams of drug substance, and similar to trabectedin, it also has a complex ring structure. Plitidepsin is mainly excreted via the faeces, it has a very long half-life and also a large volume of distribution of 2180 L in plasma and of 751 L in whole blood [12]. The volume of distribution of plitidepsin and plitidepsin metabolites (TRA) in plasma was 315 L. Ultimately, the consequence is that metabolite levels in plasma, urine and faeces are low at all times. Various reasons for this issue are proposed. As mentioned, the flat dose of 7 mg was low to begin with, but accounting for losses to the administration system, an actual mean dose of 6 mg (80 µCi) was given [12]. The ratio of cold:hot material was 4.8:2.2 mg. The amount of 14C was roughly half of the amount of 12C, but was sufficient for the determination of the routes of elimination. However, the identification of metabolites was based on the detection of protonated 12C ions (and more commonly Na+ ions) and these amounts of 12C, albeit being double the amount of 14C, was not sufficient for this purpose. One thing that can be concluded from these results is that many metabolites have been formed, but the identities of the metabolites were not always elucidated, ultimately, due to sensitivity issues and particularly low levels.
Besides the fact that metabolite levels were already very low to begin with, more issues arose during the study which contributed to the difficulty to identify plitidepsin metabolites formed in humans.
First of all, besides the expected [M + H]+ ions, the sodium and potassium adducts were also formed in the source of the mass spectrometer after ionisation in the positive ion mode. As a result, MS ions at [M + H]+ and its adducts were found in clusters around m/z + 22 and + 38, resulting in reduced sensitivity.
Second, plitidepsin contains a proline moiety in a side chain attached to the depsipeptide ring. These proline-containing peptides have more than one conformation: cis- and trans-isomers. The origin of the observed conformational isomerism in plitidepsin is slow rotation about the proline-pyruvoyl tertiary amide bond [18,19,20,21,22]. Under room temperature conditions, this conformation equilibrium reaction is slow enough that the two conformers are separated as two chromatographic peaks. As temperature increased, the conformation equilibrium reaction rate increased so the two peaks merged into one. It is unknown if under these experimental settings the conformational isomers of the metabolites are also eluting as one peak from the column. Naturally, the loss of that part of the side chain responsible for the conformational isomerism conformers would remove the conformers as well. Losses in, possibly, the side chain include metabolites Pliti-M6, Pliti-M7, Pliti-M10, Pliti-M12, Pliti-M14, Pliti-M17 and Pliti-M19. The different peaks with m/z 1071 (Pliti-M6, Pliti-M7, Pliti-M10 and Pliti-M12) could potentially be structural isomers, which could support the suggestion that the biotransformation has not taken place in the side chain, but in the Thr group instead.
Third, as is common in these types of metabolite profiling studies, sample pre-treatment of plasma and faeces samples was non-selective. This is a requirement in such studies, since a specific sample clean-up method could mean the selective loss of metabolites. To screen for metabolites, the sample pre-treatment involved addition of an organic solvent, followed by a concentration step to extract as much plitidepsin-related compounds from the matrix. As a result of the non-selective sample, pre-treatment endogenous matrix compounds were extracted too. These compounds introduce ion suppression effects and their high abundancy hampered the detection and identification of plitidepsin-related compounds, especially the metabolites that were eluting just after the dead time (F1–16). On the other hand, the non-selective sample pre-treatment resulted in a high extraction recovery.
As a consequence of these sensitivity issues, MS2 and MS3 data could not always be obtained. The development of reference standards for potential metabolites to overcome some of these problems was, unfortunately, not feasible in this study. Hence, structure elucidation of metabolites was occasionally not possible.
Similar to trabectedin, it was found that plitidepsin metabolises into a large number of metabolites, of which none is clearly predominating. In the faeces chromatograms, clear chromatographic peaks with sufficient resolution were visible, but most of the radioactivity appeared to consist of a large number of metabolites continuously eluting from the column, resulting in a smear-like appearance in the radiochromatogram. Quantification as well as identification could therefore pose problems.
Despite complications, the suitability of the assay is believed to be more than sufficient. It could be suggested that the sensitivity issues could have been overcome using ultra-performance liquid chromatography (UPLC). However, this was not a possibility since large injection volumes (50 µL) were required for TRA quantification and these large injection volumes are not compatible with UPLC. The large TRA fractions of 1 min (1 mL) were collected to compromise between chromatographic resolution and sensitivity. Moreover, sample pre-treatment methods were highly suitable, since it was non-specific, eliminating concerns about compound-specific losses.
Plitidepsin binds to red blood cells [23, 24], but no metabolite profiling was performed on whole blood samples. Nevertheless, the absolute difference between TRA and plitidepsin levels in plasma were much larger at the tmax as compared to the differences between the two in whole blood [12]. It turned out that the metabolites formed do not have higher affinity for red blood cells than plitidepsin, since TRA data are almost fully explained by the parent. When selecting time points for the screening and quantification of metabolites in plasma it was necessary to choose samples with sufficient TRA, but also samples with sufficient metabolism were to be selected. Combining these observations, it was unlikely that high concentrations of metabolites could be found in whole blood.
Finally, the position of the radioactive label could have been a drawback. The 14C-label is positioned in the side chain (Fig. 1), which means that any metabolite which has lost this side chain cannot be detected by LSC, hence not quantified.
It might have been a solution to collect smaller fractions to get a better idea of the exact retention time of the m/z value corresponding to the radioactivity in the fraction. However, this would not have improved identification/elucidation because this would not have had any effect on the MS2 and MS3 data.
This metabolite profiling study concluded that plitidepsin undergoes dealkylation (demethylation), oxidation, carbonyl reduction, and (internal) hydrolysis. N/O-demethylation has been described as a biotransformation reaction for plitidepsin. The loss of 56 Da could indicate either the net loss of C3H4O or C4H8 group as suggested by Brandon et al. [14]. However, the accurate mass data confirm the loss of C3H4O. From the metabolites which have not been identified, it can be concluded that they are of a more hydrophilic nature than plitidepsin, since they elute from the reversed phase column before the parent compound.
All in all, a substantial amount of information has been collected regarding possible structures and a concise overview of plitidepsin’s metabolism has been given.
Conclusion
Plitidepsin absorption, distribution, metabolism and excretion characteristics were investigated in a human mass balance and metabolite profiling study in which six patients received a 3-h i.v. infusion of 14C-plitidepsin. After 480 h on average 77.4% of the administered dose was recovered in urine and faeces combined. The hepatic/biliary route was the major excretory route for drug-related entities. In urine, the majority was accounted for by unchanged plitidepsin; a mere 1.5% of the total dose was excreted in urine as metabolites. Unchanged plitidepsin was excreted in small amounts (2.3% of administered dose) in faeces, indicating extensive metabolism following i.v. administration. LC-Ion Trap MS-LSC analyses were performed to identify and quantify plitidepsin metabolites in plasma, urine and faeces. No metabolites were detected in plasma. Metabolite concentrations in urine were extremely low and past the 24-h time point no metabolites were found at all. In the first 24 h after dosing, two metabolites, Pliti-M1 and Pliti-M19 were detected, together accounting for less than 1% of the administered dose. Numerous reasons for incomplete identification of metabolites have been considered, including the PK characteristics and low dose of the analyte, sensitivity issues due to ion suppression, Na+ and K+ formation, and conformational isomers. From all compounds listed in Table 2, six metabolites, Pliti-M1, Pliti-M2, Pliti-M4, Pliti-M5, Pliti-M9 and Pliti-M13 were solely identified by MS1 data, and specific biotransformation reactions and identities have been proposed for six metabolites, including Pliti-M11, Pliti-M14, Pliti-M15, Pliti-M16, Pliti-M18 and Pliti-M19. The metabolites identified in faeces included hydrolysed plitidepsin, demethylated plitidepsin, oxidated plitidepsin, oxidated demethylated plitidepsin, carbonyl-reduced plitidepsin, carbonyl-reduced demethylated plitidepsin, and various metabolites for which no structure could be elucidated, but masses were confirmed by accurate mass data, including metabolites Pliti-M3, Pliti-M6, Pliti-M7, Pliti-M8, Pliti-M10, Pliti-M12 and Pliti-M17.
References
Muñoz-Alonso MJ, González-Santiago L, Martinez TJ et al (2009) The mechanism of action of plitidepsin. Curr Opin Investig Drugs 10:536–542
Oliveira H, Thevenot J, Garanger E et al (2014) Nano-encapsulation of plitidepsin: in vivo pharmacokinetics, biodistribution, and efficacy in a renal xenograft tumor model. Pharm Res 31:983–991. https://doi.org/10.1007/s11095-013-1220-3
Alonso-Alvarez S, Pardal E, Sanchez-Nieto D et al (2017) Plitidepsin: design, development, and potential place in therapy. Drug Des Devel Ther 11:253–264. https://doi.org/10.2147/DDDT.S94165
Losada A, Muñoz-Alonso MJ, García C et al (2016) Translation elongation factor eEF1A2 is a novel anticancer target for the marine natural product plitidepsin. Sci Rep 6:1–15. https://doi.org/10.1038/srep35100
Mateos MV, Cibeira MT, Richardson PG et al (2010) Phase II clinical and pharmacokinetic study of plitidepsin 3-h infusion every two weeks alone or with dexamethasone in relapsed and refractory multiple myeloma. Clin Cancer Res 16:3260–3269. https://doi.org/10.1158/1078-0432.CCR-10-0469
Salazar R, Plummer R, Oaknin A et al (2011) Phase I study of weekly plitidepsin as 1-h infusion combined with carboplatin in patients with advanced solid tumors or lymphomas. Invest New Drugs 29:1406–1413. https://doi.org/10.1007/s10637-010-9488-1
Plummer R, Lorigan P, Brown E et al (2013) Phase I-II study of plitidepsin and dacarbazine as first-line therapy for advanced melanoma. Br J Cancer 109:1451–1459. https://doi.org/10.1038/bjc.2013.477
Aspeslagh S, Awada A, Matos-Pita SA et al (2016) Phase I dose-escalation study of plitidepsin in combination with bevacizumab in patients with refractory solid tumors. Anticancer Drugs 27:1021–1027. https://doi.org/10.1097/CAD.0000000000000409
Aspeslagh S, Stein M, Bahleda R et al (2017) Phase I dose-escalation study of plitidepsin in combination with sorafenib or gemcitabine in patients with refractory solid tumors or lymphomas. Anticancer Drugs 28:341–349. https://doi.org/10.1097/CAD.0000000000000457
US Food and Drug Administration (2008) FDA guidance for industry: safety testing of drug metabolites. US Department of Health and Human Services, Food and Drug Administration, and Center for Drug Evaluation and Research, Rockville
ICH Topic M3 (R2) (2009) Non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. doi: EMA/CPMP/ICH/286/1995
van Andel L, Fudio S, Rosing H et al (2017) Pharmacokinetics and excretion of 14C–Plitidepsin in patients with advanced cancer. Invest New Drugs 35:589–598. https://doi.org/10.1007/s10637-017-0432-5
Brandon EFA, van Ooijen RD, Sparidans RW et al (2005) Structure elucidation of aplidine metabolites formed in vitro by human liver microsomes using triple quadrupole mass spectrometry. J Mass Spectrom 40:821–831. https://doi.org/10.1002/jms.863
Brandon EFA, Sparidans RW, van Ooijen RD et al (2006) In vitro characterization of the human biotransformation pathways of aplidine, a novel marine anti-cancer drug. Invest New Drugs 25:9–19. https://doi.org/10.1007/s10637-006-7589-7
Ngoka LCM, Gross ML, Toogood PL (1999) Sodium-directed selective cleavage of lactones: a method for structure determination of cyclodepsipeptides. Int J Mass Spectrom 182–183:289–298. https://doi.org/10.1016/S1387-3806(98)14248-3
Nijenhuis CM, Schellens JHM, Beijnen JH (2016) Regulatory aspects of human radiolabeled mass balance studies in oncology: concise review. Drug Metab Rev 48:266–280. https://doi.org/10.1080/03602532.2016.1181081
Beumer JH, Rademaker-Lakhai JM, Rosing H et al (2007) Metabolism of trabectedin (ET-743, Yondelis) in patients with advanced cancer. Cancer Chemother Pharmacol 59:825–837. https://doi.org/10.1007/s00280-006-0342-2
Sparidans RW, Kettenes-Van Den Bosch JJ, Van Tellingen O et al (1999) Bioanalysis of aplidine, a new marine antitumoral depsipeptide, in plasma by high-performance liquid chromatography after derivatization with trans-4′-hydrazino-2-stilbazole. J Chromatogr B Biomed Sci Appl 729:43–53. https://doi.org/10.1016/S0378-4347(99)00118-8
Cárdenas F, Thormann M, Feliz M et al (2001) Conformational analysis of dehydrodidemnin B (aplidine) by NMR spectroscopy and molecular mechanics/dynamics calculations. J Org Chem 66:4580–4584. https://doi.org/10.1021/jo010123h
Nuijen B, Rodrigues-Campos I, Noain C et al (2001) HPLC-UV method development and impurity profiling of the marine anticancer agent aplidine in raw drug substance and pharmaceutical dosage form. J Liq Chromatogry Relat Technol 24:3119–3139
Cárdenas F, Caba JM, Feliz M et al (2003) Analysis of conformational equilibria in aplidine using selective excitation 2D NMR spectroscopy and molecular mechanics/dynamics calculations. J Org Chem 68:9554–9562. https://doi.org/10.1021/jo034798r
Yin J, Aviles P, Lee W et al (2003) Development of a liquid chromatography/tandem mass spectrometry assay for the quantification of aplidine, a novel marine-derived antineoplastic agent, in human plasma. Rapid Commun Mass Spectrom 17:1909–1914. https://doi.org/10.1002/rcm.1133
Faivre S, Chièze S, Delbaldo C et al (2005) Phase I and pharmacokinetic study of aplidine, a new marine cyclodepsipeptide in patients with advanced malignancies. J Clin Oncol 23:7871–7880. https://doi.org/10.1200/JCO.2005.09.357
Nalda-Molina R, Valenzuela B, Ramon-Lopez A et al (2009) Population pharmacokinetics meta-analysis of plitidepsin (aplidin) in cancer subjects. Cancer Chemother Pharmacol 64:97–108. https://doi.org/10.1007/s00280-008-0841-4
Funding
This study was funded by Pharma Mar, S.A.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
RL, PA, AF and SF are currently employees at Pharma Mar, S.A. LA, HR, MMT, LL, AG, MJXH, JHMS and JHB are employed at the Netherlands Cancer Institute.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the participating institution and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Rights and permissions
About this article

Cite this article
van Andel, L., Rosing, H., Tibben, M.M. et al. Metabolite profiling of the novel anti-cancer agent, plitidepsin, in urine and faeces in cancer patients after administration of 14C-plitidepsin. Cancer Chemother Pharmacol 82, 441–455 (2018). https://doi.org/10.1007/s00280-018-3637-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-018-3637-1