
Abstract
Purpose
Dose adjustment of 5-fluorouracil (FU) based on pharmacokinetic monitoring has been shown to reduce toxicities and increase efficacy compared with dosing based on body surface area in patients with metastatic colorectal cancer (mCRC). We evaluated the efficacy and safety of pharmacokinetic dose adjustment of FU in a modified FOLFOX7 (mFOLFOX7) plus bevacizumab regimen in Japanese patients with previously untreated mCRC.
Methods
This single-arm, multicenter phase II trial enrolled 48 patients with mCRC. Treatment consisted of 5 mg/kg intravenous bevacizumab followed by mFOLFOX7 (oxaliplatin 85 mg/m2 on day 1, infused leucovorin 200 mg/m2, followed by a 2400 mg/m2 infusion of FU for 46 h starting on day 1), repeated every 2 weeks. FU concentrations were measured by immunoassay between 18 and 36 h after the start of continuous FU infusion, and the FU dose was then adjusted if required in subsequent cycles. The primary endpoint was response rate.
Results
The median initial area under the concentration–time curve for FU was 23 mg h/L. Twenty-nine patients (60%) achieved the target concentration at the first cycle, and all 48 achieved it within the fourth cycle. The overall frequency of grade 3/4 adverse effects was 38%, with no significant difference between patients who did and not require dose adjustments. The overall response rate was 48% (95% confidence intervals = 34–62%). The median progression-free and overall survival rates were 11.3 and 24.1 months, respectively.
Conclusions
Pharmacokinetic dose adjustment of FU in mFOLFOX7 plus bevacizumab can optimize FU concentrations promptly and is safe in Japanese patients with mCRC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer is one of the most common malignancies in humans [1, 2]. Although it is highly treatable and curable when localized, 50% of patients experience metastatic progression associated with a poor prognosis, and complete cure, in general, remains difficult even by an aggressive attempt to eradicate all the metastatic sites [3–5]. In this setting, FOLFOX regimens consisting of 5-fluorouracil (FU), oxaliplatin (L-OHP), and leucovorin (LV) have been the mainstay of combination chemotherapy over the past decade [6]. Furthermore, bevacizumab, a humanized monoclonal antibody that inhibits vascular endothelial growth factor, improved response rates and patient survival when combined with standard chemotherapy treatments, including FOLFOX, in phase III randomized trials in patients with metastatic colorectal cancer (mCRC) [7, 8]. Treatment guidelines accordingly recommend that first-line treatment for mCRC should include doublet cytotoxic anticancer agents plus one of the molecular targeting agents [6].
Therapeutic drug monitoring involves measuring drug concentrations in biological samples to individualize the drug dosage and thereby improve its efficacy and reduce related toxicities [9]. Although FU has been the cornerstone of colorectal cancer treatment since 1960s, with numerous refinements and modifications of the regimens in order to increase its efficacy, the standard method of FU dosing remains based on body surface area (BSA) [10]. However, BSA-based dosing is associated with several limitations, including wide interpatient variability in FU levels associated with individual differences in activities of FU-metabolizing enzymes such as thymidylate synthase and dihydropyrimidine dehydrogenase (DPD) [11, 12]. Several clinical trials have been conducted by implementing FU dose adjustment in mCRC patients, and documented reduced toxicities and increased efficacy [12–15]. Gamelin et al. [16] conducted a pivotal phase III randomized trial in 208 patients and demonstrated that a regimen comprising individual FU dose adjustment based on pharmacokinetic monitoring, plus LV, significantly improved the objective response rate and reduced severe toxicities compared with BSA-based dosing. However, whether or not similar interpatient differences in FU pharmacokinetics exist for currently used regimens (doublet cytotoxic anticancer agents plus a targeted agent) in Japanese patients with mCRC remains unclear.
In the present study, a single-arm, multicenter phase II trial was conducted in order to evaluate the efficacy and safety of pharmacokinetics-guided dose adjustment of FU in Japanese mCRC patients treated by the modified FOLFOX7 (mFOLFOX7) plus bevacizumab.
Materials and methods
Patient eligibility
This single-arm, multicenter phase II trial was approved by an internal review board at each participating facility after review of the scientific and ethical validity of the protocol. This study was conducted in accordance with the Declaration of Helsinki (2008) and registered with the University Hospital Medical Information Network Clinical Trial Registry as UMIN000007194 (http://www.umin.ac.jp/ctr/index.htm). Signed, written informed consent was obtained from each patient.
Patients from 11 institutions were included if they met the following eligibility criteria: (1) histologically confirmed colorectal adenocarcinoma; (2) no prior chemotherapy (adjuvant chemotherapy including FU and/or oxaliplatin was allowed, but the last course of adjuvant chemotherapy must have concluded more than 6 months prior to colorectal cancer recurrence); (3) placement of central venous line; (4) one or more target lesions present according to Response Evaluation Criteria In Solid Tumors (RECIST) version 1.1 [17]; (5) Eastern Cooperative Oncology Group (ECOG) Performance Status 0 or 1; (6) age ≥ 20 years when informed consent was granted; (7) adequate function of vital organs; and (8) ≥12 weeks life expectancy. Key exclusion criteria included: severe drug allergy; uncontrolled pleural effusion or ascites; brain metastasis; presence of other active malignancies; present or past (within the past 1 year) clinically significant cerebrovascular disease or thromboembolism; coagulation disorder; nephropathy requiring medication or transfusion; uncontrolled diarrhea; and impaired peripheral nerve function.
Treatment
On the first day of the 14-day treatment cycle, patients received 5 mg/kg bevacizumab followed by mFOLFOX7 (L-OHP 85 mg/m2 on day 1, infused for 2 h; LV 200 mg/m2, infused for 2 h; followed by a 2400 mg/m2 infusion of 5- FU for 46 h starting on day 1) one hour after the initial administration of bevacizumab. Treatment was repeated every 2 weeks until disease progression or termination of the study. The dose of FU was adjusted according to plasma FU concentrations, by setting 3000 mg/m2 as the upper limit. The protocol treatment was discontinued in the event of disease progression, severe adverse effects (AEs), treatment interval longer than 14 days, conversion to surgery, or patient refusal.
In the event of AEs, the dose of each drug was reduced as specified in the study protocol according to detailed algorithms designed to manage drug-specific toxicities such as FU-related diarrhea, hand–foot syndrome, L-OHP-related neuropathy, bevacizumab-related thromboembolism, and other treatment-related toxicities. The criteria for dose reduction or cessation of drugs because of AEs was defined based on hematological toxicity (grade 4 neutropenia, grade 3 febrile neutropenia, or ≥ grade 3 decrease in platelets) and grade 3 non-hematological toxicity.
Pharmacokinetics-guided FU dose adjustment
Venous blood samples were collected from all the patients between 18 and 36 h after the start of continuous FU infusion once for each treatment course. Plasma samples were sent to FALCO Biosystems Ltd. (Kyoto, Japan) for the analysis. FU measurements were performed using the My5-FU® assay, a competitive homogeneous two-reagent nanoparticle agglutination immunoassay, under patent license from Saladax Biomedical, Inc. (Bethlehem, PA, USA) [10]. The quantitative target range for FU exposure, expressed as area under the blood concentration–time curve (AUC), was calculated from the measured concentration of FU. FU doses were adjusted during the first to third cycles of mFOLFOX7 treatment, according to the protocol shown in Table 1.
Endpoints
The primary objective of this trial was to determine the overall response rate. Response rate, confirmation of response, and disease progression were determined according to the RECIST version 1.1 [17]. Computed tomography scans were performed approximately every 8 weeks during treatment to assess tumor status. Response-rate assessments based on the target lesions were performed at each institution.
Secondary endpoints were FU concentration, relative dose intensity of L-OHP, time to treatment failure, disease-control rate, progression-free survival (PFS), overall survival (OS), and AEs. Toxicities during chemotherapy were evaluated according to the findings of physical examinations and laboratory tests (hematology, chemistry, electrolytes, and urinalysis) and graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.0) [18].
Statistical analysis
The expected response rate with confirmation, the primary endpoint of this trial, was set as 55% with reference to previous clinical trials [19–21]. We calculated that 43 patients would be necessary to keep the 95% confidence interval (CI) within ±15%, and 48 patients were required estimating a loss as high as 10% from the final subject population. Overall response rate was a proportion of patients with complete response (CR) or partial response (PR), and 95% CI was calculated based on Wald type estimator. Survival probability was estimated using the Kaplan–Meier method, and CIs were calculated using the Cox proportional hazards model. PFS was defined as the interval from the date of enrollment to the date of first documented detection of disease progression or death from any cause. OS was defined as the date from enrollment until the date of death from any cause. Quantitative differences in categorical and continuous variables between groups were compared using χ2 and Mann–Whitney tests, respectively [22]. A statistically significant difference was defined as p < 0.05. Analyses were performed using SAS version 9.4 (SAS Institute, Inc., Cary, NC).
Results
Baseline patient characteristics
Forty-eight patients (male 77%, median age 67 years, colon cancer 67%) who met the eligibility criteria were recruited from the 11 institutions between April 2012 and June 2013. All patients underwent at least one plasma FU measurement and four courses of the planned treatment. Baseline patient characteristics are shown (Table 2). The most frequent metastatic site was liver (71%). The baseline BSA was 1.59 m2 (range 1.32–2.11 m2).
Pharmacokinetics
Plasma FU concentrations were evaluated during the first course of planned treatment in all 48 patients. The median initial AUC value was 23 mg h/L (range 10–34 mg h/L). Twenty-nine patients (60%) achieved the target concentration (‘Target’ group) (Table 1). Three patients (6%) with higher AUC values (‘Above’ group) and 16 patients (33%) with lower AUC values (‘Below’ group) underwent dose adjustment of FU at the second course. There was no significant difference in initial AUC between males and females. Six patients (13%) required dose adjustments twice according to the study protocol, and all 48 patients eventually achieved the target concentration of FU. Dose reduction in FU was required in five patients (10%) because of AEs as follows: nausea (n = 2), fatigue (n = 1), stomatitis (n = 1), and diarrhea (n = 1).
Treatment characteristics
Patients received a median of 11 cycles of mFOLFOX7 plus bevacizumab (range 4–33 cycles). The median relative dose intensities in the fourth cycle of the protocol treatment were all 100% for FU (range 4–144%), L-OHP (range 4–102%), and bevacizumab (range 0–100%). The median time to treatment failure was 6.2 months (95% CI 5.1–7.1 months). The reasons for discontinuation of the treatment were disease progression (n = 22), AEs (n = 9), conversion to surgery (n = 10), patient withdrawal (n = 4), and physician’s discretion (n = 3).
Toxicities of chemotherapy
Forty-eight (100%) and 18 (38%) patients experienced at least one AE of any grade or ≥grade 3, respectively (Table 3). Frequent AEs (any grade) included anemia, (n = 44, 92%), neuropathy (n = 42, 88%), fatigue (n = 34, 71%), neutropenia (n = 33, 67%), anorexia (n = 28, 58%), thrombocytopenia (n = 27, 56%), and nausea (n = 24, 50%). Frequent AEs ≥ grade 3 were neutropenia (n = 13, 27%), neuropathy (n = 4, 8%), fatigue (n = 4, 8%), and anorexia (n = 3, 6%). There was no significant differences in the frequency of AEs ≥ grade 3 among the Below, Target, and Above groups.
Treatment responses
The best radiographic responses based on shrinkage of the target lesions in each patient are presented (Fig. 1a). There was no significant differences in the best radiographic responses among the three groups (p = 0.537). The overall rates of CR, PR, stable disease, and progressive disease were 0, 48, 52, and 0%, respectively, and the response and disease-control rates were 48% (95% CI 34–62%) and 100%, respectively (Table 4). Response rates in the Below, Target, and Above groups were 38, 55, and 33%, respectively.

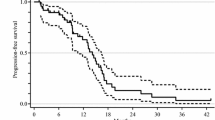
a Waterfall plot of maximum percentage of target-lesion shrinkage. b Progression-free survival time in 48 patients. c Overall survival time in 48 patients
The median PFS was 11.3 months (Fig. 1b), and the median OS was 24.1 months (Fig. 1c). Subgroup analysis according to the initial AUC of FU revealed similar PFS in all groups (Fig. 2a), but the Below group tended to have shorter OS than the other groups (Fig. 2b).

Kaplan–Meier curves of a progression-free and b overall survival according to initial plasma FU concentrations
Discussion
Therapeutic drug monitoring is rarely used in cancer therapy [23]. However, given the relatively narrow therapeutic index and substantial interpatient pharmacokinetic variabilities associated with cytotoxic and targeted agents, the concept of therapeutic drug monitoring represents a clinically relevant strategy for incorporation into cancer therapy [23, 24]. Previous pharmacokinetic dosing studies used high-performance liquid chromatography to measure plasma FU concentrations, but this technique is not widely available and is difficult to use in the clinical setting because of time and cost limitations [9, 13, 16]. The advent of the My5-FU® assay allows physicians to measure plasma FU concentrations rapidly by using widely available biochemical autoanalyzers [10, 23, 25].
Individual FU dose adjustment based on pharmacokinetic monitoring, together with LV, has been shown to allow dose intensification and has demonstrated favorable results in terms of improved efficacy and toxicity in patients with mCRC [3, 16, 25]. However, standard practice for first-line treatment of mCRC has shifted toward combination therapy including doublet cytotoxic anticancer agents plus a targeted agent, and we therefore conducted the current phase II clinical trial to extend the dose-adjustment approach in patients treated with mFOLFOX7 plus bevacizumab.
In this trial, the median initial AUC value was 23 mg h/L, which was within the target range and was achieved in 60% of the patients during the first cycle, that have been administered according to the BSA-based dose. After the two opportunities for dose modification, all the 48 patients met the target FU concentration. Saam et al. [10] examined FU concentrations in 357 patients receiving FU 2400 mg/m2 (FOLFIRI/FOLFOX6 with or without bevacizumab) for mCRC and found a wide range of AUCs (1–69 mg h/L) at the initial measurement. The mean AUC was 20.4 mg h/L, and only 21.3% of patients achieved the AUC target range of 20–24 mg h/L. Interestingly, the distributions of AUCs were similar between patients receiving FOLFOX and FOLFIRI, patients treated with and without bevacizumab, and between patients treated in the metastatic or adjuvant setting. Among 62 patients who required dose adjustment and were followed over four cycles, only 23 patients (37%) achieved the AUC target range of 20–24 mg h/L [10]. In contrast to the Saam’s results, we found that a relatively high proportion of patients (60%) had achieved the target range at the initial FU dose, and that plasma FU concentrations were easily modifiable by dose adjustment in the Japanese patients.
With respect to safety, Capitain et al. [3] conducted a phase II clinical trial and evaluated the safety of pharmacokinetically guided FU dose adjustment in a FOLFOX regimen in 118 patients with mCRC. The frequencies of ≥grade 3 toxicities were 18% for neutropenia, 2% for diarrhea, and 1% for stomatitis, which were comparable with our results of 27% for neutropenia, 2% for diarrhea, and 4% for stomatitis [3]. Notably, the frequency of AEs was not higher in the Below group in the current study, even though they received higher doses of FU than the initial BSA-based dose. This suggests that dose elevation could be achieved safely in the Japanese patients when guided by individual pharmacokinetic data. In Caucasian and African American populations, the frequency of low DPD activity, which is recognized as one of the reasons for increased FU-related AEs, was estimated to be approximately 4% [26–28]. On the other hand, Ogura et al. [29] conducted a population study of DPD activity in peripheral blood mononuclear cells in 150 Japanese healthy volunteers and found only one (0.7%) had a low DPD activity due to heterozygosity for a mutant allele of the DPYD gene. Thus, when interpreting pharmacokinetic data of FU, the race differences in FU metabolism should be taken into consideration.
The overall response rate in the current study was 48%, which failed to reach the expected value of 55%. However, the response rate in the Target group was 55%, compared with 38% in the Below group. The Below group also tended to have a shorter OS compared with the other groups, even though their plasma FU concentrations were adjusted within three cycles. These results thus emphasize the clinical problem of how best to enhance the therapeutic effect in the Below group. Overall, this study demonstrated the following points in the Japanese patients with mCRC: (1) the distribution of initial FU AUCs was different between the Japanese and previously reported Western populations; (2) plasma FU concentrations were quickly optimized by the dose adjustment; (3) dose elevation based on pharmacokinetic data did not increase severe AEs; and (4) a low FU AUC at the initial dose might be associated with a poorer response rate and shorter OS. The effect of pharmacological adjustment to the intended target AUC has had a limited impact in relapsed free or overall survival, but the methodology is robust and could allow targeting distinct AUCs with the intent of having on impact patient outcomes. A large-scale clinical trial is warranted to expand and validate these findings and to clarify the benefits of pharmacokinetic dose adjustment of FU in this setting.
The current study had some limitations. The relatively small sample size precluded subgroup analyses according to initial AUC levels. Furthermore, a central review system for assessing response rates might help to reduce potential bias. Our discussion about FU pharmacokinetics might be limited by a lack of information on the activities of thymidylate synthase and DPD [30, 31]. Finally, the survival benefit of FU dose adjustment could not be determined because of the single-arm nature of the study.
In conclusion, pharmacokinetically guided dosage adjustment of FU for mFOLFOX7 plus bevacizumab can optimize FU concentrations promptly with no increase in toxicity and might, to some extent, be able to improve prognosis of the Japanese patients with mCRC.
References
Siegel RL, Miller KD, Jemal A (2015) Cancer statistics. CA Cancer J Clin 65(1):5–29
Munemoto Y, Kanda M, Ishibashi K, Hata T, Kobayashi M, Hasegawa J, Fukunaga M, Takagane A, Otsuji T, Miyake Y, Nagase M, Sakamoto J, Matsuoka M, Oba K, Mishima H (2015) Capecitabine and oxaliplatin combined with bevacizumab are feasible for treating selected Japanese patients at least 75 years of age with metastatic colorectal cancer. BMC Cancer 15:786
Capitain O, Asevoaia A, Boisdron-Celle M, Poirier AL, Morel A, Gamelin E (2012) Individual fluorouracil dose adjustment in FOLFOX based on pharmacokinetic follow-up compared with conventional body-area-surface dosing: a phase II, proof-of-concept study. Clin Colorectal Cancer 11(4):263–267
de Jong MC, Pulitano C, Ribero D, Strub J, Mentha G, Schulick RD, Choti MA, Aldrighetti L, Capussotti L, Pawlik TM (2009) Rates and patterns of recurrence following curative intent surgery for colorectal liver metastasis: an international multi-institutional analysis of 1669 patients. Ann Surg 250(3):440–448
Abdalla EK (2011) Resection of colorectal liver metastases. J Gastrointest Surg 15(3):416–419
Brenner H, Kloor M, Pox CP (2014) Colorectal cancer. Lancet 383(9927):1490–1502
Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F (2004) Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 350(23):2335–2342
Kabbinavar FF, Hambleton J, Mass RD, Hurwitz HI, Bergsland E, Sarkar S (2005) Combined analysis of efficacy: the addition of bevacizumab to fluorouracil/leucovorin improves survival for patients with metastatic colorectal cancer. J Clin Oncol 23(16):3706–3712
Ychou M, Duffour J, Kramar A, Debrigode C, Gourgou S, Bressolle F, Pinguet F (2003) Individual 5-FU dose adaptation in metastatic colorectal cancer: results of a phase II study using a bimonthly pharmacokinetically intensified LV5FU2 regimen. Cancer Chemother Pharmacol 52(4):282–290
Saam J, Critchfield GC, Hamilton SA, Roa BB, Wenstrup RJ, Kaldate RR (2011) Body surface area-based dosing of 5-fluoruracil results in extensive interindividual variability in 5-fluorouracil exposure in colorectal cancer patients on FOLFOX regimens. Clin Colorectal Cancer 10(3):203–206
Milano G, Etienne MC, Cassuto-Viguier E, Thyss A, Santini J, Frenay M, Renee N, Schneider M, Demard F (1992) Influence of sex and age on fluorouracil clearance. J Clin Oncol 10(7):1171–1175
Gamelin E, Boisdron-Celle M, Guerin-Meyer V, Delva R, Lortholary A, Genevieve F, Larra F, Ifrah N, Robert J (1999) Correlation between uracil and dihydrouracil plasma ratio, fluorouracil (5-FU) pharmacokinetic parameters, and tolerance in patients with advanced colorectal cancer: a potential interest for predicting 5-FU toxicity and determining optimal 5-FU dosage. J Clin Oncol 17(4):1105
Di Paolo A, Lencioni M, Amatori F, Di Donato S, Bocci G, Orlandini C, Lastella M, Federici F, Iannopollo M, Falcone A, Ricci S, Del Tacca M, Danesi R (2008) 5-fluorouracil pharmacokinetics predicts disease-free survival in patients administered adjuvant chemotherapy for colorectal cancer. Clin Cancer Res 14(9):2749–2755
Kaldate RR, Haregewoin A, Grier CE, Hamilton SA, McLeod HL (2012) Modeling the 5-fluorouracil area under the curve versus dose relationship to develop a pharmacokinetic dosing algorithm for colorectal cancer patients receiving FOLFOX6. Oncologist 17(3):296–302
Freeman K, Connock M, Cummins E, Gurung T, Taylor-Phillips S, Court R, Saunders M, Clarke A, Sutcliffe P (2015) Fluorouracil plasma monitoring: systematic review and economic evaluation of the My5-FU assay for guiding dose adjustment in patients receiving fluorouracil chemotherapy by continuous infusion. Health Technol Assess 19(91):1–321
Gamelin E, Delva R, Jacob J, Merrouche Y, Raoul JL, Pezet D, Dorval E, Piot G, Morel A, Boisdron-Celle M (2008) Individual fluorouracil dose adjustment based on pharmacokinetic follow-up compared with conventional dosage: results of a multicenter randomized trial of patients with metastatic colorectal cancer. J Clin Oncol 26(13):2099–2105
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45(2):228–247
Liu YJ, Zhu GP, Guan XY (2012) Comparison of the NCI-CTCAE version 4.0 and version 3.0 in assessing chemoradiation-induced oral mucositis for locally advanced nasopharyngeal carcinoma. Oral Oncol 48(6):554–559
Hochster HS, Hart LL, Ramanathan RK, Childs BH, Hainsworth JD, Cohn AL, Wong L, Fehrenbacher L, Abubakr Y, Saif MW, Schwartzberg L, Hedrick E (2008) Safety and efficacy of oxaliplatin and fluoropyrimidine regimens with or without bevacizumab as first-line treatment of metastatic colorectal cancer: results of the TREE Study. J Clin Oncol 26(21):3523–3529
Saltz LB, Clarke S, Diaz-Rubio E, Scheithauer W, Figer A, Wong R, Koski S, Lichinitser M, Yang TS, Rivera F, Couture F, Sirzen F, Cassidy J (2008) Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 26(12):2013–2019
Sogabe S, Komatsu Y, Yuki S, Kusumi T, Hatanaka K, Nakamura M, Kato T, Miyagishima T, Hosokawa A, Iwanaga I, Sakata Y, Asaka M (2011) Retrospective cohort study on the safety and efficacy of bevacizumab with chemotherapy for metastatic colorectal cancer patients: the HGCSG0801 study. Jpn J Clin Oncol 41(4):490–497
Kanda M, Shimizu D, Tanaka H, Shibata M, Iwata N, Hayashi M, Kobayashi D, Tanaka C, Yamada S, Fujii T, Nakayama G, Sugimoto H, Koike M, Fujiwara M, Kodera Y (2016) Metastatic pathway-specific transcriptome analysis identifies MFSD4 as a putative tumor suppressor and biomarker for hepatic metastasis in patients with gastric cancer. Oncotarget 7(12):13667–13679
Lee JJ, Beumer JH, Chu E (2016) Therapeutic drug monitoring of 5-fluorouracil. Cancer Chemother Pharmacol
Beumer JH, Chu E, Salamone SJ (2012) Body-surface area-based chemotherapy dosing: appropriate in the 21st century? J Clin Oncol 30(31):3896–3897
Yang R, Zhang Y, Zhou H, Zhang P, Yang P, Tong Q, Lyu Y, Han Y (2016) Individual 5-fluorouracil dose adjustment via pharmacokinetic monitoring versus conventional body-area-surface method: a meta-analysis. Ther Drug Monit 38(1):79–86
Lu Z, Zhang R, Diasio RB (1993) Dihydropyrimidine dehydrogenase activity in human peripheral blood mononuclear cells and liver: population characteristics, newly identified deficient patients, and clinical implication in 5-fluorouracil chemotherapy. Cancer Res 53(22):5433–5438
Fleming RA, Milano GA, Gaspard MH, Bargnoux PJ, Thyss A, Plagne R, Renee N, Schneider M, Demard F (1993) Dihydropyrimidine dehydrogenase activity in cancer patients. Eur J Cancer 29(5):740–744
Etienne MC, Lagrange JL, Dassonville O, Fleming R, Thyss A, Renee N, Schneider M, Demard F, Milano G (1994) Population study of dihydropyrimidine dehydrogenase in cancer patients. J Clin Oncol 12(11):2248–2253
Ogura K, Ohnuma T, Minamide Y, Mizuno A, Nishiyama T, Nagashima S, Kanamaru M, Hiratsuka A, Watabe T, Uematsu T (2005) Dihydropyrimidine dehydrogenase activity in 150 healthy Japanese volunteers and identification of novel mutations. Clin Cancer Res 11(14):5104–5111
Salonga D, Danenberg KD, Johnson M, Metzger R, Groshen S, Tsao-Wei DD, Lenz HJ, Leichman CG, Leichman L, Diasio RB, Danenberg PV (2000) Colorectal tumors responding to 5-fluorouracil have low gene expression levels of dihydropyrimidine dehydrogenase, thymidylate synthase, and thymidine phosphorylase. Clin Cancer Res 6(4):1322–1327
Bai W, Wu Y, Zhang P, Xi Y (2015) Correlations between expression levels of thymidylate synthase, thymidine phosphorylase and dihydropyrimidine dehydrogenase, and efficacy of 5-fluorouracil-based chemotherapy for advanced colorectal cancer. Int J Clin Exp Pathol 8(10):12333–12345
Acknowledgements
This study was supported, in part, by the nonprofit organization Epidemiological and Clinical Research Information Network (ECRIN).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Junichi Sakamoto serves as an advisor to Takeda Pharmaceutical Company Ltd., and received lecture fees from Tsumura Co., Ltd. Hideyuki Mishima received lecture fees from Chugai Pharmaceutical Co., Ltd., and research funding from Chugai Pharmaceutical Co., Ltd. and Yakult Co., Ltd.
Additional information
Tadamichi Denda and Mitsuro Kanda have contributed equally to this work.
Rights and permissions
About this article

Cite this article
Denda, T., Kanda, M., Morita, Y. et al. Pharmacokinetic dose adjustment of 5-FU in modified FOLFOX7 plus bevacizumab for metastatic colorectal cancer in Japanese patients: a-JUST phase II clinical trial. Cancer Chemother Pharmacol 78, 1253–1261 (2016). https://doi.org/10.1007/s00280-016-3184-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-016-3184-6