
Abstract
Lymphomas, complex and heterogeneous malignant tumors, originate from the lymphopoietic system. These tumors are notorious for their high recurrence rates and resistance to treatment, which leads to poor prognoses. As ongoing research has shown, epigenetic modifications like DNA methylation, histone modifications, non-coding RNA regulation, and RNA modifications play crucial roles in lymphoma pathogenesis. Epigenetic modification–targeting drugs have exhibited therapeutic efficacy and tolerability in both monotherapy and combination lymphoma therapy. This review discusses pathogenic mechanisms and potential epigenetic therapeutic targets in common lymphomas, offering new avenues for lymphoma diagnosis and treatment. We also discuss the shortcomings of current lymphoma treatments, while suggesting potential areas for future research, in order to improve the prediction and prognosis of lymphoma.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Lymphoma, a malignant tumor group, originates from lymph nodes and extralymphatic tissues [1]. There are two categories: Hodgkin lymphoma (HL) and non-Hodgkin lymphoma (NHL). HL, which constitutes approximately 5 to 10% of lymphomas, branches into classical and nodular lymphocyte-predominant types. In contrast, NHL is further divided into B cell, T cell, and natural killer (NK) cell types [2]. The most prevalent type of aggressive NHL is diffuse large B cell lymphoma (DLBCL) [3], accounting for approximately 40% of adult NHL in China [4]. The most common type of low-grade lymphoma, follicular lymphoma (FL), represents 2.5 to 6.6% of NHL in China [5]. According to a survey [6], in 2020, there were 544,352 new NHL cases globally, ranking it 13th in terms of new malignant tumor cases worldwide, with 55.9% of cases in males and 44.1% in females. Its overall mortality rate was 28.1%. There were 83,087 HL cases (males: 59.1%; females: 40.9%), with an overall mortality rate of 47.7%. Each lymphoma type differs in pathogenesis, clinical manifestations, treatment modalities, and prognosis assessment [7]. Therefore, lymphoma treatments need to be tailored according to the patient’s age, physical condition, clinical stage, pathological type, and molecular genetic characteristics [8].
Recently, targeted therapies for lymphoma have seen significant expansion [9]. B cell lymphoma (BCL)–targeted therapies include rituximab targeting CD20, chimeric antigen receptor–modified T cell (CAR-T) therapy targeting CD19, targeted small-molecule drugs such as ibrutinib [10], and therapies targeting the tumor microenvironment like bortezomib and lenalidomide [11]. The first-line treatment for common lymphomas remains cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) or a CHOP-like regimen (CHOEP) [12]. However, due to drug resistance, some patients do not respond well, leading to high failure and relapse rates [13]. Hematopoietic stem cell transplantation (SCT) has become a vital tool for treating relapsed/refractory (R/R) lymphoma. Notwithstanding, the source and quality of hematopoietic stem cells, along with their post-transplantation developmental and functional status, are still limiting outcome factors [14]. Therefore, finding new lymphoma treatment targets and developing new, more effective drug combinations are critical for reducing lymphoma relapse and mortality rates.
Epigenetic modifications play a significant role in hematological tumor cell regulation [15]. Epigenetics encompasses heritable modifications that alter gene activation, independent of nucleotide sequence changes. These include DNA methylation, histone modifications, non-coding RNA regulation, and RNA modifications. They mediate gene transcription and translation changes, regulate gene expression, and play a role in trait inheritance and disease pathology [28].
The term “epigenetic landscape,” introduced by Waddington in 1939, describes the mechanisms of translating genetic traits into phenotypes [29]. Recently, epigenetics has become a key research area, aiming to understand how social conditions, the environment, psychosocial factors, and nutrition can impact gene expression [30]. Numerous studies have demonstrated that epigenetics is crucial to lymphoma development and progression. DNA methylation and histone modifications have been implicated in tumor pathogenesis and are considered important targets for treating different types of lymphoma and many other tumors (Fig. 1) [31]. Furthermore, epigenetic targets have shown promising therapeutic results in clinical treatment [32]. For instance, DNA methylation–blocking antibodies have been developed, and combinations of drugs like decitabine, a DNA methylation inhibitor, and chidamide, a histone acetylase inhibitor, can significantly inhibit lymphoma progression [9]. Current therapeutic approaches for the cancer epigenome include FDA-approved therapies and investigational agents in clinical trials. These target regulators of histone acetylation, histone methylation, DNA methylation, and histone phosphorylation (Table 1). This review focuses on the research progress on epigenetic modifications, their roles in the pathogenesis, and targeted therapy of common clinical lymphomas.

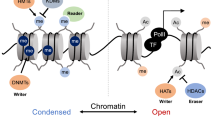
Epigenetic mechanisms and treatment schemes in lymphoma. The pathogenesis of lymphoma is multi-disciplinary. Here, four epigenetic abnormalities associated with the lymphoma development are displayed: histone modifications (mainly including histone methylation and histone acetylation), DNA methylation, non-coding RNA regulation, and RNA modifications. Among them, DNA methylation mainly involves the selective addition of methyl to cytosines at specific sites on the DNA sequence, catalyzed by DNA methyltransferases (DNMTs), to form 5-methylcytosine. It regulates gene expression by recruiting proteins involved in gene repression or inhibiting the binding of transcription factors to DNA, causing further development and progression of lymphoma. In the histone modification, aberrant histone modifications can be reversed by inhibiting the activity of histone methyltransferases (HMTs) and histone deacetylases (HDACs), which then further inhibit tumor cell proliferation and induce apoptosis. The red arrows represent epigenetic-based approaches for lymphoma treatment. For example, in the DNA methylation, DNMT inhibitors azacitidine and decitabine are representative drugs. In terms of histone modifications, drugs like romidepsin, chidamide, and tazemetostat have been approved by the Food and Drug Administration (FDA) for some specific types of lymphoma
DNA methylation and lymphoma
DNA methylation predominantly involves the selective addition of a methyl group to cytosine (C) at certain points within the DNA sequence. This process is facilitated by DNMTs to form 5-methylcytosine (5mC). This resultant structure, 5mC, is involved in the regulation of DNA binding, either through recruiting proteins that suppress gene expression or by preventing the expression of transcription factors [33]. The primary enzymes known for transferring methylation are DNMT3A, DNMT3B, and DNMT3L, which are responsible for the initial methylation process. DNMT1, another vital enzyme, maintains the methylation patterns [34]. DNA methylation, one of the most intensely studied epigenetic alterations in mammals, aids in precise gene control and long-term gene silencing [35]. Throughout development, the patterns of DNA methylation within the genome undergo dynamic changes through a balance of methylation and demethylation processes [36]. Therefore, differentiated cells generate stable and unique DNA methylation patterns that control gene transcription specific to certain tissues [37].
It is widely recognized that hypermethylation within promoter regions results in the inactivation of particular oncogenes, with DNMTs having a critical role in the progression of lymphoma [38]. In some lymphomas associated with the Epstein-Barr virus (EBV), there are significantly high levels of genomic DNA methylation, which are connected to a decrease in tumor suppressor gene expression. Consequently, intentional reduction of DNA methylation levels could potentially stimulate viral gene expression, exposing the cells to the immune system. Additionally, this process could enhance tumor suppressor gene expression, possibly inhibiting cancer cell growth or inducing apoptosis. As such, understanding the regulation of DNA methylation levels could pave the way for novel therapies for EBV-associated lymphoma [39]. Dalton et al. [40] have illustrated the potential of decitabine in driving viral genome hypomethylation, resulting in the re-expression of immunogenic viral genes in lymphoma cells associated with EBV. However, it is important to note that not all EBV genomes reactivate in all lymphoma cells. Epigenetic drugs are occasionally combined with T cell therapy to boost the expression of tumor antigens that bind to identical T cell receptors. For instance, decitabine and 5-azacytidine are potent stimulants of immunogenic EBV antigens in EBV-associated tumors at latent stage I. A combination of decitabine and EBV-CTL results in T cell homing to the tumor and the suppression of tumor growth in a Burkitt lymphoma (BL) xenograft model.
Guo et al. [41] conducted a genetic screen in both EBV and BL cells and discovered the UHRF1 protein, containing ubiquitin-like, PHD, and RING finger domains, and its chaperone DNMT1 to be crucial in controlling the expression of EBNA and LMP. They further found that the levels of UHRF1, DNMT1, and DNMT3B are elevated in germinal center (GC) B cells, which are the origin cells of BL. This provides a molecular connection between B cell status and EBV latency. In preclinical models of T cell lymphoma, the inhibitors of DNMTs and HDACs demonstrated synergy in vitro. The theoretical combination of azacytidine (AZA) and romidepsin (ROMI) could stimulate the expression of multiple cancer testis antigens (CTAs), thereby increasing tumor immunogenicity. In a multicenter phase II clinical trial, Falchi et al. reported that the combination therapy with AZA and ROMI was both safe and effective for treating peripheral T cell lymphoma (PTCL), particularly for patients who are treatment-naive (TN) and T follicular helper (TFH) cells. Consequently, AZA and ROMI could be adopted as a first-line combination therapy for PTCL and R/R PTCL, as well as a bridging therapy [16].
Histone modifications
The role of histone modifications in lymphoma pathogenesis
In eukaryotic cells, genetic information stored in DNA exists in a well-structured chromatin organization. The fundamental unit of chromatin is the nucleosome, comprising a histone octamer and 146–147 nucleotide base pairs. Post-translational modifications including acetylation, methylation, phosphorylation, sulfonylation, and ubiquitination can occur at both the N- and C-termini of histones [42]. These modifications, taking place at the R and K residues of histones, can be regulated by various enzymes. Typically, enzymes that add chemical groups to histone tails are termed “writers,” proteins that recognize these specific epigenetic marks are called “readers,” and other proteins that contribute to removing marks are known as “erasers” [43]. Notably, these modifications change the structure and charge of the histone tails bound to DNA, thereby modulating the state of chromatin and the signaling pathways of DNA-binding proteins, which in turn impacts gene expression [44]. Furthermore, histone modifications can create a binding platform for proteins involved in chromatin remodeling, histone chaperones, DNA/histone modifying enzymes, and general transcription factors [43]. These histone modifications are crucial for various cellular processes, and their dysregulation is linked to cancer development and developmental defects [45]. They are currently a significant area of epigenetic research [46, 47]. Histone acetylation and methylation are the most commonly studied types of histone modifications.
Histone methylation primarily occurs at residues K or R of H3 and H4. This process is instrumental in forming and maintaining heterochromatin’s structure and genome, and in regulating blotting, DNA repair, X-chromatin inactivation, and transcription. Three main factors regulate histone methylation: HMT, histone methylation recognition proteins, and histone demethylases. HMT is classified into histone lysine methyltransferase (HKMT) and protein arginine methyltransferase (PRMT). Meanwhile, histone demethylases are generally divided into two types: lysine-specific demethylase (LSD) and jumonji domain–containing histone demethylase. H3K at positions 4, 9, 27, 36, and 79 and H4K at position 20 can be methylated [48]. H3K4 and H3K9 are common modification sites. The N-terminal of L residues can undergo mono-, di-, or tri-methylation modifications, while R residues can undergo only mono- and asymmetric di-methylation modifications. Interactions between histone lysine methylation and other histone modifications may regulate gene expression. For instance, H3K4 and H3K79 methylation requires prior H2B ubiquitination in yeast. Acetylation and methylation on the same lysine residue can act as antagonists, leading to crosstalk between different histone marks [42]. Histone acetylation is a reversible process primarily maintained by histone acetyltransferases (HATs) and HDACs. Histone acetylation can occur at sites 9, 14, 18, and 23 of H3 and sites 5, 8, 12, and 16 of H4, facilitating transcription on the one hand, and weakening the interaction between histones and DNA by reconfiguring higher chromatin structure on the other [49].
Relationship between histone methylation and lymphoma development
Histone methylation, one of the most significant post-translational modifications, typically refers to the addition of methyl groups to the lysine (K) residues of histones H3 and H4 [50]. Histone lysine residues can be mono-, di-, or tri-methylated, acting as active or repressive marks for gene expression [51]. Unlike other histone modifications, which specify active or repressed chromatin states, histone lysine methylations confer active or repressive transcription depending on their positions and methylation states. Generally, H3K4, H3K36, and H3K79 methylation are often considered markers of active transcription, while H3K9, H3K27, and H4K20 methylation are associated with silent chromatin states [43]. Under normal circumstances, activation and inhibition of methylation modifications maintain a dynamic equilibrium [49].
KMT2D (also known as MLL2) triggers methylation on lysine 4 of histone H3 (H3K4me). This methylation at promoters and enhancers acts as a marker for transcriptional activation. Zhang et al. [52] discovered that mutations in the gene encoding KMT2D methyltransferase could decrease KMT2D enzyme activity. Such a reduction led to a decrease in overall H3K4 methylation in GC B cells and DLBCL cells, thereby increasing the incidence of GC-derived lymphomas. Decitabine also synergistically enhanced the interaction between KMT2D and PU.1. This interaction affected H3K4me-related signaling pathways, rendering lymphoma cells more responsive to chidamide. Dual therapy with chidamide and decitabine significantly slowed tumor growth and promoted apoptosis in a KMT2D-mutated heterozygous T cell lymphoma model. This therapeutic effect occurred via modulation of the KMT2D/H3K4me axis [53].
Moreover, SETD2 is the sole methylation transferase responsible for the trimethylation of lysine 36 on histone 3 (H3K36me3). The involvement of H3K36me3 in RNA splicing occurs not only through the recruitment of MRG15 and ZMYND11 but also through multiple pathways, such as mismatch repair (MMR), the recruitment of LEDGF, and the subsequent activation of DNA damage perception through homologous recombination (HR) [54,55,56]. In various solid tumors and leukemias, the exclusive subunit functional deficiency of SETD2 results in defective DNA damage repair and impaired transcription. However, recent findings indicate that heterozygous SETD2 deficiency in DLBCL can cause GC proliferation, increased competitive fitness, reduced DNA damage checkpoint activity, and decreased apoptosis, all of which contribute to expedited lymphoma formation [57].
PRMT serves as the catalytic subunit of the polycomb repressor complex 2 (PRC2), which represses gene expression via the trimethylation of histone H3 lysine 27 (H3k27me3). Somatic mutations in residues Y641 and A677 of the EZH2 SET domain correlate with poor prognosis in DLBCL and FL [58]. Epigenomic studies have revealed that inappropriate H3k27me3 deposition is a key determinant of aberrant transcriptomes in malignant lymphomas and various solid tumors [59]. Given that H3k27me3 is an enzymatic product, its reversibility provides an excellent foundation for the development of epigenetic drugs. Numerous preclinical studies have confirmed the efficacy of EZH2 in BCL. Tazemetostat, one of the most promising compounds, was granted accelerated approval by the FDA in June 2020. It was sanctioned for the treatment of adult patients with R/R FL, particularly those who had previously received at least two standard-of-care systemic therapies or had no suitable alternative treatment options [60, 61]. Tazemetostat, a selective EZH2 inhibitor, curbs tumor growth by reducing H3k27 methylation levels [62]. A multicenter, open-label, single-arm, phase II study involving 99 adult patients with R/R FL demonstrated that tazemetostat monotherapy exhibited good single-agent activity, a durable response, and good tolerability in patients with R/R FL [18]. With the results of this study, tazemetostat is expected to be a new promising treatment option for FL patients. Furthermore, a phase I study conducted in Japanese patients with R/R B cell NHL showed that tazemetostat, at a dosage of 800 mg twice daily, also exhibited an acceptable safety profile and promising antitumor activity [63]. Despite the underwhelming efficacy of EZH2 inhibition in the current clinical exploration of DLBCL, EZH2 inhibitors have proven to be well-tolerated. Therefore, numerous studies are investigating combination therapy options to augment the efficacy of EZH2 in DLBCL. Scholze et al. [64] discovered the synergistic effects of tazemetostat and venetoclax in DLBCL cells and 3-dimensional lymphoma organoids carrying EZH2 mutations and IGH/BCL2 translocations. Tazemetostat treatment upregulated pro-apoptotic proteins, and it is postulated that venetoclax enhances the BH3-mediated apoptosis thereby triggered. A short course of combination therapy with tazemetostat and venetoclax achieved complete remission and improved overall survival in DLBCL patient–derived xenografts (PDXs) compared to either monotherapy. Other studies have also confirmed the synergistic effects of tazemetostat in combination with R-CHOP regimens and drugs such as lenalidomide and atezolizumab for their antitumor effects in DLBCL [65,66,67].
Several ongoing clinical trials are investigating the use of tazemetostat and other EZH2 inhibitors in various malignant lymphomas (more details are shown in Table 2). Yamagishi et al. discovered that aggressive lymphomas often co-express EZH1 and EZH2, which can interfere with and compensate for each other’s function, suggesting a principle of dual targeting of EZH1/2 [68]. Moreover, Honma et al. showed that EZH1/2 dual inhibitors outperform EZH2 selective inhibitors in terms of antitumor efficacy in both in vitro and in vivo studies, without causing severe hematological side effects [69]. In September 2022, valemetostat became the first dual EZH1/2 inhibitor approved in Japan for treating aggressive adult T cell leukemia (ATL) [70]. There are ongoing phase I and II clinical trials of valemetostat for treating malignant lymphomas, such as BCL and R/R PTCL (see Table 2). These trials have shown promising safety and antitumor activity against lymphomas.
Furthermore, histone modifications and DNA methylation are functionally linked. Specifically, the removal of histone H3 lysine 4 methylation (H3K4me0) correlates with an increase in H3K9 methylation[45]. Selker et al. confirmed, using a yeast model, that DNA methylation requires histone H3K9 methylation [71]. In mammalian cells, a structural domain typically connects histone methylation to DNA methylation. For instance, the H3K4 methyltransferase MLL1 contains a CpG-interacting CXXC structural domain that couples the H3K4 methylation reaction to unmethylated DNA.
Relationship between histone acetylation and lymphoma development
Reversible acetylation, mediated by HDACs, influences a variety of physiological processes, many of which are aberrantly regulated in tumor cells [72]. In various lymphomas, HDAC inhibitors (HDACi) inhibit cell cycle progression and induce apoptosis. To safeguard and/or boost NK cell function, the specific function of individual HDACs often needs to be determined. For instance, HDAC8, a known target in T cell lymphomas, can inhibit mantle cell lymphoma (MCL) growth without changing NK cell viability, receptor expression, and antibody-dependent cell-mediated cytotoxicity (ADCC). This does not limit the therapeutic activity of rituximab in MCL patients due to impaired NK cell function [73]. Niklas et al.’s discovery that PD-404182 acts as a “molecular trigger” by forming an additional intramolecular disulfide bond between Cys102 and Cys153, thereby inhibiting HDAC8 activity, provides theoretical support for developing selective inhibitors [47].
In addition to HDACs, the HDACi target HSP72 can increase the sensitivity of Hut78 cells from the cutaneous T cell lymphoma (CTCL) cell line to vorinostat [74]. Recently, HDACi has been found to induce autophagy, increase the expression of autophagy factor LC3d, and inhibit the trophic-sensing kinase of mammalian target of rapamycin (mTOR) protein, thereby improving the drug’s therapeutic effect [75].
Additionally, HDACs regulate the G1–S transition, while Aurora A kinase (AAK) regulates mitosis through its role at the G2–M transition point. HDACi leads to AAK and Aurora B kinase (ABK) degradation and kinetochore assembly modification, providing a preclinical rationale for their interaction with AAK inhibitors. In a preclinical model of T cell NHL, Strati et al. [76] discovered that romidepsin (HDACi) and alisertib (AAKi) displayed a synergistic effect secondary to cytoplasmic fission failure, which was not observed in B cells. However, phase I clinical studies of romidepsin combined with alisertib in patients with R/R BCL (14 patients), PTCL (4 patients), and HL (7 patients) revealed that the combination treatment was inferior to the single agent regimen. HDACi combined with Janus kinase inhibitors (JAKi) showed significant antitumor effects on CTCL cells [77]. For example, resminostat (HDACi) combined with ruxolitinib (JAKi) effectively inhibits key cellular pathways in MyLa and SeAx cells, providing a new potential therapeutic approach for CTCL patients [78].
Epigenetic drugs
Romidepsin, a cyclic tetrapeptide intravenous HDACi primarily targeting the enzymatic activity of class I HDACs, induces cell cycle arrest through the upregulation of p21. This substance has shown synergistic effects with novel agents such as bortezomib [79]. In 2011, the FDA gave its approval for romidepsin to be used in the treatment of patients with PTCL who had received at least one prior treatment. Subsequent clinical trials revealed that romidepsin provided durable responses and long-term tolerance in patients with R/R PTCL. Accordingly, the National Comprehensive Cancer Network recommends romidepsin for the second-line and subsequent treatment of patients, whether or not they intend to receive high-dose therapy or SCT [80]. Combination regimens with romidepsin and other drugs such as pralatrexate [81], bendamustine [82], or azacitidine [83] have demonstrated better effectiveness and manageable toxicity for PTCL treatment compared to romidepsin monotherapy. To enhance treatment regimens, phase II studies of lenalidomide and romidepsin are currently being conducted for patients with untreated PTCL. These studies are driven by the promising results of lenalidomide monotherapy in similar patient cohorts, including longer median progression-free survival (PFS), overall survival (OS), and duration of response (DOR). Phase II clinical trials in refractory CTCL implied the clinical efficacy and safety of romidepsin. In patients with CTCL who have received at least one systemic therapy, romidepsin responses were observed in all stages of the disease and in all sites, including the blood, lymph nodes, and skin [84]. However, a phase III study of untreated PTCL indicated that the combination of romidepsin and CHOP failed to significantly improve PFS and remission rates compared to the CHOP regimen alone. This combination was also associated with an increased frequency of grade ≥ 3 treatment-emergent adverse events [23].
Belinostat, a pan-HDACi derived from hydroxypentanoic acid, broadly inhibits all zinc independent HDAC enzymes. This includes class I HDACs (HDAC1, HDAC2, and HDAC3), class II HDACs (HDAC6, HDAC9, and HDAC10), and the class IV HDAC (HDAC11). In 2014, the FDA approved belinostat for the treatment of PTCL. A phase II study involving 129 patients with R/R PTCL showed an objective response rate (ORR) of approximately 26% and a complete response (CR) rate of 11% following belinostat administration [85]. Twelve patients subsequently received a hematopoietic SCT, 10 of whom were still alive at the time of statistical data cutoff (OS range 9.4 to 22.9 months). Furthermore, the occurrence of grade 3–4 hematologic toxicity made it possible to include belinostat in combination regimens with other agents (thrombocytopenia, 7%; neutropenia, 6.2%; anemia, 10.9%). Johnston et al. [86] conducted a trial with belinostat combined with the CHOP regimen, yielding promising results. Out of 23 patients, the overall ORR was 86%, including an 89% response in patients with angioimmunoblastic T cell lymphoma (AITL) and a 90% response in patients with bone marrow involvement. The CR at the maximum tolerated dose was 71%. The optimal dose of belinostat in the belinostat-CHOP regimen was found to be the same as that for belinostat monotherapy, with no additional toxicity identified. Although combination regimens with belinostat appear promising, further validation is needed to establish their efficacy and safety.
Vorinostat, another hydroxyvalerate derivative, was approved by the FDA in 2006 for the treatment of CTCL. It is a broad inhibitor of class I and class II HDACs. A phase I study of vorinostat combined with CHOP in PTCL patients reported an OS of 82%, a median PFS of 79%, and a median response duration of 29 months. However, further studies are needed to confirm the effectiveness of this combination and its impact on patient survival [87]. In a phase I/II clinical study, vorinostat combined with rituximab-CHOP achieved an ORR of 81% (n = 63) in patients with R/R DLBCL. However, due to high rates of febrile neutropenia (38%) and sepsis (19%), this regimen is not recommended for routine clinical use [88]. Other substances such as abexinostat and fimepinostat have been granted fast track designation (FTD) by the FDA for patients with R/R FL and R/R DLBCL respectively. Another HDACi, panobinostat, was approved by the FDA in 2015 for use in combination with bortezomib and dexamethasone in the treatment of multiple myeloma in patients who had received at least two prior therapies. A phase I clinical trial showed good tolerability of panobinostat in patients with CTCL [89].
In 2017, the China Food and Drug Administration (CFDA) authorized chidamide, an independently developed Chinese drug, as a standalone therapy for R/R PTCL. Selectively inhibiting the activity of HDAC1, 2, 3, and 10, chidamide blocks the AKT/mTOR and MAPK signaling pathways while activating the ATM-Chk2-p53-p21 pathway, thus exerting antitumor effects in NK/TCL cells [90]. Additionally, chidamide inhibits the HDACs/STAT3/Bcl-2 pathway, which triggers apoptosis in DLBCL cells [91]. Chidamide enhances OS in DLBCL patients by upregulating OTUD7B expression and synergizing with low-dose adriamycin [92]. A clinical study involving 383 PTCL patients validated the safety and effectiveness of chidamide monotherapy, particularly for AITL patients with an international prognostic index (IPI) score of 2 or more. Furthermore, combining chidamide with chemotherapy—specifically CHOP-like or platinum-containing regimens—yielded higher ORR and DOR in AITL patients [93]. This indicates that the combination of chidamide with chemotherapy is effective in R/R PTCL, especially in young patients with a high IPI who are negative for CD30 expression [94]. Combining chidamide with the CPET regimen (prednisone, etoposide, thalidomide) in a multicenter phase II clinical trial involving 71 newly diagnosed AITL patients (with 51 completing eight cycles) resulted in a 90.2% ORR and a median PFS of 42.6 months [95]. In a retrospective study, Wang et al. [96] evaluated the combination of chidamide with the PEL regimen (prednisone, etoposide, lenalidomide) for R/R DLBCL patients who were not eligible for aggressive chemotherapy or autologous SCT. Furthermore, a patient with EZB/C3 subtype DLBCL who relapsed post-R-CHOP treatment achieved complete remission following the combined treatment with lenalidomide, chidamide, and R-CHOP. This case report supports the feasibility of combining chidamide with the R2-CHOP regimen. In addition to the findings about chidamide’s potential use in treating NK/T cell lymphoma (NKTCL) [90], Zhou et al. also highlighted its potential as a therapeutic target against the EBV.
Beyond its direct anticancer activity, HDACi can influence immune regulation [97]. Evidence indicates that epigenetic factors can enhance the efficacy of anti-programmed cell death protein (PD-1) or anti-PD1-ligand 1 (PD-L1) therapy [98]. Numerous studies have shown that HDACi can amplify the antitumor activity and persistence of PD-1 antibodies by mediating immune recognition and upregulating NKG2D ligand and HSP70 genes [99]. Moreover, HDACi can promote tumor suppression by activating p53 acetylation to stimulate PD-1, inherent in cancer cells [100]. Combining sintilimab and chidamide yielded a durable response with minimal toxicity in a patient with R/R NKTCL resistant to pegaspargase and immunotherapy [101]. Additional case reports endorse the potential of combining anti-PD-1 antibodies with HDACi for treating R/R lymphoma [102,103,104]. The side effects of PD-L1 antibodies are less significant compared to those of PD-1 antibodies. Mocetinostat (MGCD0103), a specific benzamide histone deacetylase inhibitor [24], can elevate PD-L1 levels by reducing BCL-2 levels. One patient with refractory extranodal NKTCL responded well to a treatment regimen that combined radiotherapy, chidamide, and the PD-L1 antibody atezolizumab [105]. Thus, combining HDACi with PD-L1 shows promise for R/R lymphoma treatment. Moreover, Bissonnette et al. highlighted in preclinical studies that chidamide can trigger epigenetic modifications in the tumor microenvironment to amplify the effectiveness of immune checkpoint inhibitors (ICIs) [106]. This finding provides a theoretical basis for combining chidamide and ICIs in treating various tumors, including lymphoma. Consequently, HDACi may be most beneficial when combined with other drugs—such as protein kinase inhibitors, Bcl-2 inhibitors, and other epigenetic agents—or with other therapies like immunotherapy, as is being examined in ongoing clinical trials (Table 3).
Non-coding RNA regulation
Non-coding RNAs (ncRNAs), which do not code for proteins, constitute a significant portion of the human genome. High-throughput genome sequencing and array-based studies estimate that around 90% of the human genome can potentially be transcribed [107]. However, only about 2% of this transcribed genome encodes proteins, totaling around 20,000 proteins [108]. For a long time, this small protein-coding part of the genome was the primary focus of medical research, and ncRNAs were disregarded as “evolutionary junk” [109]. Recent studies have highlighted the vital role ncRNAs can play in various human diseases, notably cancer, as well as in regulating fundamental biological processes such as growth, development, and organ function [110,111,112]. ncRNAs, due to their smaller molecular weight, ability to carry multiple negative charges, and excellent histocompatibility, are promising candidates as therapeutic targets [113]. ncRNAs can be classified into small non-coding RNAs (sncRNA, 18–200 nt) and long-stranded non-coding RNAs (lncRNA, > 200 nt) based on their length. sncRNAs include types such as rRNA, tRNA, miRNA, and piRNA, all of which play a critical role in gene expression regulation [114]. Long-stranded non-coding RNAs (lncRNAs) often function similarly to miRNAs, regulating cancer progression. Cyclic RNA, another type of ncRNA, differs structurally from linear RNA and performs complex functions that are still being explored [115].
miRNAs, the most studied type of ncRNA, are small (18–24 nucleotide) molecules that mainly bind recognition sites in the 3ʹUTRs of genes, thereby limiting post-transcriptional gene expression [116]. They can also down-regulate protein levels through sequence-specific binding to target mRNAs, leading to translation inhibition or mRNA degradation [117]. Additionally, miRNAs can regulate tumor cell interactions with the microenvironment, contributing to lymphoma progression [118]. Table 4 presents various dysregulated miRNAs reported in lymphoma. Lin et al. [119] identified 20 potential miRNA biomarkers in PTCL-not otherwise specified (PTCL-NOS) using PCR alignment and gene ontology analysis. Thirteen miRNAs were upregulated, and 7 miRNAs were downregulated. Notably, the overexpression of miR187 was linked to PTCL-NOS tumor progression and poor prognosis in patients by regulating the Ras-mediated ERK/AKT/MYC axis. The proteasome inhibitor bortezomib suppressed miR187 overexpression during tumor progression [120]. For instance, miR-340-5p enhances the infiltration and antitumor function of CD8 + tumor-infiltrating T lymphocytes by regulating lysine methyltransferase 5A and constitutive photomorphogenesis protein 1, which activate CD73 ubiquitination. This miRNA also directly regulates DLBCL cells [121], suggesting a potential target for DLBCL immunotherapy. miR155, a BCL biomarker, is associated with poor prognosis in DLBCL patients and is implicated in tumor progression by regulating PD-1/PD-L1-mediated interactions with CD8 + T cells in the tumor microenvironment [118]. Other miRNAs like let-7f, miR-9, and miR-27a are specifically expressed and consistently upregulated in classical HL [122]. Furthermore, miR-21 is regarded as a biomarker for HIV-associated lymphoma [123].
lncRNAs represent the most diverse group of ncRNAs. They typically contain long sequences that exceed 200 nucleotides. Unlike short ncRNAs, primarily associated with gene regulation, lncRNAs demonstrate a broad array of mechanistic functions. For instance, the EBV supports BCL progression by suppressing the novel p53-responsive lncRNA, IGFBP7-AS1. However, the exogenous introduction of IGFBP7 or wt-p53 can restore the tumorigenic properties affected by the deletion of IGFBP7-AS1 [147]. One study involving DLBCL patients and healthy controls employed bioinformatics analysis to construct a lymphoma, drug, and lncRNA protein–protein interaction network. This study suggested lncRNAs HOTAIR, GAS5, and XIST as potential diagnostic tools for DLBCL, and HOTAIR and GAS5 as indicators for evaluating treatment efficacy [148]. Zhu et al. [149] conducted a comprehensive genome-wide analysis of lncRNA expression in five DLBCL cell lines and normal B cells. The goal was to identify potential DLBCL-related targets via gene ontology and pathway analysis. The study revealed that a total of 1053 lncRNAs and 4391 mRNAs were aberrantly regulated in DLBCL cells compared to normal B cells. Pathways highly relevant included cell cycle/apoptosis/B cell receptors and the nuclear factor B signaling pathway. Table 5 lists specific dysregulated lncRNAs and their functions in various prevalent lymphoma subtypes. The differing lncRNA activity profiles align with the classification of PTCL subtypes [150].
RNA modification
While the triplet code of the open reading frame is well understood for mRNAs, some functions of non-coding RNAs remain unknown, and advances in genome sequencing technologies have revolutionized our understanding of the genome and its transcription [164]. Many studies have begun to provide a clearer picture of RNA modifications, and more than 160 RNA modifications, including N7-methyluracil (m7G), N6-methyladenosine (m6A), and 5-methylcytosine (m5C), have been identified [165,166,167].
In eukaryotes, m6A is the most abundant mRNA modification, accounting for more than 80% of all RNA methylation modifications. M6A is mainly regulated by methyltransferases (METTL3, METTL14, WTAP, and VIRMA), demethylase proteins (FTO, ALKBH5), and reading proteins (YTH family proteins) [168, 169] that are expressed in a variety of tumors and play a major part in promoting tumor progression, especially in lymphomas [170]. In DLBCL, the m6 A regulatory genes piRNA-30473 and WTAP prolong DLBCL patient survival and it has been demonstrated that the piRNA-30473/WTAP/HK2 axis promotes tumorigenesis by regulating m6A RNA methylation [171]. In addition, m6A-modified mRNAs mediate multiple cellular and viral functions. In lymphoma, m6A-modified EBV transcripts are disrupted by the UTUDF1 protein, leading to the downregulation of m6A-dependent EBV infection and replication, which could provide a new target for the treatment of EBV-associated lymphoma [172].
Furthermore, m6A mRNA modification was associated with histone modification. m6A modification increased H3K36me3 levels, and m6A was significantly reduced when H3K36me3 was depleted in cells. H3K36me3 levels showed the importance of m6A in terms of specificity and dynamic deposition in mRNA and revealed the interaction between histone and RNA methylation during gene expression regulation [173]. In DLBCL tumors and cell lines, the m6A and methyltransferase 3, N6-adenosine-methyltransferase complex catalytic subunit (METTL3) levels were upregulated, and silencing METTL3 decreased cell proliferation and reduced m6A methylation and pigment epithelium-derived factor (PEDF) total mRNA levels. High levels of PEDF eliminated the inhibitory effect of METTL3 silencing on DLBCL cell proliferation, demonstrating that METTL3 promotes DLBCL development via moderating m6A levels of PEDF mRNA [174]. The m6A methyltransferase complex, which brings the m6A methyltransferases METTL3 and METTL14 to their respective mRNA targets and is involved in catalyzing the formation of m6A, is made up in key part of the nuclear protein known as WT1-associated protein (WTAP). WTAP is overexpressed in various types of malignancies, and it functions as an oncogene. It has been demonstrated that WTAP is upregulated in human NKTCL and silencing WTAP inhibits NK/T cell proliferation while enhancing apoptosis of tumor cells. WTAP also enhances the chemoresistance of NK/T cells to cisplatin by increasing dual specificity phosphatase 6 mRNA levels in an m6A-dependent manner. Sustained upregulation of WTAP promoted the proliferative capacity of DLBCL cells and improved tumor resistance to apoptosis, and downregulation of WTAP resulted in a significant increase in apoptosis after etoposide treatment [175].
There are two classes of modifying enzymes involved in m5C: the methyltransferase family (DNMT2/TRDMT1) and the NOP2/Sun domain (NSUN) family (NSUN1-7) [176]. DNMT2 is one of the first identified RNA m5C methyltransferases, and it was considered a DNA methyltransferase because it has all the structural features of a DNA methyltransferase, except a specific nucleic acid binding region. DNMT2 has important regulatory roles in tissue and organ development, hematopoiesis, and external stress responses [177]. In addition, synergistic interactions between m5C and m6A may regulate protein expression. In tumor cells, NSUN2 catalyzes the m5C modification, and METTL3 and METTL14 catalyze the m6A modification, which synergistically increases p21 translation, leading to elevated p21 expression and oxidative stress–induced cellular senescence [178]. NSUN2 is a nucleoprotein that plays a significant role in tissue homeostasis, spindle stability, and early embryogenesis. NSUN2 promotes mRNA stability, regulates miRNA expression, enhances protein synthesis and translation, and influences the expression and translation of key cell cycle regulators [179]. NSUN2 and METTL1 silencing in tumor cells significantly enhanced their sensitivity to 5-fluorouracil. NSUN2 and METTL1 were phosphorylated by ABK and AKT, tRNA modification activity was inhibited by phosphorylation, and overexpression reduced 5-FU sensitivity. Thus, interfering with tRNA methylation may provide a new direction for treatment with 5-FU [180].
Conclusion and outlook
Lymphoma represents a group of malignant tumors that originate from the lymphopoietic system. These tumors are characterized by high recurrence rates, high mortality, and short survival time. DLBCL, the most common clinical malignant lymphoma, is traditionally treated with the R-CHOP regimen as the standard first-line chemotherapy. Although the treatment has high response rates, 30–40% of DLBCL patients ultimately progress to R/R DLBCL and face a fatal outcome. Therefore, identifying new targets for lymphoma treatment and developing new drugs and more effective combination regimens are of clinical importance.
Epigenetics has provided a novel perspective compared to classical genetic theories, paving the way for precision medicine in oncology. Currently, substantial progress has been made in studying lymphoma pathogenesis and targeted drugs. Beyond genetic variants, primarily gene mutations, epigenetic changes play an essential role in lymphoma pathogenesis. Epigenetics-based targeted treatments, such as decitabine and chidamide, have been well received clinically. Additionally, while some epigenetic drugs like pan-HDACi vorinostat and EZH2 selective inhibitor tazemetostat have limited efficacy alone, their combination with other drugs such as immunostimulatory monoclonal antibodies, proteasome inhibitors, or multiple epigenetic drugs has demonstrated high response rates and good tolerability. Nevertheless, in clinical and preclinical studies, investigators should carefully monitor whether combination regimens can manage the incidence of adverse effects.
In summary, the evolution and investigation of epigenetics are crucial to the future realization of more effective precision therapy. However, epigenetic studies alone are insufficient to fully comprehend lymphoma pathogenesis. Integration of multidimensional clinical data with transcriptomics, proteomics, metabolomics, and epigenomics datasets could unveil superior treatment targets and therapeutic regimens with enhanced clinical efficacy. Minimizing the toxic side effects of chemotherapy and other treatments will enhance patient’s quality of life.
Data Availability
The datasets analysed during the current study are available to download from https://clinicaltrials.gov/.
Abbreviations
- HL:
-
Hodgkin’s lymphoma
- NHL:
-
Non-Hodgkin’s lymphoma
- DLBCL:
-
Diffuse large B cell lymphoma
- CAR-T:
-
Chimeric antigen receptor-modified T cell
- CHOP:
-
Cyclophosphamide, doxorubicin, vincristine, and prednisone
- CHOEP:
-
CHOP-like regimen
- SCT:
-
Stem cell transplantation
- DNMT:
-
DNA methyltransferases
- 5mC:
-
5-Methylcytosine
- EBV:
-
Epstein-Barr virus
- BL:
-
Burkitt lymphoma
- UHRF1:
-
Ubiquitin-like, containing PHD and RING finger domains, 1
- AZA:
-
5-Azacytidine
- ROMI:
-
Romidepsin
- CTAs:
-
Cancer testis antigens
- PTCL:
-
Peripheral T cell lymphoma
- TN:
-
Treatment-naïve
- TFH:
-
T follicular helper cell
- GC:
-
Germinal center
- PRC2:
-
Polycomb repressor complex 2
- HDAC:
-
Histone deacetylase
- HDACi:
-
Histone deacetylase inhibitors
- MCL:
-
Mantle cell lymphoma
- ADCC:
-
Antibody-dependent cell-mediated cytotoxicity
- MTCL:
-
Mature T cell lymphoma
- CTCL:
-
Cutaneous T cell lymphoma
- mTOR:
-
Mammalian target of rapamycin
- AAK:
-
Aurora A kinase
- ABK:
-
Aurora B kinase
- AAKi:
-
Aurora A kinase inhibitors
- JAKi:
-
Janus kinase inhibitors
- C:
-
Completed
- R:
-
Recruiting
- ANR:
-
Active, not recruiting
- DLT:
-
Dose-limiting toxicity
- CRR:
-
Complete response rate
- MTD:
-
Maximum tolerable dose
- ORR:
-
Objective response rate
- PRR:
-
Partial response rate
- PFS:
-
Progression-free survival
- PTCL:
-
Previously untreated peripheral T cell lymphoma
- R/R:
-
Relapsed/refractory
- ENKTCL:
-
Extranodal natural killer cell/T cell lymphoma
- CTL:
-
Cutaneous T cell lymphoma
- ATCL:
-
Angioimmunoblastic T cell lymphoma
- TCL:
-
T cell lymphoma
- MCL:
-
Mature cell lymphoma
- AITL:
-
Angioimmunoblastic T cell lymphoma
- CFDA:
-
The China Food and Drug Administration
- IPI:
-
International prognostic index
- CPET regimen:
-
Prednisone, etoposide, and thalidomide
- PEL regimen:
-
Prednisone, etoposide, lenalidomide
- PD-1:
-
Programmed death-1
- PD-L1:
-
Anti-PD1-ligand 1
- ICIs:
-
Immune checkpoint inhibitors
- ncRNAs:
-
Non-coding RNAs
- sncRNA:
-
Small non-coding RNAs
- lncRNA:
-
Long-stranded non-coding RNAs
- PTCL-NOS:
-
PTCL-not otherwise specified
- BCL:
-
B cell lymphoma
- m7G:
-
N7-methyluracil
- m6A:
-
N6-methyladenosine
- m5C:
-
5-Methylcytosine
- METTL3:
-
Methyltransferase 3, N6-adenosine-methyltransferase complex catalytic subunit
- PEDF:
-
Pigment epithelium-derived factor
- WTAP:
-
WT1-associated protein
References
Matasar MJ, Zelenetz AD (2008) Overview of lymphoma diagnosis and management. Radiol Clin North Am 46(175–198):vii. https://doi.org/10.1016/j.rcl.2008.03.005
Mugnaini EN, Ghosh N (2016) Lymphoma. Prim Care 43:661–675. https://doi.org/10.1016/j.pop.2016.07.012
Jaffe ES (2019) Diagnosis and classification of lymphoma: impact of technical advances. Semin Hematol 56:30–36. https://doi.org/10.1053/j.seminhematol.2018.05.007
Shi Y, Han Y, Yang J, Liu P, He X, Zhang C, Zhou S, Zhou L, Qin Y, Song Y et al (2019) Clinical features and outcomes of diffuse large B-cell lymphoma based on nodal or extranodal primary sites of origin: analysis of 1,085 WHO classified cases in a single institution in China. Chin J Cancer Res 31:152–161. https://doi.org/10.21147/j.issn.1000-9604.2019.01.10
Wang XM, Bassig BA, Wen JJ, Li GD, Liu ZB, Yao WX, Hu W, Wang Y, Li JM, Wang XD et al (2016) Clinical analysis of 1629 newly diagnosed malignant lymphomas in current residents of Sichuan province, China. Hematol Oncol 34:193–199. https://doi.org/10.1002/hon.2202
China Anti-Cancer Association Lymphoma Committee, Chinese Association for Clinical Oncologists, Medical Oncology Branch of Chinese International Exchange and Promotion Association for Medical and Healthcare (2021) Clinical practice guideline for multi-disciplinary treatment strategy of lymphoma in China. Chin J Oncol 43(02):163–166. https://doi.org/10.3760/cma.j.cn112152-20201109-00971
Bakhshi TJ, Georgel PT (2020) Genetic and epigenetic determinants of diffuse large B-cell lymphoma. Blood Cancer J 10:123. https://doi.org/10.1038/s41408-020-00389-w
China Anti-cancer Association Lymphoma Committee, Chinese Association for Clinical Oncologists, Medical Oncology Branch of Chinese International Exchange and Promotion Association for Medical and Healthcare (2021) Clinical practice guideline for lymphoma in China (2021 Edition). Chin J Oncol 43(07):707–735. https://doi.org/10.3760/cma.j.cn112152-20210516-00382
Miao Zhaoyi, Zhao Zhigang (2020) Advances in epigenetic modulation-based therapies for lymphoma. Chinese J. Clin. Oncol. 47(7):359–364. https://doi.org/10.3969/j.issn.1000-8179.2020.07.330
Chung C (2019) Current targeted therapies in lymphomas. Am J Health Syst Pharm 76:1825–1834. https://doi.org/10.1093/ajhp/zxz202
Hopken UE, Rehm A (2019) Targeting the tumor microenvironment of leukemia and lymphoma. Trends Cancer 5:351–364. https://doi.org/10.1016/j.trecan.2019.05.001
Godfrey J, Leukam MJ, Smith SM (2018) An update in treating transformed lymphoma. Best Pract Res Clin Haematol 31:251–261. https://doi.org/10.1016/j.beha.2018.07.008
Melani C, Wilson WH (2022) Front-Line treatment of diffuse large B-cell lymphoma in patients with cardiovascular comorbidities; omission of anthracycline reduces cure. Leuk Lymphoma 63:511–513. https://doi.org/10.1080/10428194.2021.2002323
Bhatt VR, Vose JM (2014) Hematopoietic stem cell transplantation for non-Hodgkin lymphoma. Hematol Oncol Clin North Am 28:1073–1095. https://doi.org/10.1016/j.hoc.2014.08.015
Yang H, Green MR (2020) Harnessing lymphoma epigenetics to improve therapies. Hematol Am Soc Hematol Educ Program 2020:95–100. https://doi.org/10.1182/hematology.2020006908
Falchi L, Ma H, Klein S, Lue JK, Montanari F, Marchi E, Deng C, Kim HA, Rada A, Jacob AT et al (2021) Combined oral 5-azacytidine and romidepsin are highly effective in patients with PTCL: a multicenter phase 2 study. Blood 137:2161–2170. https://doi.org/10.1182/blood.2020009004
Nieto Y, Valdez BC, Thall PF, Jones RB, Wei W, Myers A, Hosing C, Ahmed S, Popat U, Shpall EJ et al (2016) Double epigenetic modulation of high-dose chemotherapy with azacitidine and vorinostat for patients with refractory or poor-risk relapsed lymphoma. Cancer 122:2680–2688. https://doi.org/10.1002/cncr.30100
Morschhauser F, Tilly H, Chaidos A, McKay P, Phillips T, Assouline S, Batlevi CL, Campbell P, Ribrag V, Damaj GL et al (2020) Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol 21:1433–1442. https://doi.org/10.1016/S1470-2045(20)30441-1
Maruyama D, Tobinai K, Makita S, Ishida T, Kusumoto S, Ishitsuka K, Yoshimitsu M, Imaizumi Y, Sawayama Y, Takeuchi S et al (2017) First-in-human study of the EZH1/2 dual inhibitor DS-3201b in patients with relapsed or refractory non-Hodgkin lymphomas — preliminary results. Blood 130:4070–4070. https://doi.org/10.1182/blood.V130.Suppl_1.4070.4070
Izutsu K, Makita S, Nosaka K, Yoshimitsu M, Utsunomiya A, Kusumoto S, Morishima S, Tsukasaki K, Kawamata T, Ono T et al (2023) An open-label, single-arm phase 2 trial of valemetostat for relapsed or refractory adult T-cell leukemia/lymphoma. Blood 141:1159–1168. https://doi.org/10.1182/blood.2022016862
Amorim S, Stathis A, Gleeson M, Iyengar S, Magarotto V, Leleu X, Morschhauser F, Karlin L, Broussais F, Rezai K et al (2016) Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: a dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol 3:e196-204. https://doi.org/10.1016/S2352-3026(16)00021-1
Liu W, Zhao D, Liu T, Niu T, Song Y, Xu W, Jin J, Cai Q, Huang H, Li Z et al (2021) A multi-center, real-world study of chidamide for patients with relapsed or refractory peripheral T-cell lymphomas in China. Front Oncol 11:750323. https://doi.org/10.3389/fonc.2021.750323
Bachy E, Camus V, Thieblemont C, Sibon D, Casasnovas RO, Ysebaert L, Damaj G, Guidez S, Pica GM, Kim WS et al (2022) Romidepsin plus CHOP versus CHOP in patients with previously untreated peripheral T-cell lymphoma: results of the Ro-CHOP Phase III study (conducted by LYSA). J Clin Oncol 40:242–251. https://doi.org/10.1200/JCO.21.01815
Batlevi CL, Crump M, Andreadis C, Rizzieri D, Assouline SE, Fox S, van der Jagt RHC, Copeland A, Potvin D, Chao R et al (2017) A phase 2 study of mocetinostat, a histone deacetylase inhibitor, in relapsed or refractory lymphoma. Br J Haematol 178:434–441. https://doi.org/10.1111/bjh.14698
Campbell P, Thomas CM (2017) Belinostat for the treatment of relapsed or refractory peripheral T-cell lymphoma. J Oncol Pharm Pract 23:143–147. https://doi.org/10.1177/1078155216634178
Gui L, Cao J, Ji D, Zhang H, Fan Q, Zhu J, Song Y, Jiang S, Ning Z, Yu J et al (2021) Chidamide combined with cyclophosphamide, doxorubicin, vincristine and prednisone in previously untreated patients with peripheral T-cell lymphoma. Chin J Cancer Res 33:616–626. https://doi.org/10.21147/j.issn.1000-9604.2021.05.08
Amengual JE, Lichtenstein R, Lue J, Sawas A, Deng C, Lichtenstein E, Khan K, Atkins L, Rada A, Kim HA et al (2018) A phase 1 study of romidepsin and pralatrexate reveals marked activity in relapsed and refractory T-cell lymphoma. Blood 131:397–407. https://doi.org/10.1182/blood-2017-09-806737
Jeffries MA (2020) The development of epigenetics in the study of disease pathogenesis. Adv Exp Med Biol 1253:57–94. https://doi.org/10.1007/978-981-15-3449-2_2
Zhang L, Lu Q, Chang C (2020) Epigenetics in health and disease. Adv Exp Med Biol 1253:3–55. https://doi.org/10.1007/978-981-15-3449-2_1
Werner RJ, Kelly AD, Issa JJ (2017) Epigenetics and precision oncology. Cancer J 23:262–269. https://doi.org/10.1097/PPO.0000000000000281
Villanueva L, Alvarez-Errico D, Esteller M (2020) The contribution of epigenetics to cancer immunotherapy. Trends Immunol 41:676–691. https://doi.org/10.1016/j.it.2020.06.002
Miranda Furtado CL, Dos Santos Luciano MC, Silva Santos RD, Furtado GP, Moraes MO, Pessoa C (2019) Epidrugs: targeting epigenetic marks in cancer treatment. Epigenetics 14:1164–1176. https://doi.org/10.1080/15592294.2019.1640546
Garsuault D, Bouyer C, Nguyen E, Kandhari R, Prochazkova-Carlotti M, Chevret E, Forgez P, Segal-Bendirdjian E (2020) Complex context relationships between DNA methylation and accessibility, histone marks, and hTERT gene expression in acute promyelocytic leukemia cells: perspectives for all-trans retinoic acid in cancer therapy. Mol Oncol 14:1310–1326. https://doi.org/10.1002/1878-0261.12681
Zhang H, Ying H, Wang X (2020) Methyltransferase DNMT3B in leukemia. Leuk Lymphoma 61:263–273. https://doi.org/10.1080/10428194.2019.1666377
Kulis M, Esteller M (2010) DNA methylation and cancer. Adv Genet 70:27–56. https://doi.org/10.1016/B978-0-12-380866-0.60002-2
Moore LD, Le T, Fan G (2013) DNA methylation and its basic function. Neuropsychopharmacology 38:23–38. https://doi.org/10.1038/npp.2012.112
Mervis JS, McGee JS (2020) DNA methylation and inflammatory skin diseases. Arch Dermatol Res 312:461–466. https://doi.org/10.1007/s00403-019-02005-9
Leoni C, Montagner S, Rinaldi A, Bertoni F, Polletti S, Balestrieri C, Monticelli S (2017) Dnmt3a restrains mast cell inflammatory responses. Proc Natl Acad Sci U S A 114:E1490–E1499. https://doi.org/10.1073/pnas.1616420114
Sinclair AJ (2021) Could changing the DNA methylation landscape promote the destruction of Epstein-Barr virus-associated cancers? Front Cell Infect Microbiol 11:695093. https://doi.org/10.3389/fcimb.2021.695093
Dalton T, Doubrovina E, Pankov D, Reynolds R, Scholze H, Selvakumar A, Vizconde T, Savalia B, Dyomin V, Weigel C et al (2020) Epigenetic reprogramming sensitizes immunologically silent EBV+ lymphomas to virus-directed immunotherapy. Blood 135:1870–1881. https://doi.org/10.1182/blood.2019004126
Guo R, Zhang Y, Teng M, Jiang C, Schineller M, Zhao B, Doench JG, O’Reilly RJ, Cesarman E, Giulino-Roth L et al (2020) DNA methylation enzymes and PRC1 restrict B-cell Epstein-Barr virus oncoprotein expression. Nat Microbiol 5:1051–1063. https://doi.org/10.1038/s41564-020-0724-y
Zhang Y, Sun Z, Jia J, Du T, Zhang N, Tang Y, Fang Y, Fang D (2021) Overview of histone modification. Adv Exp Med Biol 1283:1–16. https://doi.org/10.1007/978-981-15-8104-5_1
Hyun K, Jeon J, Park K, Kim J (2017) Writing, erasing and reading histone lysine methylations. Exp Mol Med 49:e324. https://doi.org/10.1038/emm.2017.11
Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N et al (2011) Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146:1016–1028. https://doi.org/10.1016/j.cell.2011.08.008
Allis CD, Jenuwein T (2016) The molecular hallmarks of epigenetic control. Nat Rev Genet 17:487–500. https://doi.org/10.1038/nrg.2016.59
Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, Johnson NA, Severson TM, Chiu R, Field M et al (2011) Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 476:298–303. https://doi.org/10.1038/nature10351
Jansch N, Sugiarto WO, Muth M, Kopranovic A, Desczyk C, Ballweg M, Kirschhofer F, Brenner-Weiss G, Meyer-Almes FJ (2020) Switching the switch: ligand induced disulfide formation in HDAC8. Chemistry 26:13249–13255. https://doi.org/10.1002/chem.202001712
Kim JJ, Lee SY, Miller KM (2019) Preserving genome integrity and function: the DNA damage response and histone modifications. Crit Rev Biochem Mol Biol 54:208–241. https://doi.org/10.1080/10409238.2019.1620676
Nichol JN, Dupere-Richer D, Ezponda T, Licht JD, Miller WH Jr (2016) H3K27 Methylation: a focal point of epigenetic deregulation in cancer. Adv Cancer Res 131:59–95. https://doi.org/10.1016/bs.acr.2016.05.001
Lawrence M, Daujat S, Schneider R (2016) Lateral thinking: how histone modifications regulate gene expression. Trends Genet 32:42–56. https://doi.org/10.1016/j.tig.2015.10.007
Francis M, Gopinathan G, Foyle D, Fallah P, Gonzalez M, Luan X, Diekwisch TGH (2020) Histone methylation: Achilles heel and powerful mediator of periodontal homeostasis. J Dent Res 99:1332–1340. https://doi.org/10.1177/0022034520932491
Zhang J, Dominguez-Sola D, Hussein S, Lee JE, Holmes AB, Bansal M, Vlasevska S, Mo T, Tang H, Basso K et al (2015) Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med 21:1190–1198. https://doi.org/10.1038/nm.3940
Ji MM, Huang YH, Huang JY, Wang ZF, Fu D, Liu H, Liu F, Leboeuf C, Wang L, Ye J et al (2018) Histone modifier gene mutations in peripheral T-cell lymphoma not otherwise specified. Haematologica 103:679–687. https://doi.org/10.3324/haematol.2017.182444
Baubec T, Colombo DF, Wirbelauer C, Schmidt J, Burger L, Krebs AR, Akalin A, Schubeler D (2015) Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 520:243–247. https://doi.org/10.1038/nature14176
Aymard F, Bugler B, Schmidt CK, Guillou E, Caron P, Briois S, Iacovoni JS, Daburon V, Miller KM, Jackson SP et al (2014) Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat Struct Mol Biol 21:366–374. https://doi.org/10.1038/nsmb.2796
Li F, Mao G, Tong D, Huang J, Gu L, Yang W, Li GM (2013) The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell 153:590–600. https://doi.org/10.1016/j.cell.2013.03.025
Leung W, Teater M, Durmaz C, Meydan C, Chivu AG, Chadburn A, Rice EJ, Muley A, Camarillo JM, Arivalagan J et al (2022) SETD2 haploinsufficiency enhances germinal center-associated AICDA somatic hypermutation to drive B-cell lymphomagenesis. Cancer Discov 12:1782–1803. https://doi.org/10.1158/2159-8290.CD-21-1514
McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A 3rd, Diaz E et al (2012) EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492:108–112. https://doi.org/10.1038/nature11606
Pfister SX, Ashworth A (2017) Marked for death: targeting epigenetic changes in cancer. Nat Rev Drug Discov 16:241–263. https://doi.org/10.1038/nrd.2016.256
Morin RD, Arthur SE, Assouline S (2021) Treating lymphoma is now a bit EZ-er. Blood Adv 5:2256–2263. https://doi.org/10.1182/bloodadvances.2020002773
Mondello P, Ansell SM (2022) Tazemetostat: a treatment option for relapsed/refractory follicular lymphoma. Expert Opin Pharmacother 23:295–301. https://doi.org/10.1080/14656566.2021.2014815
Italiano A, Soria JC, Toulmonde M, Michot JM, Lucchesi C, Varga A, Coindre JM, Blakemore SJ, Clawson A, Suttle B et al (2018) Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol 19:649–659. https://doi.org/10.1016/S1470-2045(18)30145-1
Munakata W, Shirasugi Y, Tobinai K, Onizuka M, Makita S, Suzuki R, Maruyama D, Kawai H, Izutsu K, Nakanishi T et al (2021) Phase 1 study of tazemetostat in Japanese patients with relapsed or refractory B-cell lymphoma. Cancer Sci 112:1123–1131. https://doi.org/10.1111/cas.14822
Scholze H, Stephenson RE, Reynolds R, Shah S, Puri R, Butler SD, Trujillo-Alonso V, Teater MR, van Besien H, Gibbs-Curtis D et al (2020) Combined EZH2 and Bcl-2 inhibitors as precision therapy for genetically defined DLBCL subtypes. Blood Adv 4:5226–5231. https://doi.org/10.1182/bloodadvances.2020002580
Tong KI, Yoon S, Isaev K, Bakhtiari M, Lackraj T, He MY, Joynt J, Silva A, Xu MC, Prive GG et al (2021) Combined EZH2 inhibition and IKAROS degradation leads to enhanced antitumor activity in diffuse large B-cell lymphoma. Clin Cancer Res 27:5401–5414. https://doi.org/10.1158/1078-0432.CCR-20-4027
Palomba ML, Cartron G, Popplewell L, Ribrag V, Westin J, Huw LY, Agarwal S, Shivhare M, Hong WJ, Raval A et al (2022) Combination of atezolizumab and tazemetostat in patients with relapsed/refractory diffuse large B-cell lymphoma: results from a phase Ib study. Clin Lymphoma Myeloma Leuk 22:504–512. https://doi.org/10.1016/j.clml.2021.12.014
Sarkozy C, Morschhauser F, Dubois S, Molina T, Michot JM, Cullieres-Dartigues P, Suttle B, Karlin L, Le Gouill S, Picquenot JM et al (2020) A LYSA phase Ib study of tazemetostat (EPZ-6438) plus R-CHOP in patients with newly diagnosed diffuse large B-cell lymphoma (DLBCL) with poor prognosis features. Clin Cancer Res 26:3145–3153. https://doi.org/10.1158/1078-0432.CCR-19-3741
Yamagishi M, Hori M, Fujikawa D, Ohsugi T, Honma D, Adachi N, Katano H, Hishima T, Kobayashi S, Nakano K et al (2019) Targeting excessive EZH1 and EZH2 activities for abnormal histone methylation and transcription network in malignant lymphomas. Cell Rep 29:2321–2337. https://doi.org/10.1016/j.celrep.2019.10.083. e2327
Honma D, Kanno O, Watanabe J, Kinoshita J, Hirasawa M, Nosaka E, Shiroishi M, Takizawa T, Yasumatsu I, Horiuchi T et al (2017) Novel orally bioavailable EZH1/2 dual inhibitors with greater antitumor efficacy than an EZH2 selective inhibitor. Cancer Sci 108:2069–2078. https://doi.org/10.1111/cas.13326
Dou F, Tian Z, Yang X, Li J, Wang R, Gao J (2022) Valemetostat: first approval as a dual inhibitor of EZH1/2 to treat adult T-cell leukemia/lymphoma. Drug Discov Ther 16:297–299. https://doi.org/10.5582/ddt.2022.01085
Hashimoto H, Vertino PM, Cheng X (2010) Molecular coupling of DNA methylation and histone methylation. Epigenomics 2:657–669. https://doi.org/10.2217/epi.10.44
Khan O, La Thangue NB (2012) HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol Cell Biol 90:85–94. https://doi.org/10.1038/icb.2011.100
Watters JM, Wright G, Smith MA, Shah B, Wright KL (2021) Histone deacetylase 8 inhibition suppresses mantle cell lymphoma viability while preserving natural killer cell function. Biochem Biophys Res Commun 534:773–779. https://doi.org/10.1016/j.bbrc.2020.11.001
Fujii K, Idogawa M, Suzuki N, Iwatsuki K, Kanekura T (2021) Functional depletion of HSP72 by siRNA and quercetin enhances vorinostat-induced apoptosis in an HSP72-overexpressing cutaneous T-cell lymphoma cell line, Hut78. Int J Mol Sci 22 https://doi.org/10.3390/ijms222011258
Gammoh N, Lam D, Puente C, Ganley I, Marks PA, Jiang X (2012) Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc Natl Acad Sci U S A 109:6561–6565. https://doi.org/10.1073/pnas.1204429109
Strati P, Nastoupil LJ, Davis RE, Fayad LE, Fowler N, Hagemeister FB, Kwak L, Oki Y, Wang M, Westin J et al (2020) A phase 1 trial of alisertib and romidepsin for relapsed/refractory aggressive B-cell and T-cell lymphomas. Haematologica 105:e26–e28. https://doi.org/10.3324/haematol.2019.220012
Karagianni F, Piperi C, Mpakou V, Spathis A, Foukas PG, Dalamaga M, Pappa V, Papadavid E (2021) Ruxolitinib with resminostat exert synergistic antitumor effects in cutaneous T-cell lymphoma. PLoS One 16:e0248298. https://doi.org/10.1371/journal.pone.0248298
Karagianni F, Piperi C, Casar B, de la Fuente-Vivas D, Garcia-Gomez R, Lampadaki K, Pappa V, Papadavid E (2022) Combination of resminostat with ruxolitinib exerts antitumor effects in the chick embryo chorioallantoic membrane model for cutaneous T cell lymphoma. Cancers (Basel) 14 https://doi.org/10.3390/cancers14041070
Yazbeck VY, Grant S (2015) Romidepsin for the treatment of non-Hodgkin’s lymphoma. Expert Opin Investig Drugs 24:965–979. https://doi.org/10.1517/13543784.2015.1041586
Barbarotta L, Hurley K (2015) Romidepsin for the treatment of peripheral T-cell lymphoma. J Adv Pract Oncol 6:22–36
Ma H, Davarifar A, Amengual JE (2018) The future of combination therapies for peripheral T cell lymphoma (PTCL). Curr Hematol Malig Rep 13:13–24. https://doi.org/10.1007/s11899-018-0432-3
Nachmias B, Shaulov A, Lavie D, Goldschmidt N, Gural A, Saban R, Lebel E, Gatt ME (2019) Romidepsin-bendamustine combination for relapsed/refractory T cell lymphoma. Acta Haematol 141:216–221. https://doi.org/10.1159/000498905
O’Connor OA, Falchi L, Lue JK, Marchi E, Kinahan C, Sawas A, Deng C, Montanari F, Amengual JE, Kim HA et al (2019) Oral 5-azacytidine and romidepsin exhibit marked activity in patients with PTCL: a multicenter phase 1 study. Blood 134:1395–1405. https://doi.org/10.1182/blood.2019001285
Kim M, Thompson LA, Wenger SD, O’Bryant CL (2012) Romidepsin: a histone deacetylase inhibitor for refractory cutaneous T-cell lymphoma. Ann Pharmacother 46:1340–1348. https://doi.org/10.1345/aph.1R036
O’Connor OA, Horwitz S, Masszi T, Van Hoof A, Brown P, Doorduijn J, Hess G, Jurczak W, Knoblauch P, Chawla S et al (2015) Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: results of the pivotal phase II BELIEF (CLN-19) study. J Clin Oncol 33:2492–2499. https://doi.org/10.1200/JCO.2014.59.2782
Johnston PBCA, Nikolinakos PG, Beaven AW, Barta SK, Bhat G, Hasal SJ, De Vos S, Oki Y, Deng C, Foss FM (2021) Belinostat in combination with standard cyclophosphamide, doxorubicin, vincristine and prednisone as first-line treatment for patients with newly diagnosed peripheral T-cell lymphoma. Exp Hematol Oncol 10(1):15. https://doi.org/10.1186/s40164-021-00203-8
Fukutomi A, Hatake K, Matsui K, Sakajiri S, Hirashima T, Tanii H, Kobayashi K, Yamamoto N (2012) A phase I study of oral panobinostat (LBH589) in Japanese patients with advanced solid tumors. Invest New Drugs 30:1096–1106. https://doi.org/10.1007/s10637-011-9666-9
Oki Y, Younes A, Copeland A, Hagemeister F, Fayad LE, McLaughlin P, Shah J, Fowler N, Romaguera J, Kwak LW et al (2013) Phase I study of vorinostat in combination with standard CHOP in patients with newly diagnosed peripheral T-cell lymphoma. Br J Haematol 162:138–141. https://doi.org/10.1111/bjh.12326
Persky DO, Li H, Rimsza LM, Barr PM, Popplewell LL, Bane CL, Von Gehr A, LeBlanc M, Fisher RI, Smith SM et al (2018) A phase I/II trial of vorinostat (SAHA) in combination with rituximab-CHOP in patients with newly diagnosed advanced stage diffuse large B-cell lymphoma (DLBCL): SWOG S0806. Am J Hematol 93:486–493. https://doi.org/10.1002/ajh.25010
Zhou J, Zhang C, Sui X, Cao S, Tang F, Sun S, Wang S, Chen B (2018) Histone deacetylase inhibitor chidamide induces growth inhibition and apoptosis in NK/T lymphoma cells through ATM-Chk2-p53-p21 signalling pathway. Invest New Drugs 36:571–580. https://doi.org/10.1007/s10637-017-0552-y
Zhang H, Chi F, Qin K, Mu X, Wang L, Yang B, Wang Y, Bai M, Li Z, Su L, et al. (2021) Chidamide induces apoptosis in DLBCL cells by suppressing the HDACs/STAT3/Bcl2 pathway. Mol Med Rep 23 https://doi.org/10.3892/mmr.2021.11947
Qiu S, Liu Y, Gui A, Xia Z, Liu W, Gu JJ, Zuo J, Yang L, Zhang Q (2022) Deubiquitinase OTUD7B is a potential prognostic biomarker in diffuse large B-cell lymphoma. J Cancer 13:998–1004. https://doi.org/10.7150/jca.65835
Shi Y, Jia B, Xu W, Li W, Liu T, Liu P, Zhao W, Zhang H, Sun X, Yang H et al (2017) Chidamide in relapsed or refractory peripheral T cell lymphoma: a multicenter real-world study in China. J Hematol Oncol 10:69. https://doi.org/10.1186/s13045-017-0439-6
Wang J, Fang Y, Ma S, Su N, Zhang Y, Huang H, Li Z, Huang H, Tian X, Cai J et al (2021) Comparison of chidamide-contained treatment modalities versus chemotherapy in the second-line treatment for relapsed or refractory peripheral T-cell lymphoma. Leuk Res 111:106705. https://doi.org/10.1016/j.leukres.2021.106705
Wang Y, Zhang M, Song W, Cai Q, Zhang L, Sun X, Zou L, Zhang H, Wang L, Xue H (2022) Chidamide plus prednisone, etoposide, and thalidomide for untreated angioimmunoblastic T-cell lymphoma in a Chinese population: a multicenter phase II trial. Am J Hematol. https://doi.org/10.1002/ajh.26499
Wang Y, Xue H, Song W, Xiao S, Jing F, Dong T, Wang L (2022) Chidamide with PEL regimen (prednisone, etoposide, lenalidomide) for elderly or frail patients with relapsed/refractory diffuse large B-Cell lymphoma -results of a single center, retrospective cohort in China. Hematol Oncol https://doi.org/10.1002/hon.2979
Zhao LM, Zhang JH (2019) Histone deacetylase inhibitors in tumor immunotherapy. Curr Med Chem 26:2990–3008. https://doi.org/10.2174/0929867324666170801102124
Chen X, Pan X, Zhang W, Guo H, Cheng S, He Q, Yang B, Ding L (2020) Epigenetic strategies synergize with PD-L1/PD-1 targeted cancer immunotherapies to enhance antitumor responses. Acta Pharm Sin B 10:723–733. https://doi.org/10.1016/j.apsb.2019.09.006
Burke B, Eden C, Perez C, Belshoff A, Hart S, Plaza-Rojas L, Delos Reyes M, Prajapati K, Voelkel-Johnson C, Henry E et al (2020) Inhibition of histone deacetylase (HDAC) enhances checkpoint blockade efficacy by rendering bladder cancer cells visible for T cell-mediated destruction. Front Oncol 10:699. https://doi.org/10.3389/fonc.2020.00699
Cao Z, Kon N, Liu Y, Xu W, Wen J, Yao H, Zhang M, Wu Z, Yan X, Zhu WG, et al. (2021) An unexpected role for p53 in regulating cancer cell-intrinsic PD-1 by acetylation. Sci Adv 7 https://doi.org/10.1126/sciadv.abf4148
Yan Z, Yao S, Liu Y, Zhang J, Li P, Wang H, Chu J, Zhao S, Yao Z (2020) Durable response to sintilimab and chidamide in a patient with pegaspargase- and immunotherapy-resistant NK/T-cell lymphoma: case report and literature review. Front Oncol 10:608304. https://doi.org/10.3389/fonc.2020.608304
Chen C, Zhang W, Zhou D, Zhang Y (2021) Sintilimab and chidamide for refractory transformed diffuse large B cell lymphoma: a case report and a literature review. Front Oncol 11:757403. https://doi.org/10.3389/fonc.2021.757403
Zheng R, Chen X, Wang C, Qin P, Tan H, Luo X (2020) Triplet therapy with PD-1 blockade, histone deacetylase inhibitor, and DNA methyltransferase inhibitor achieves radiological response in refractory double-expressor diffuse large B-cell lymphoma with 17p deletion. Case Rep Hematol 2020:8879448. https://doi.org/10.1155/2020/8879448
Xu J, Xu X, Chen J, Wang J, Jiang C, Lv C, Chen B (2021) Sustained remission of multi-line relapsed extranodal NK/T-cell lymphoma, nasal type, following sintilimab and chidamide: a case report. Medicine (Baltimore) 100:e24824. https://doi.org/10.1097/MD.0000000000024824
Wang J, Gao YS, Xu K, Li XD (2022) Combination of atezolizumab and chidamide to maintain long-term remission in refractory metastatic extranodal natural killer/T-cell lymphoma: a case report. World J Clin Cases 10:1609–1616. https://doi.org/10.12998/wjcc.v10.i5.1609
Bissonnette RP, Cesario RM, Goodenow B, Shojaei F, Gillings M (2021) The epigenetic immunomodulator, HBI-8000, enhances the response and reverses resistance to checkpoint inhibitors. BMC Cancer 21:969. https://doi.org/10.1186/s12885-021-08702-x
Ye B, Shi J, Kang H, Oyebamiji O, Hill D, Yu H, Ness S, Ye F, Ping J, He J et al (2020) Advancing pan-cancer gene expression survial analysis by inclusion of non-coding RNA. RNA Biol 17:1666–1673. https://doi.org/10.1080/15476286.2019.1679585
Misso G, Zarone MR, Grimaldi A, Di Martino MT, Lombardi A, Kawasaki H, Stiuso P, Tassone P, Tagliaferri P, Caraglia M (2017) Non coding RNAs: a new avenue for the self-tailoring of blood cancer treatment. Curr Drug Targets 18:35–55. https://doi.org/10.2174/1389450117666160606104208
Esteller M (2011) Non-coding RNAs in human disease. Nat Rev Genet 12:861–874. https://doi.org/10.1038/nrg3074
Slack FJ, Chinnaiyan AM (2019) The role of non-coding RNAs in oncology. Cell 179:1033–1055. https://doi.org/10.1016/j.cell.2019.10.017
Benetatos L, Benetatou A, Vartholomatos G (2020) Long non-coding RNAs and MYC association in hematological malignancies. Ann Hematol 99:2231–2242. https://doi.org/10.1007/s00277-020-04166-4
Roisman A, Castellano G, Navarro A, Gonzalez-Farre B, Perez-Galan P, Esteve-Codina A, Dabad M, Heath S, Gut M, Bosio M et al (2019) Differential expression of long non-coding RNAs are related to proliferation and histological diversity in follicular lymphomas. Br J Haematol 184:373–383. https://doi.org/10.1111/bjh.15656
Li Y, Li G, Guo X, Yao H, Wang G, Li C (2020) Non-coding RNA in bladder cancer. Cancer Lett 485:38–44. https://doi.org/10.1016/j.canlet.2020.04.023
Fuchs S, Naderi J, Meggetto F (2019) Non-coding RNA networks in ALK-positive anaplastic-large cell lymphoma. Int J Mol Sci 20 https://doi.org/10.3390/ijms20092150
Dahl M, Kristensen LS, Gronbaek K (2018) Long non-coding RNAs guide the fine-tuning of gene regulation in B-cell development and malignancy. Int J Mol Sci 19 https://doi.org/10.3390/ijms19092475
Sole C, Larrea E, Di Pinto G, Tellaetxe M, Lawrie CH (2017) miRNAs in B-cell lymphoma: molecular mechanisms and biomarker potential. Cancer Lett 405:79–89. https://doi.org/10.1016/j.canlet.2017.07.020
Labi V, Schoeler K, Melamed D (2019) miR-17 approximately 92 in lymphocyte development and lymphomagenesis. Cancer Lett 446:73–80. https://doi.org/10.1016/j.canlet.2018.12.020
Zheng Z, Sun R, Zhao HJ, Fu D, Zhong HJ, Weng XQ, Qu B, Zhao Y, Wang L, Zhao WL (2019) MiR155 sensitized B-lymphoma cells to anti-PD-L1 antibody via PD-1/PD-L1-mediated lymphoma cell interaction with CD8+T cells. Mol Cancer 18:54. https://doi.org/10.1186/s12943-019-0977-3
Lin Y, Chen WM, Wang C, Chen XY (2017) MicroRNA profiling in peripheral T-cell lymphoma, not otherwise specified. Cancer Biomark 18:339–347. https://doi.org/10.3233/CBM-160126
Yan ZX, Wu LL, Xue K, Zhang QL, Guo Y, Romero M, Leboeuf C, Janin A, Chen SJ, Wang L et al (2014) MicroRNA187 overexpression is related to tumor progression and determines sensitivity to bortezomib in peripheral T-cell lymphoma. Leukemia 28:880–887. https://doi.org/10.1038/leu.2013.291
Xu Y, Liu Z, Lv L, Li P, Xiu B, Qian W, Liang A (2020) MiRNA-340-5p mediates the functional and infiltrative promotion of tumor-infiltrating CD8(+) T lymphocytes in human diffuse large B cell lymphoma. J Exp Clin Cancer Res 39:238. https://doi.org/10.1186/s13046-020-01752-2
Paczkowska J, Giefing M (2021) MicroRNA signature in classical Hodgkin lymphoma. J Appl Genet 62:281–288. https://doi.org/10.1007/s13353-021-00614-7
Sekar D, Hairul Islam VI, Thirugnanasambantham K, Saravanan S (2014) Relevance of miR-21 in HIV and non-HIV-related lymphomas. Tumour Biol 35:8387–8393. https://doi.org/10.1007/s13277-014-2068-9
Han BWS, Zhao H (2020) MicroRNA-21 and microRNA-155 promote the progression of Burkitt’s lymphoma by the PI3K/AKT signaling pathway. Int J Clin Exp Pathol 13:89–98
Sun R, Zhang PP, Weng XQ, Gao XD, Huang CX, Wang L, Hu XX, Xu PP, Cheng L, Jiang L et al (2022) Therapeutic targeting miR130b counteracts diffuse large B-cell lymphoma progression via OX40/OX40L-mediated interaction with Th17 cells. Signal Transduct Target Ther 7:80. https://doi.org/10.1038/s41392-022-00895-2
Chang Y, Cui M, Fu X, Zhang L, Li X, Li L, Wu J, Sun Z, Zhang X, Li Z et al (2019) MiRNA-155 regulates lymphangiogenesis in natural killer/T-cell lymphoma by targeting BRG1. Cancer Biol Ther 20:31–41. https://doi.org/10.1080/15384047.2018.1504721
Zheng X, Rui H, Liu Y, Dong J (2020) Proliferation and apoptosis of B-cell lymphoma cells under targeted regulation of FOXO3 by miR-155. Mediterr J Hematol Infect Dis 12:e2020073. https://doi.org/10.4084/MJHID.2020.073
Niu F, Dzikiewicz-Krawczyk A, Koerts J, de Jong D, Wijenberg L, Fernandez Hernandez M, Slezak-Prochazka I, Winkle M, Kooistra W, van der Sluis T, et al. (2020) MiR-378a-3p is critical for Burkitt lymphoma cell growth. Cancers (Basel) 12 https://doi.org/10.3390/cancers12123546
Wu W, Chen L, Chen C, Yu L, Zheng J (2021) miRNA-425-5p enhances diffuse large B cell lymphoma growth by targeting PTEN. Transl Cancer Res 10:4905–4913. https://doi.org/10.21037/tcr-21-2394
Wang CC, Han L, Hou YH, Ying XY (2020) MiRNA-584 suppresses the progression of NK/T-cell lymphoma by targeting FOXO1. Eur Rev Med Pharmacol Sci 24:4404–4411. https://doi.org/10.26355/eurrev_202004_21022
Lin L, Huang Y, Zhuang W, Lin P, Ma X (2020) miR-100 inhibits cell proliferation in mantle cell lymphoma by targeting mTOR. Exp Hematol Oncol 9:25. https://doi.org/10.1186/s40164-020-00182-2
Huang Y, Zou Y, Lin L, Ma X, Zheng R (2019) miR-101 regulates cell proliferation and apoptosis by targeting KDM1A in diffuse large B cell lymphoma. Cancer Manag Res 11:2739–2746. https://doi.org/10.2147/CMAR.S197744
Wei H, Liu R, Guo X, Zhou Y, Sun B, Wang J (2019) miRNA-135a regulates Hut78 cell proliferation via the GATA-3/TOX signaling pathway. Mol Med Rep 19:2361–2367. https://doi.org/10.3892/mmr.2019.9885
Gao Y, Ding X (2021) miR-145-5p exerts anti-tumor effects in diffuse large B-cell lymphoma by regulating S1PR1/STAT3/AKT pathway. Leuk Lymphoma 62:1884–1891. https://doi.org/10.1080/10428194.2021.1894642
Wang QM, Lian GY, Song Y, Peng ZD, Xu SH, Gong Y (2019) Downregulation of miR-152 contributes to DNMT1-mediated silencing of SOCS3/SHP-1 in non-Hodgkin lymphoma. Cancer Gene Ther 26:195–207. https://doi.org/10.1038/s41417-018-0057-7
Sun JR, Zhang X, Zhang Y (2019) MiR-214 prevents the progression of diffuse large B-cell lymphoma by targeting PD-L1. Cell Mol Biol Lett 24:68. https://doi.org/10.1186/s11658-019-0190-9
Tian Y, Wang L, Zhang Y, Li L, Fei Y, Zhang X, Lin G (2021) Association between miR-212-3p and SOX11, and the effects of miR-212-3p on cell proliferation and migration in mantle cell lymphoma. Oncol Lett 22:709. https://doi.org/10.3892/ol.2021.12970
Xia L, Wu L, Xia H, Bao J, Li Q, Chen X, Xia R (2019) miR-337 suppresses cutaneous T-cell lymphoma via the STAT3 pathway. Cell Cycle 18:1635–1645. https://doi.org/10.1080/15384101.2019.1629789
Xu B, Jiang L, Cui JL, Zhu XL, Bai YJ, Chen J, Diao YQ (2022) MiR-363 suppresses the tumor growth of natural killer/T-cell lymphoma via the SIRT6/PI3K/AKT axis. Ann Transl Med 10:1276. https://doi.org/10.21037/atm-22-5649
Zhou W, Xu Y, Zhang J, Zhang P, Yao Z, Yan Z, Wang H, Chu J, Yao S, Zhao S et al (2022) MiRNA-363-3p/DUSP10/JNK axis mediates chemoresistance by enhancing DNA damage repair in diffuse large B-cell lymphoma. Leukemia 36:1861–1869. https://doi.org/10.1038/s41375-022-01565-6
Jafarzadeh A, Noori M, Sarrafzadeh S, TamehriZadeh SS, Nemati M, Chatrabnous N, Jafarzadeh S, Hamblin MR, Jafari Najaf Abadi MH, Mirzaei H (2022) MicroRNA-383: a tumor suppressor miRNA in human cancer. Front Cell Dev Biol 10:955486. https://doi.org/10.3389/fcell.2022.955486
Chen LY, Han BQ, Zhang XM, Yu XB, Yao DD, Yu LQ (2021) MicroRNA-383-5p predicts favorable prognosis and inhibits the progression of diffuse large B-cell lymphoma. Oncol Lett 22:515. https://doi.org/10.3892/ol.2021.12776
Matsuda Y, Ikeda S, Abe F, Takahashi Y, Kitadate A, Takahashi N, Wakui H, Tagawa H (2022) Downregulation of miR-26 promotes invasion and metastasis via targeting interleukin-22 in cutaneous T-cell lymphoma. Cancer Sci 113:1208–1219. https://doi.org/10.1111/cas.15296
Liu Y, Li Q, Dai Y, Jiang T, Zhou Y (2020) miR-532-3p inhibits proliferation and promotes apoptosis of lymphoma cells by targeting beta-catenin. J Cancer 11:4762–4770. https://doi.org/10.7150/jca.45684
Wang Y, Guo D, Li B, Wang Y, Wang B, Wang Z, Wang M, Teng Q (2022) MiR-665 suppresses the progression of diffuse large B cell lymphoma (DLBCL) through targeting LIM and SH3 protein 1 (LASP1). Leuk Res 112:106769. https://doi.org/10.1016/j.leukres.2021.106769
Zhang MY, Wang LQ, Chim CS (2021) miR-1250-5p is a novel tumor suppressive intronic miRNA hypermethylated in non-Hodgkin’s lymphoma: novel targets with impact on ERK signaling and cell migration. Cell Commun Signal 19:62. https://doi.org/10.1186/s12964-021-00707-0
Dang W, Cao P, Yan Q, Yang L, Wang Y, Yang J, Xin S, Zhang J, Li J, Long S et al (2021) IGFBP7-AS1 is a p53-responsive long noncoding RNA downregulated by Epstein-Barr virus that contributes to viral tumorigenesis. Cancer Lett 523:135–147. https://doi.org/10.1016/j.canlet.2021.10.006
Senousy MA, El-Abd AM, Abdel-Malek RR, Rizk SM (2021) Circulating long non-coding RNAs HOTAIR, Linc-p21, GAS5 and XIST expression profiles in diffuse large B-cell lymphoma: association with R-CHOP responsiveness. Sci Rep 11:2095. https://doi.org/10.1038/s41598-021-81715-5
Zhu D, Fang C, Li X, Geng Y, Li R, Wu C, Jiang J, Wu C (2017) Predictive analysis of long non-coding RNA expression profiles in diffuse large B-cell lymphoma. Oncotarget 8:23228–23236. https://doi.org/10.18632/oncotarget.15571
Yu X, Li Z, Liu J (2015) MiRNAs in primary cutaneous lymphomas. Cell Prolif 48:271–277. https://doi.org/10.1111/cpr.12179
Zhang MY, Calin G, Deng MD, Au-Yeung RKH, Wang LQ, Chim CS (2022) Epigenetic silencing of tumor suppressor lncRNA NKILA: Implication on NF-kappaB Signaling in non-Hodgkin’s lymphoma. Genes (Basel) 13 https://doi.org/10.3390/genes13010128
Li Y, Lv Y, Wang J, Zhu X, Chen J, Zhang W, Wang C, Jiang L (2022) LncRNA NORAD mediates the proliferation and apoptosis of diffuse large-B-cell lymphoma via regulation of miR-345–3p/TRAF6 axis. Arch Med Res https://doi.org/10.1016/j.arcmed.2022.01.004
Xing X, Xu T, Liu B, Guo Q (2022) LncRNA SNHG5 can regulate the proliferation and migration of diffuse large B cell lymphoma progression via targeting miR-181-5p/XIAP. J Cancer 13:784–792. https://doi.org/10.7150/jca.60521
Zhao L, Liu Y, Zhang J, Liu Y, Qi Q (2019) LncRNA SNHG14/miR-5590-3p/ZEB1 positive feedback loop promoted diffuse large B cell lymphoma progression and immune evasion through regulating PD-1/PD-L1 checkpoint. Cell Death Dis 10:731. https://doi.org/10.1038/s41419-019-1886-5
Zhao CC, Jiao Y, Zhang YY, Ning J, Zhang YR, Xu J, Wei W, Kang-Sheng G (2019) Lnc SMAD5-AS1 as ceRNA inhibit proliferation of diffuse large B cell lymphoma via Wnt/beta-catenin pathway by sponging miR-135b-5p to elevate expression of APC. Cell Death Dis 10:252. https://doi.org/10.1038/s41419-019-1479-3
Zhao J, Su L, Jiang J (2020) Long non-coding RNA paternally expressed imprinted gene 10 (PEG10) elevates diffuse large B-cell lymphoma progression by regulating kinesin family member 2A (KIF2A) via targeting MiR-101–3p. Med Sci Monit 26:e922810. https://doi.org/10.12659/MSM.922810
Song W, Fei F, Qiao F, Weng Z, Yang Y, Cao B, Yue J, Xu J, Zheng M, Li J (2022) ALKBH5-mediated N(6)-methyladenosine modification of TRERNA1 promotes DLBCL proliferation via p21 downregulation. Cell Death Discov 8:25. https://doi.org/10.1038/s41420-022-00819-7
Li J, Chen Y, Guo X, Bai X, Xu X, Han T, Tan A, Liu N, Xia Y, Sun Q et al (2022) lncNBAT1/APOBEC3A is a mediator of HBX-induced chemoresistance in diffuse large B cell lymphoma cells. Mol Ther Nucleic Acids 27:1064–1077. https://doi.org/10.1016/j.omtn.2022.01.015
Jathar S, Kumar V, Srivastava J, Tripathi V (2017) Technological developments in lncRNA biology. Adv Exp Med Biol 1008:283–323. https://doi.org/10.1007/978-981-10-5203-3_10
Wang Y, Wang L, Sui M (2019) Long non-coding RNA H19 promotes proliferation of Hodgkin’s lymphoma via AKT pathway. J BUON 24:763–769
Fan CB, Yan XH, Tian M, Zhang S, Liu JL, Sheng YX, Dong L, Zhang WL (2020) Long non-coding RNA NEAT1 regulates Hodgkin’s lymphoma cell proliferation and invasion via miR-448 mediated regulation of DCLK1. Eur Rev Med Pharmacol Sci 24:6219–6227. https://doi.org/10.26355/eurrev_202006_21518
Wang L, Yang J, Wang HN, Fu RY, Liu XD, Piao YS, Wei LQ, Wang JW, Zhang L (2021) LncRNA BCYRN1-induced autophagy enhances asparaginase resistance in extranodal NK/T-cell lymphoma. Theranostics 11:925–940. https://doi.org/10.7150/thno.46655
Zhang H, Huang Q (2021) TP73-AS1 promotes malignant progression of NK/T cell lymphoma by regulating DKK1 methylation. J Buon 26:1530–1535
Roundtree IA, Evans ME, Pan T, He C (2017) Dynamic RNA modifications in gene expression regulation. Cell 169:1187–1200. https://doi.org/10.1016/j.cell.2017.05.045
Jonkhout N, Tran J, Smith MA, Schonrock N, Mattick JS, Novoa EM (2017) The RNA modification landscape in human disease. RNA 23:1754–1769. https://doi.org/10.1261/rna.063503.117
Barbieri I, Kouzarides T (2020) Role of RNA modifications in cancer. Nat Rev Cancer 20:303–322. https://doi.org/10.1038/s41568-020-0253-2
Zheng HX, Zhang XS, Sui N (2020) Advances in the profiling of N(6)-methyladenosine (m(6)A) modifications. Biotechnol Adv 45:107656. https://doi.org/10.1016/j.biotechadv.2020.107656
Coker H, Wei G, Brockdorff N (2019) m6A modification of non-coding RNA and the control of mammalian gene expression. Biochim Biophys Acta Gene Regul Mech 1862:310–318. https://doi.org/10.1016/j.bbagrm.2018.12.002
Zhu ZM, Huo FC, Pei DS (2020) Function and evolution of RNA N6-methyladenosine modification. Int J Biol Sci 16:1929–1940. https://doi.org/10.7150/ijbs.45231
Ma S, Chen C, Ji X, Liu J, Zhou Q, Wang G, Yuan W, Kan Q, Sun Z (2019) The interplay between m6A RNA methylation and noncoding RNA in cancer. J Hematol Oncol 12:121. https://doi.org/10.1186/s13045-019-0805-7
Han H, Fan G, Song S, Jiang Y, Qian C, Zhang W, Su Q, Xue X, Zhuang W, Li B (2021) piRNA-30473 contributes to tumorigenesis and poor prognosis by regulating m6A RNA methylation in DLBCL. Blood 137:1603–1614. https://doi.org/10.1182/blood.2019003764
Xia TL, Li X, Wang X, Zhu YJ, Zhang H, Cheng W, Chen ML, Ye Y, Li Y, Zhang A et al (2021) N(6)-methyladenosine-binding protein YTHDF1 suppresses EBV replication and promotes EBV RNA decay. EMBO Rep 22:e50128. https://doi.org/10.15252/embr.202050128
Huang H, Weng H, Zhou K, Wu T, Zhao BS, Sun M, Chen Z, Deng X, Xiao G, Auer F et al (2019) Histone H3 trimethylation at lysine 36 guides m(6)A RNA modification co-transcriptionally. Nature 567:414–419. https://doi.org/10.1038/s41586-019-1016-7
Cheng Y, Fu Y, Wang Y, Wang J (2020) The m6A methyltransferase METTL3 is functionally implicated in DLBCL development by regulating m6A modification in PEDF. Front Genet 11:955. https://doi.org/10.3389/fgene.2020.00955
Ma H, Shen L, Yang H, Gong H, Du X, Li J (2021) m6A methyltransferase Wilms’ tumor 1-associated protein facilitates cell proliferation and cisplatin resistance in NK/T cell lymphoma by regulating dual-specificity phosphatases 6 expression via m6A RNA methylation. IUBMB Life 73:108–117. https://doi.org/10.1002/iub.2410
Bohnsack KE, Hobartner C, Bohnsack MT (2019) Eukaryotic 5-methylcytosine (m(5)C) RNA methyltransferases: mechanisms, cellular functions, and links to disease. Genes (Basel) 10 https://doi.org/10.3390/genes10020102
Jeltsch A, Ehrenhofer-Murray A, Jurkowski TP, Lyko F, Reuter G, Ankri S, Nellen W, Schaefer M, Helm M (2017) Mechanism and biological role of Dnmt2 in nucleic acid methylation. RNA Biol 14:1108–1123. https://doi.org/10.1080/15476286.2016.1191737
Li Q, Li X, Tang H, Jiang B, Dou Y, Gorospe M, Wang W (2017) NSUN2-mediated m5C methylation and METTL3/METTL14-mediated m6A methylation cooperatively enhance p21 translation. J Cell Biochem 118:2587–2598. https://doi.org/10.1002/jcb.25957
Chellamuthu A, Gray SG (2020) The RNA methyltransferase NSUN2 and its potential roles in cancer. Cells 9 https://doi.org/10.3390/cells9081758
Okamoto M, Fujiwara M, Hori M, Okada K, Yazama F, Konishi H, Xiao Y, Qi G, Shimamoto F, Ota T et al (2014) tRNA modifying enzymes, NSUN2 and METTL1, determine sensitivity to 5-fluorouracil in HeLa cells. PLoS Genet 10:e1004639. https://doi.org/10.1371/journal.pgen.1004639
Acknowledgements
The work was supported by the Department of Hematology of Linyi People’s Hospital and Weifang Medical University, and I would like to show great gratitude to them all.
Funding
This study was supported by Shandong Provincial Postdoctoral Innovation Project (201903077) and Shandong Provincial Natural Science Foundation (ZR2018PH014).
Author information
Authors and Affiliations
Contributions
All authors searched for the literature and wrote and edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhuang, S., Yang, Z., Cui, Z. et al. Epigenetic alterations and advancement of lymphoma treatment. Ann Hematol 103, 1435–1454 (2024). https://doi.org/10.1007/s00277-023-05395-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-023-05395-z