
Abstract
Purpose
The hydrazide backbone is a well-known structural core system found in a broad range of biologically activated compounds. Among which, the compounds with anticancer activity have been cited in a number of studies. With this object in mind, we focused on the in vitro and in vivo anticancer potential of two novel hydrazide derivatives bearing furan or thiophen substituents (compounds 1 and 2).
Methods
The cytotoxic property was evaluated using MTT assay against MCF-7 human breast adenocarcinoma cell line, while the in vivo antitumor activity was investigated in BALB/c mice bearing 4T1 mammary carcinoma cells. Flow cytometry was used for cell cycle analysis, and detection of apoptosis was examined by Annexin-V-FLUOS/PI assay. Protein expression of Bax, Bcl-2 and procaspase-3 was estimated by Western blotting.
Results
Compounds 1 and 2 were found to be cytotoxic towards breast cancer cells presenting IC50 values of 0.7 and 0.18 µM, respectively, and selectivity over normal fibroblast cells. Our findings further indicated that 2 × IC50 concentrations of both compounds induce early stage apoptosis and increase percentage of sub-G1 population in MCF-7 cells at 48 h. An elevation in Bax/Bcl-2 ratio and caspase-3 cleavage suggested that apoptosis induced by the two compounds is through a caspase- and mitochondrial-dependent pathway. In the in vivo study, compounds 1 and 2 at doses of 10 and 1 mg/Kg/day, respectively, significantly hindered the growth of tumor after 3 weeks of i.p. administration, when compared to vehicle-treated mice.
Conclusion
Collectively, the great potential exhibited by compound 2 could make it a promising chemotherapeutic candidate for human cancers, especially for breast cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Breast cancer is one of the most frequent carcinomas among women with more than half a million cases reported every year and it is estimated that 1.7 million women will be diagnosed with breast cancer in 2020 [1, 2]. One of the growing problems of cancers, including breast cancer, leading to high mortality is the undesirable side effects and tumor resistance to the current cancer chemotherapeutics [3, 4]. Thus, exploring the novel synthetic anticancer agents remains a significant and intriguing goal in pharmacological research, and there is always a need towards developing new and more effective compounds, which could be less cytotoxic for normal cells and overcoming tumor multidrug resistance.
A large number of synthetic compounds, possessing remarkable anticancer effects toward breast cancer, target different cellular and molecular factors that contribute to apoptosis. Apoptosis is an essential process in maintaining tissue homeostasis in which the cells that are unnecessary or threaten the organism are eradicated. Imperfect regulation of this process, also known as cell suicide, leads to tumor resistance [5, 6]. It has proved difficult to induce apoptosis in breast cancer cells; thus, search for new agents effective in activating apoptosis appears to be of great interest in chemotherapy [7].
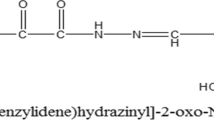
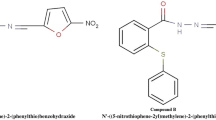
Hydrazide-hydrazones were reported as good candidates for different pharmaceutical applications, and their therapeutic importance has been well established [8,9,10,11,12,13,14]. In addition, hydrazide–hydrazones have been the subject of intense studies due to their different bioactivities, including anti-malarial, anti-tuberculotic, and anti-HIV activities [15,16,17,18,19,20,21]. Among the various activities, there are also reports of anticancer property for hydrazide derivatives [22, 23]. These reports motivated us to screen a subset of synthesized hydrazide derivatives, previously evaluated for antimycobacterial activity [24], against different cancer cell lines including breast, colon, oral and leukemic cells (data not published) to identify molecules with a desirable potency as well as the most sensitive cell lines. From this screening, we recognized compounds 1 and 2 (Fig. 1) as novel anticancer lead compounds possessing furan and thiophen moieties, which increased cell toxicity by >50% in the leukemic and breast cancer cells. In this regard, these two compounds revealed a pronounced broad spectrum anticancer activity against leukemia cell line through induction of apoptosis in a mitochondrial-dependent pathway [25]. These findings along with the knowledge that MCF-7 cell is chemo-resistant to chemical compounds [26] prompted us herein to evaluate the in vitro as well as in vivo anticancer property of these two compounds in breast cancer.

Chemical structures of compounds 1 and 2
Materials and methods
Cells and chemicals
Human breast adenocarcinoma (MCF-7, C135), mouse mammary tumor (4T1, C604) and human gingival fibroblast (HGF-1 PI1, C165) cell lines were obtained from National Cell Bank of Pasteur Institute of Iran (NCBI). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco-BRL, Rockville, IN) supplemented with 10% fetal bovine serum (FBS) (Gibco-BRL, Rockville, IN), and 1% Penicillin/Streptomycin (Gibco-BRL, Rockville, IN). Polyclonal anti-Bcl-2 (1:500), anti-caspase-3 (1:500), anti-GAPDH (1:1000) and anti-Bax (1:500) antibodies were purchased from Abcam (Cambridge, MA), whereas anti-rabbit IgG horseradish peroxidase (HRP) antibody (1:5000) was from Cell Signaling Technology (Beverly, MA). Apoptosis assay was performed using Annexin-V-FLUOS commercial kit obtained from Roche Applied Science (Indianapolis, IN, USA). ECL advance Western blotting detection kit was purchased from General Electric Health Care Life Sciences (Buckinghamshire, UK). Compounds 1 and 2 were synthesized by the Medicinal Chemistry Research Laboratory at the Faculty of Pharmacy, Pharmaceutical Sciences Branch, Islamic Azad University [24]. The remaining chemicals used, otherwise indicated, were from Merck (Darmstadt, Germany), and Sigma-Aldrich (St Louis, MO, USA).
Cytotoxicity assay
The cytotoxic activity of compounds 1 and 2 was evaluated by employing MTT assay. MCF-7 cells were seeded at a concentration of 5–7 × 103 cells per well in 96 well plates and incubated for 24 h at 37 °C in a humidified 5% CO2 incubator. Cells were then treated by different concentrations (0.001–20.0 µM) of the test compounds for 24, 48 and 72 h. Untreated cells, 0.3% DMSO- and doxorubicin-treated cells served as negative, vehicle and positive control cells, respectively. Following addition of 20 μL of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide, 5 mg/mL) to each well, cells were further incubated at 37 °C for 4 h. The supernatants were then aspirated, and 200 µL of dimethyl sulfoxide (DMSO) were used to dissolve purple formazan in each well. The plates were then shaken for an additional 15 min and using a Microplate Reader (Star Fax-2100, ST. Louis, USA) the absorbance values were read at 545 nm.
Cell cycle analysis
Cell cycle phase distribution carried out using PARTEC flow cytometer (Partec GmbH, Munster, Germany) with Flowjo Software. Cell cycle phases were determined by recording the peak area of FL3-A on a linear scale. MCF-7 cells (3 × 105) were seeded in 6-well plates and treated with compounds 1 and 2 at their 2 × IC50 concentrations. Following 48-h treatment, the cells were washed with PBS and fixed in ice cold 70% ethanol. The plates were finally stained using PI (Propidium Iodide, 1 mg/mL), Triton X-100 (0.1%), and RNAse (100 mg/mL) and incubated for 20 min in dark at 37 °C and then analyzed for DNA content using flow cytometer. Samples were prepared in triplicates with at least three repetitions for each experiment. To exclude cell debris and cell clumps from the analysis, suitable gating was employed.
Annexin-V/PI double staining for apoptosis identification
After incubation of MCF-7 cells with compounds 1 and 2 at their 2 × IC50 concentrations for 48 h, cells were trypsinized and washed with PBS. Following adjustment of concentration of cells to 106 cells/mL, MCF-7 cells were stained using 100 μL of annexin-V-FLUOS labeling solution containing 2 μL annexin-V-FLUOS labeling agent, 2 μL PI solution and 1 mL incubation buffer. Cells were then subjected to flow cytometry, following an incubation period of 15 min at 37 °C. The percentages of viable (Annexin-V−/PI−), early apoptotic (Annexin-V+/PI−), late apoptotic (Annexin-V+/PI+) and necrotic (Annexin-V−/PI +) cells were finally analyzed.
Western blot analysis
Proteins were extracted from untreated control and compounds 1 and 2-treated cells at their 2 × IC50 concentrations after 48 h using lysis buffer (Tris 62.5 mM (pH 6.8), DTT 50 mM, SDS 10%, glycerol) with the protease inhibitor. Following determination of protein concentration with Bradford protein assay, the extracted proteins were electrophoresed in 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto PVDF membrane. Membranes were then blocked with TBST (50 mmoL/L Tris–HCl, pH 7.6, 150 mmoL/L NaCl and 0.1% Tween 20) containing 1% (w/v) casein for 2 h, and further incubated with primary antibodies overnight. After washing the membranes with the HRP conjugated goat anti-rabbit IgG for 2 h, blots were developed using ECL advance Western blotting detection kit (GE health care, Buckinghamshire, UK). Protein marker (Prestained Protein Ladder, SinaClon) was used to determine the size of the targeted proteins and the density of the bands were analyzed using Image J software (Version 1.48, National Institute of Health, USA) by normalizing against GAPDH (Glyceraldehyde Phosphate Dehydrogenase).
In vivo study
Female BALB/c mice aged between 6 and 8 weeks (18–20 g) were prepared from the National Animal Center (Pasteur Institute of Karaj) and maintained in a 12/12-h light–dark cycle, with food and water supplied ad libitum. Animals were treated in accordance with the guidelines approved by the animal ethics committee of Pasteur Institute of Iran. Following trypsinization of exponentially 4T1 cells, 10 6 cells were re-suspended and inoculated into the mammary fat pad of the mice. Seven days after inoculation, once the tumor masses were palpable, tumors-bearing mice were randomly divided into eight groups (10 mice per group). To explore the tumor growth inhibitory property of compounds 1 and 2, treatments were daily administered by intra-peritoneal injection of vehicle alone and various doses of compounds 1 and 2 (1, 10 and 50 mg/Kg/day) for 3 weeks. The tumors growth was monitored every other day and the tumors volume was measured thrice a week in two dimensions using a digital caliper. The tumor volume (mm3) was determined through the following formula: (length × width2)/2.
Statistical analysis
The data are expressed as mean ± SEM of at least triplicate determinations, and comparisons were based on ANOVA followed by the Tukey’s post test using GraphPad Prism 6.0 Software. A p value lower than 0.05 was considered as significant.
Results
Compounds 1 and 2 effectively increased cytotoxicity
Two new synthesized compounds (1 and 2) were investigated for their cytotoxicity against human breast adenocarcinoma (MCF-7) as cancerous and human fibroblast (HGF) as normal cell lines using the MTT assay. Doxorubicin-treated cells were used as the positive control and the results were summarized in Table 1. IC50 values reported in Table 1 revealed a potent cytotoxic activity against MCF-7 cells after 72 h of compounds 1 and 2 treatment with IC50 values of 0.7 and 0.18 µM, respectively. Considering the IC50 values, the two compounds were superior to doxorubicin which showed an IC50 value of 2.1 µM. The highest cytotoxic activity was for compound 2, showing a approximately four-fold potency more than compound 1 after 72 h of treatment. We also evaluated the effect of these two compounds on normal human fibroblast (HGF) cytotoxicity. Interestingly, compound 2 had little effect on gingival fibroblast cell viability (IC50 >10 µM) indicating the selectivity of compound 2 towards normal and cancer cell lines, whereas compound 1 demonstrated less selectivity compared to compound 2 (IC50 >7.5 µM).
Effect of compounds 1 and 2 on MCF-7 cell cycle progression
Since the induction of cancer cell death can be due to necrosis, apoptosis, cell-cycle arrest, or a combination of these processes, and considering the promising cytotoxic properties of compounds 1 and 2, we further evaluated the potential of compounds 1 and 2 to induce apoptosis in breast cancer MCF-7 cells. In this regard, we used flow cytometry to assess the effects of both compounds on cell cycle progression. As shown in Fig. 2, treatment of MCF-7 cells with both compounds at their 2 × IC50 concentrations for 48 h caused a significant cell population increase in the sub-G1 phase from 16.6% in control to 22.67 and 27.05% in compounds 1 and 2-treated cells, respectively, indicating a cell apoptosis induced by the two compounds. Accumulation of cells in sub-G1 phase was more significant in compound 2- than in compound 1-treated cells; however, neither change in the percentage of cells in S phase nor cell cycle arrest was detected after 48 h of treatment of MCF-7 cells with compounds 1 and 2.

Monitoring of MCF-7 cell cycle phase distribution using flow cytometry after staining with propidium iodide (PI). The data presented are the mean ± SE of three independent experiments. **p < 0.01, ***p < 0.001 compared with control after treatment of MCF-7 cells with compounds 1 (1.4 µM) and 2 (0.4 µM) for 48 h
Compounds 1 and 2 induced cell death via the apoptotic pathway
To confirm whether the cell cytotoxicity was due to apoptosis, we analyzed apoptosis following treatment of breast cancer cells with compounds 1 and 2 at their 2 × IC50 concentrations for 48 h using Annexin-V staining. Flow cytometry using FLUOS conjugated annexin-V (FL1-H) and PI (FL2-H) double staining differentiates between alive normal cells (Annexin-V−/P−), early apoptotic (Annexin-V+/PI−), late apoptotic (Annexin-V+/PI+) and necrotic cells (Annexin-V−/PI+). Our results revealed that MCF-7 treatment with 2 × IC50 concentrations of compounds 1 and 2 results in 4.63 ± 0.93 and 7.06 ± 0.38% early apoptosis, respectively, whereas MCF-7 control cells (untreated cells) caused 0.34 ± 0.18% of apoptosis (Table 2). Additionally, no significant change in population of late apoptotic and necrotic cells was observed in the compound 1 and 2-treated cells compared to the vehicle-treated cells (Table 2). These results support the idea that compounds 1 and 2 most certainly induce early apoptosis in the human breast cancer cell line MCF-7.
Once a cell is committed to apoptosis, a series of reactions occurs lastly resulting in the activation of caspase-3. Thus, the effect of compounds 1 and 2 on procaspase-3 cleavage was investigated and found that caspase-3 was activated after 48 h of treatment with compounds 1 and 2 at 2 × IC50 concentrations (Fig. 3). Cell death signaling pathways such as apoptosis are usually triggered by changes in the mitochondria [27]. Proteins of Bcl-2 family regulate the mitochondrial-dependent apoptotic pathway through both pro-apoptotic (Bax) and anti-apoptotic (Bcl-2) effects in cancer cells. The final consequence of Bax/Bcl-2 ratio change is the release of cytochrome c from mitochondria into the cytosol and the activation of caspases, which leads to apoptosis induction [28]. To evaluate if the apoptotic induction occurred via the mitochondrial pathway, the effect of compounds 1 and 2 on the expression levels of Bax and Bcl-2 was analyzed by Western blotting. As shown in Fig. 4a, treatment of MCF-7 cells with 2 × IC50 concentrations of compounds 1 and 2 led to a significantly elevated level of Bax (Fig. 4b) and reduced level of Bcl-2 (Fig. 4c). The Bax/Bcl-2 ratio was elevated by 1.5- and 2-fold for compounds 1 and 2, respectively, compared to the control (Fig. 4d). Thus, mitochondria have an important role in apoptosis, induced by compounds 1 and 2.

Western blot analysis of the procaspase-3 expression in MCF-7 cells treated with compounds 1 (1.4 μM) and 2 (0.4 μM) for 48 h. GAPDH was used as a loading control. Protein band intensities were quantified by Image J. Results were considered significant if *p < 0.05, **p < 0.01

Expression analysis of apoptosis-associated proteins by Western blot analysis. MCF-7 cells were treated with compounds 1 (1.4 μM) and 2 (0.4 μM). a Immunoblot showing the expression levels of Bax, Bcl-2 and GADPH. Statistical analysis of b Bax, c Bcl-2 and d Bax/Bcl-2 after 48 h of treatment. The results of 3 independent experiments are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 indicate significance differences of each group with the control group
Compounds 1 and 2 suppressed primary tumor growth
To validate the anticancer activity of compounds 1 and 2 in vivo, BALB/c mice were subcutaneously (s.c.) inoculated with 4T1 murine tumor cells. Following the evaluation of mammary tumor volume, tumors were developed on day 7 after cell inoculation. Then, the mice were separated in three groups and intraperitoneally (i.p.) treated with either the vehicle or compounds 1 and 2 (1, 10, 50 mg/Kg/day) for 21 days. The tumor volume was measured and compared to the vehicle-treated mice thrice a week from the beginning of the treatment. As illustrated in Fig. 5a, 1 mg/Kg/day i.p. administration of compound 1 failed to significantly inhibit mammary tumor growth on the first and second week compared to the vehicle-control group. However, tumor volume reduction was significant after 3 weeks of treatment with 1 mg/Kg/day of compound 1. With an increase in the dose of compound 1 to 10 mg/Kg/day, tumor growth was inhibited significantly (p < 0.05) compared to the control animals, even on week 1. An upward trend in tumor volume reduction was observed during the 3-week treatment with 10 mg/Kg of compound 1 per day. Paradoxically, once the dose of daily administration reached 50 mg/Kg, compound 1 revealed a lower efficiency in mammary tumor growth inhibition compared to a dose of 10 mg/Kg/day throughout the experiment. Importantly, 1 mg/Kg of compound 2 daily i.p. administration for 3 consecutive weeks diminished the tumor volume much more significantly than 10 mg/Kg/day i.p. administration of compound 1, when compared to control mice at the third week (p < 0.001) (Fig. 5a, b). It is worth mentioning that the significant effect of compound 2 on tumor volume reduction at dose of 1 mg/Kg/day was first observed on the second week. Surprisingly, the larger tumor volumes were associated with the higher doses of compound 2 administration (10 and 50 mg/Kg/day) (Fig. 5b). At the end of experiment, a significant inhibition of tumor growth was observed with 10 mg/Kg/day of compound 1 and 1 mg/Kg/day of compound 2 from the second week of treatment (Fig. 5). The efficacy in reduction of tumor volume was highest for compound 2 in the third week (Fig. 6). No weight loss, drug-related death or discomfort was observed during the treatment period at all three doses, indicating that both compounds were well tolerated by the animals at 10 and 1 mg/Kg per day.

Tumor volume (mm3) in BALB/c mice with mammary cancer (4T1 breast cancer model) treated by a compound 1 and b compound 2 at doses of 1,10 and 50 mg/Kg/day for 3 weeks. One-way ANOVA test (post-Tukey test) done for n = 10 mice per group. Error bar indicates SEM, (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001)

Comparison of the effect of compounds 1 and 2 at different doses (1, 10, 50 mg/Kg/day) on tumor growth during the 3 weeks treatment. Data are expressed as mean ± SEM, n = 10 mice per group
Discussion
Our results demonstrated that compounds 1 and 2, the most active molecules among the synthesized hydrazide derivatives, reduced breast cancer cell (MCF-7) viability, with little or no cytotoxicity against normal fibroblast cells. Interestingly, the IC50 values determined for compounds 1 and 2 on MCF-7 cells following 72 h of incubation were much lower than that of doxorubicin. Notably, the IC50 value obtained for doxorubicin on MCF-7 cells was relative similar to that reported in the literature [29]. Both compounds caused the accumulation of breast cancer cells in sub-G1 phase; however, this effect was more significant in MCF-7 cells treated with compound 2 at the 2 × IC50 concentrations after 48 h.
One of the prominent events occurring in apoptosis is the activation of caspases leading to the activation of caspase-activated deoxyribonuclease (CAD), which subsequently degrades genomic DNA into oligomers [30, 31]. Cells containing a lower DNA content constitute the sub-G1 cell population. In our study, compound 2 caused a more significant accumulation of cells in sub-G1 phase revealing that compound 2 induces more caspase mediated apoptosis, a finding confirmed through the Annexin-V/PI staining assay and pro-caspase-3 cleavage experiment. In this regard, our results demonstrated that both compounds amplify procaspase-3 cleavage and induce cell apoptosis mainly through early stage, with a greater effect exerted by compound 2. These results are in line with our previous study [25] demonstrating a strong anti-breast cancer property of compound 2 in leukemic cells; however, MCF-7 cell line was less sensitive to apoptosis induced by both compounds compared to leukemic cells.
Evidence indicates that MCF-7 cells are the most resistant towards treatment with the hydrazide compounds, with cell viability reduction ranging from 5 to 33% [21]. Interestingly, compounds 1 and 2 were observed to have a great effect on cell cytotoxicity and apoptosis induction. Notably, compound 2, bearing a thiophen substituent, was found to be the most active agent with an IC50 value of 0.2 µM against MCF-7 cell line. The significance of the thiophen moiety was previously indicated by Bedia et al., showing a stronger activity of hydrazide compounds bearing thiophen substituent [32], and was further confirmed by our study [25]. Probably, the increase in lipophilicity, resulting in a better uptake by the cells, could be a qualitative description of advantageous changes in the cytotoxic efficiency of compound 2, which is in line with a previous report by Matysiak et al. [33]. It is also worth mentioning that compound 1 with an IC50 value of 0.7 µM was found to be moderately active against the above-mentioned cells; however, it displayed a ~13-fold potency in early stage induction of apoptosis compared to untreated cells.
Accumulated evidence indicates that Bax is vital for the onset of mitochondrial apoptosis via the intrinsic pathway. The balance between the expression levels of anti-apoptotic Bcl-2 and pro-apoptotic Bax plays a key role in cell survival or death, wherein an elevated Bax/Bcl-2 ratio will result in the activation of mitochondrial dysfunction and subsequently triggers the intrinsic apoptotic pathway [34]. Also, Bax to Bcl-2 ratio is important in supporting drug-induced apoptosis via the mitochondrial-mediated apoptotic pathway [35]. Given that hydrazide derivates most likely activate apoptosis via the mitochondrial pathway [25, 36] and to define the related mechanism associated with apoptosis induction by compounds 1 and 2, we determined Bax (pro-apoptotic) and Bcl-2 (anti-apoptotic) protein expression levels in MCF-7 cells treated with compounds 1 and 2 and compared them to the untreated cells. We observed that Bcl-2 levels decreased while that of Bax increased in the MCF-7 cells treated with compounds 1 and 2, suggesting that both compounds induced apoptosis through modulation of the Bcl-2 family. However, compound 2 was more potent in Bax to Bcl-2 ratio elevation compared to compound 1. Considering the results obtained from our in vitro assessment, compound 2 was more efficacious as an anti-breast cancer reagent.
Due to the effectiveness of compounds 1 and 2 in apoptosis induction in breast cancer cells in our in vitro model, we performed and confirmed their influence on mammary tumor volume reduction in an in vivo model. Statistically significant (p < 0.05) tumor growth inhibition was observed in compounds 1- and 2-treated groups compared to untreated group following 3 weeks of treatment. Importantly, compound 2 administered at 1 mg/Kg/day revealed a greater reduction in tumor volume compared to compound 1 at a dose of 10 mg/Kg/day. In this regard, treatment of mice with compound 1 resulted in more than two-fold decrease in tumor volume after 3 weeks, while compound 2 caused a five-fold reduction in tumor volume. Moreover, the tumor volume began to diminish at the first week in compound 2-treated group at doses of 1 and 10 mg/Kg/day and lasted for 3 weeks. This occurrence was observed in the group treated with 10 mg/Kg/day of compound 1 following a 3-week time period, suggesting that compound 2 is potent and that it rapidly reaches tumor site probably due to its more lipophilic characteristic. This result is consistent with the in vitro studies described above in suggesting a better uptake of compound 2 in mammary tumor cells.
In conclusion, owing to the in vitro evidence mentioned above, compound 2 induced breast cancer cell cytotoxicity through induction of apoptosis. The apoptosis-inducing activity of compound 2 was further verified by the accumulation of cells in sub-G1 phase, up-regulation of Bax expression, down-regulation of Bcl-2, and the activation of caspase-3. In addition, compound 2 consistently prevented the growth of mammary tumor in BALB/c mice. Collectively, this study may open a new door for design and synthesis of novel anticancer agents serving as effective therapeutic drugs.
References
Bray F, Ren JS, Masuyer E, Ferlay J (2013) Global estimates of cancer prevalence for 27 sites in the adult population in 2008. Int J Cancer 132:1133–1145
Forouzanfar MH, Foreman KJ, Delossantos AM, Lozano R, Lopez AD, Murray CJL, Naghavi M (2011) Breast and cervical cancer in 187 countries between 1980 and 2010: a systematic analysis. The Lancet 378(9801):1461–1484
Hajrezaie M, Paydar MJ, Looi CY, Moghadamtousi S, Hassandarvish P, Salga MS, Karimian H, Shams K, Zahedifard M, Majid NA (2015) Apoptotic effect of novel Schiff Based CdCl2 (C14H21N3O2) complex is mediated via activation of the mitochondrial pathway in colon cancer cells. Sci Rep 5:90–97
Marsh S, McLeod HL (2007) Pharmacogenetics and oncology treatment for breast cancer. Expert Opin Pharmacother 8:119–127
Kennedy SG, Wanger AJ, Conzen SD, Jordan J, Bellacosa A, Tsichlis PN, Hay N (1997) The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev 11:701–713
Low SW, Lin AW (2000) Apoptosis in cancer. Carcinogenesis 21(3):485–495
Wardakhan WW, EL-Sayed NN, Mohareb RM (2013) Synthesis and anti-tumor evaluation of novel hydrazide-hydrazone derivatives. Acta Pharmaceut 63:45–57
Bharti SK, Nath G, Tilak R, Singh SK (2010) Synthesis, anti-bacterial and anti-fungal activities of some novel Schiff bases containing 2, 4-disubstituted thiazole ring. Eur J Med Chem 45:651–660
Loncle C, Brunel JM, Vidal N, Dherbomez M, Letourneux Y (2004) Synthesis and antifungal activity of cholesterol-hydrazone derivatives. Eur J Med Chem 39:1067–1071
Papakonstantinou-Garoufalias S, Pouli N, Marakos P, Chytyroglou-ladas A (2002) Synthesis antimicrobial and antifungal activity of some new 3-substituted derivatives of 4-(2, 4-dichlorophenyl)-5-adamantyl-1H-1, 2, 4-triazole. Il Farmaco 57:973–977
Vicini P, Zani F, Cozzini P, Doytchinova I (2002) Hydrazones of 1, 2-benzisothiazole hydrazides: synthesis, antimicrobial activity and QSAR investigations. Eur J Med Chem 37:553–564
Sridhar SK, Pandeya SN, Stables JP, Ramesh A (2002) Anticonvulsant activity of hydrazones, schiff and mannich bases of isatin derivatives. Eur J Pharm Sci 16:129–132
Almasirad A, Hosseini R, Jalalizadeh H, Rahimi-Moghaddam Z, Abaeian N, Janafrooz M, Abbaspour M, Ziaee V, Dalvandi A, Shafiee A (2006) Synthesis and analgesic activity of 2-phenoxybenzoic acid N-phenylanthranilic acid hydrazones. Biol Pharm Bull 29(6):1180–1185
Kaymakçıoğlu BK, Rollas S (2002) Synthesis, characterization and evaluation of antituberculosis activity of some hydrazones. Il Farmaco 57:595–599
Vicini P, Incerti M, La Colla P, Lodda R (2009) Anti-HIV evaluation of benzo [d] isothiazole hydrazones. Eur J Med Chem 44:1801–1807
Rahman VM, Mukhtar S, Ansari WH, Lemiere G (2005) Synthesis stereochemistry and biological activity of some novel long alkyl chain substituted thiazolidin-4-ones and thiazan-4-one from 10-undecenoic acid hydrazide. Eur J Med Chem 40:173–184
Dimmock JR, Vashishtha SC, Stables JP (2000) Anticonvulsant properties of various acetylhydrazones, oxamoylhydrazones and semicarbazones derived from aromatic and unsaturated carbonyl compounds. Eur J Med Chem 35:241–248
Kaushik D, Khan SA, Chawla G, Kumar S (2010) N′-[(5-chloro-3-methyl-1-phenyl-1H-pyrazol-4-yl) methylene] 2/4-substituted hydrazides: synthesis and anticonvulsant activity. Eur J Med Chem 45:3943–3949
KüçükgüzelSG Mazi A, Sahin F, Ozturk S, Stables J (2003) Synthesis and biological activities of diflunisal hydrazide–hydrazones. Eur J Med Chem 38:1005–1013
Melnyk P, Leroux V, Sergheraerta C, Grellier P (2006) Design, synthesis and in vitro antimalarial activity of an acylhydrazone library. Bioorg Med Chem Lett 16(1):31–35
Menendez C, Chollet A, Rodriguez F, Inard C, Pasca MR, Lherbet C, Baltas M (2012) Chemical synthesis and biological evaluation of triazole derivatives as inhibitors of InhA and antituberculosis agents. Eur J Med Chem 52:275–283
Bingul M, Tan O, Gardner CR, Sutton SK, Arndt GM, Marshall GM, Cheung BB, Kumar N, Black DS (2016) Synthesis, characterization and anti-cancer activity of hydrazide derivatives incorporating a quinoline moiety. Molecules 21(7):916
Cihan-Üstündağ G, Şatana D, Özhan G, Çapan G (2016) Indole-based hydrazide–hydrazones and 4-thiazolidinones: synthesis and evaluation as antitubercular and anticancer agents. J Enzyme Inhib Med Chem 31(3):369–380
Almasirad A, Samiee-Sadr S, Shafiee A (2011) Synthesis and antimycobacterial activity of 2-(Phenylthio) benzoylarylhydrazone derivatives. Iran J Pharm Res 10(4):727–731
Tavakolfar S, Musavi E, Almasirad A, Amanzadeh A, Atyabi SM, Yaghamii P, Samiee-sadr S, Salimi M (2016) In vitro anticancer effects of two new potent hydrazide compounds in leukemic cells. Anti-Cancer Agents Med Chem 16:1646–1651
Devarajan E, Sahin AA, Chen JS, Krishnamurthy RR, Aggarwal N, Brun AM, Sapino A, Zhang F, Sharma D, Yang XH (2002) Down-regulation of caspase 3 in breast cancer: a possible mechanism for chemoresistance. Oncogene 21:8843–8851
Kroemer G, Reed JC (2000) Mitochondrial control of cell death. Nat Med 6:513–519
Breckenridge DG, Xue D (2004) Regulation of mitochondrial membrane permeabilization by BCL-2 family proteins and caspases. Curr Opin Cell Biol 16:647–652
Abdul Aziz MY, Abu N, Yeap SK, Ho WY, Omar AR, Ismail NH, Ahmad S, Pirozyan MR, Akhtar NM, Alitheen NB (2016) Combinatorial cytotoxic effects of damnacanthal and doxorubicin against human breast cancer MCF-7 cells in vitro. Molecules 21(9):E1228
Enari M, Sakahiro H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S (1998) A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 391:43–50
Roopashree R, Mohan CD, Swaroop TR, Jagadish S, Raghava B, Balaji KS, Jayarama S, Rangappa KS (2015) Novel synthetic bisbenzimidazole that targets angiogenesis in Ehrlich ascites carcinoma bearing mice. Bioorg Med Chem Lett 25:2589–2593
Bedia KK, Elcin O, Seda U, Fatma K, Nathaly S, Sevin R, Dimoglo A (2006) Synthesis and characterization of novel hydrazide–hydrazones and the study of their structure–antituberculosis activity. Eur J Med Chem 41(11):1253–1261
Matysiak J, Juszczak M, Karpinska MM, Langer E, Walczak K, Lemieszek MK, Skrzypek A, Niewiadomy A, Rzeski W (2015) Synthesis of 2-(2, 4-dihydroxyphenyl) thieno-1, 3-thiazin-4-ones, their lipophilicity and anticancer activity in vitro. Mol Divers 19(4):725–736
Wu S, Liu B, Zhang Q, Liu J, Zhou W, Wang C, Li M, Bao S, Zhu R (2013) Dihydromyricetin reduced Bcl-2 expression via p53 in human hepatoma HepG2 cells. PLoS One 8(11):e76886
Miyashita T, Krajewski S, Krajewska M, Wang HG, Lin HK, Liebermann DA, Hoffman B, Reed JC (1994) Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene 9:1799–1805
Das Mukherjee D, Kumar NM, Tantak MP, Das A, Ganguli A, Datta S, Kumar D, Chakrabarti G (2016) Development of novel bis (indolyl)-hydrazide–hydrazone derivatives as potent microtubule-targeting cytotoxic agents against A549 lung cancer cells. Biochemistry 55:3020–3035
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This research was financially supported by Pasteur Institute of Iran.
Conflict of interest
The authors have declared that there is no conflict of interests to disclose.
Ethical approval
All applicable national and institutional guidelines for the care and use of animals were followed. This article does not contain any studies with human participants performed by any of the authors.
Rights and permissions
About this article

Cite this article
Mousavi, E., Tavakolfar, S., Almasirad, A. et al. In vitro and in vivo assessments of two novel hydrazide compounds against breast cancer as well as mammary tumor cells. Cancer Chemother Pharmacol 79, 1195–1203 (2017). https://doi.org/10.1007/s00280-017-3318-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-017-3318-5