
Abstract
Non-Hodgkin lymphoma is a diverse group of lymphocyte-derived neoplasms. Although a heterogeneous group of malignancies, it has become apparent that epigenetic alterations, such as disturbances of DNA methylation and histone modification, are a common occurrence in both B cell and T cell lymphomas, contributing to lymphomagenesis. As a result, the use of epigenetic targeted therapy has been incorporated into various pre-clinical and clinical studies, demonstrating significant efficacy in lymphoma, with vorinostat becoming the first epigenetic therapy to receive FDA approval in any malignancy. The role of epigenetic drugs is evolving, with its potential use in combination therapy as well as a means of overcoming chemotherapy resistance. In this review, we discuss the epigenetic alterations in non-Hodgkin lymphomas as well as provide an overview of current epigenetic drugs and their role in clinical practice, and on-going clinical trials.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The term epigenetics refers to the reversible modification of gene expression independent of DNA sequence. This concept of an “epigenetic landscape” was first introduced by C.H. Waddington in the late 1930s to describe the interactions of genes and their cellular environment to produce a phenotype [1]. Normal human cells depend on epigenetic modifications to control gene activity and nuclear architecture. For instance, X-chromosome inactivation in females and genomic imprinting of genes are dependent upon DNA hypermethylation and histone modification [2, 3]. Furthermore, hypermethylation of repetitive genomic sequences (CpG islands) provides chromosomal stability by preventing translocations and reactivation of transposable elements [4].
Epigenetic alterations are complex and dependent on several biochemical mechanisms including: (1) acetylation and deacetylation of histones catalyzed by histone acetyltransferases (HATs) and histone deacetylases (HDACs), respectively; (2) post translational modifications including methylation, phosphorylation and ubiquination of histones executed by histone methylases, kinases, and ubiquitin ligases, respectively; (3) DNA methylation carried out by DNA methyltransferases; and (4) the activity of non-coding RNAs, such as micro-RNAs. The subversion of these epigenetic processes contributes to and drives various malignancies through the silencing of tumor-suppressor genes, DNA repair proteins, and cell cycle control enzymes (Fig. 1) [5, 6].

Epigenetic modifications influence gene expression. Histone and DNA modification influences transcriptional state. Silencing of tumor-suppressor genes, DNA repair proteins, and cell cycle control enzymes contributes to lymphomagenesis. Methylation of CpG islands and histones tails by DMNT and EZH2 or MLL, respectively, lead to a transcriptionally repressed state. Histone acetylation leads to transcriptional activation as it disrupts histone–DNA interaction, exposing DNA to transcription factors. Epigenetic drugs, such as DNMT inhibitors, EZH2 inhibitors, and HDAC inhibitors lead to a transcriptionally active state and can induce the re-expression of previously repressed genes, such as tumor suppressors
In general, histone acetylation is associated with transcriptional activation as it causes the disruption of histone–DNA interaction [7, 8]. Acetylation of the lysine tails of histone 3 and 4 (H3 and H4) induces chromatin decondensation, allowing access of transcription factors to the DNA, and thus transcriptional activation. Based on several studies, HAT proteins, specifically CBP and p300, have been implicated as tumor suppressors in B cell lymphomas. CBP and p300 function as co-activators of transcription factors and acetylate various proteins including p53, HSP90, and NFƙB [9–11]. P300 is a direct target of BCL6, an essential oncoprotein, driving various B cell lymphomagenesis. In vivo and in vitro use of BCL6 inhibitors in diffuse large B cell lymphoma (DLBCL) can stimulate p300 protein function, leading to activation of tumor-suppressor activity [11]. Lymphoma cells with mutant p300 are resistant to BCL6 inhibitors, further supporting the role of p300 in preserving normal cell function [11].
The role of HDACs in cancer biology has become a critical area of research, leading to the incorporation of HDAC inhibitors into new treatment paradigms for patients with lymphoma. HDACs are a large family of chromatin-modifying proteins that remove acetyl groups from lysine residues on histones, which enhances chromatin condensation (or compaction) and leads to transcriptional repression. HDACs can be classified into four distinct classes based on homology to yeast counterparts. Class I (HDAC1, HDAC2, HDAC3, and HDAC8), class IIa (HDAC4, HDAC5, HDAC 7, and HDAC9), class IIb (HDAC6 and HDAC10), and class IV (HDAC11) HDACs which contain zinc-dependent deaceytlase domains. The sirtuin family, also known as the class III histone proteins, is made up of seven members, each having distinct cellular localizations and targets. The sirtuins are dependent on NAD+ as a co-factor to carry out deacetylase activity [12]. Although there is overlap between the HDAC classes, in general, each class is found in distinct compartments of the cell and is involved in different cellular processes [13, 14].
Aberrancy in histone methyltransferases, such as EZH2 and MLL, are thought to play a role in the pathogenesis of follicular lymphoma (FL) and DLBCL [15, 16]. Loss of function mutations in MLL are seen in 89 % of FLs and 32 % of DLBCLs [16], while mutations in EZH2 are found in 7–12 % of FLs, and 22 % of Germinal Center (GC) DLBCLs [17, 18]. EZH2 is a histone methyltransferase and is a component of polycomb repression complex 2 (PCR2), which is responsible for tri-methylation of histone 3 lysine 27 (H3K27) [19]. In early B cell development, EZH2 is required for VDJ recombination and is then subsequently down-regulated in mature B cells [20]. Similar to BCL6, EZH2 activity is highly expressed in actively proliferating germinal center B cells, with a direct influence on genes that are involved in differentiation and inhibitors of cell growth and proliferation, such as CDKN1A, CDKN1B, and CDKN2 [15]. Activating mutations of EZH2 promotes tri-methylation of H3K27, and in turn, leads to the inhibition of tumor-suppressor genes, promoting lymphomagenesis.
Hypermethylation of histone 3 lysine 4 (H3K4) opposes the actions of EZH2. Methylation of H3K4 is partially mediated by the MLL family of histone methyltransferases. MLL also forms a complex with UTX, which is an H3K27 demethylase [21]. Another important function of MLL involves DNA damage surveillance during S-phase, as inactivating mutations of MLL lead to DNA damage-independent DNA synthesis and tolerance of genetic instability [22]. Together, mutations in EZH2 and MLL have illustrated the significance of abnormal epigenetic activity underlying lymphomagenesis, particularly those that are GC derived.
DNA methylation serves an essential role in gene activity and transcription. DNA methylation occurs at CpG-rich regions (CpG islands), which are located at the 5′ regulatory regions of many genes [6]. Both hypomethylation and hypermethylation have been linked to the pathogenesis of multiple cancers [23]. For example, CpG hypermethylation of the promoter region for CDKN2A/p16 has been found to be associated with more aggressive forms of non-Hodgkin lymphoma (NHL) [24].
Interestingly, mutations of epigenetic genes in angioimmunoblastic T cell lymphoma (AITL) and peripheral T cell lymphomas, not otherwise specified (PTCL-NOS) have centered on pathways that result in abnormal DNA methylation. Mutations in TET2, IDH2, and DNMT3A have been identified in both PTCL-NOS and AITL [25–28]. Isocitrate dehydrogenase 2 (IDH2) is responsible for the conversion of isocitrate to alpha-ketoglutarate. The mutated form of IDH2 results in the inadvertent production of 2-hydroxyglutarate (2HG), which inhibits TET2, a DNA methylase. Therefore, mutations in IDH2 and TET2 cause DNA hypermethylation and subsequent gene silencing [29, 30]. Additionally, 2HG prevents the action of lysine-specific demethylases, thereby inducing additional histone methylation [29]. Inhibitors of IDH are now in early clinical development (NCT02071862, NCT02364206) [31, 32].
Due to the robust link between epigenetic modification and carcinogenesis, multiple drug targets, including HDAC inhibitors, DNA methylation inhibitors (DNMTi) and EZH2 inhibitors have shown efficacy in both pre-clinical and clinical studies, with T cell lymphomas (TCL) being the first lymphoma subtype for which an epigenetic therapy has been FDA approved.
Drugs Targeting Epigenetic Operations
HDAC Inhibitors
C.W. Friend, renowned for the discovery that a virus (Friend leukemia virus) could cause erythroleukemia, serendipitously observed that adding DMSO to erythroleukemic cells induced differentiation into mature erythrocytes [33, 34]. For a long period of time, the mechanism of DMSO stimulating differentiation of leukemic cells was not well understood; however, the accumulation of hyperacetylated histones was prominently observed and thought to be linked. It was not until 1996 when Taunton and colleagues developed an assay that identified HDACs as the targets of DMSO that the etiology became clear [35].
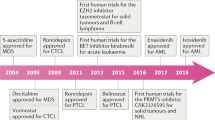
The first successful application of epigenetic targeting was realized with HDAC inhibitors in relapsed/refractory TCL [36–44]. HDAC inhibitors are thought to exert their therapeutic action by modulating gene expression, inducing cell differentiation, apoptosis, and cell cycle control [45, 46]. Vorinostat, also known as suberoylanilide hydroxamic acid (SAHA), is an orally available HDAC inhibitor with activity against class I and class II HDACs [43, 44]. In a panel of cutaneous T cell lymphoma (CTCL) cell lines, exposure to vorinostat led to acetylation of histones, BAX, STAT6, and caspase-3, and triggered higher rates of apoptosis in peripheral blood mononuclear cells isolated from CTCL patients as compared to healthy donors [47]. Vorinostat was first approved in CTCL after phase II and phase IIa studies demonstrated an objective response rate (ORR) of 24.2 and 29.5 %, respectively, with a time to progression (TTP) of 12 to 20 weeks [36, 37]. It was the first HDAC inhibitor approved for the treatment of any malignancy.
Vorinostat has also shown efficacy in other subtypes of lymphoma including FL and marginal zone lymphoma (MZL). In a phase II trial of patients with relapsed/refractory FL, MZL, and mantle cell lymphoma (MCL), the ORR was 29 %, with a complete response (CR) of 14.5 % after treatment with vorinostat. Upon further subgroup analysis, the response rates for FL and MZL were 47 and 22 %, respectively, with no responders among the MCL patients [48].
In a phase II clinical trial, vorinostat was used to treat 18 patients with relapsed DLBCL and demonstrated limited single agent activity (1 CR, 2 stable disease) [49]. However, Amengual and colleagues demonstrated that the combination of niacinamide, a sirtuin inhibitor, and pan-HDAC inhibitors (romidepsin, vorinostat, belinostat or panobinostat) led to synergistic cytoxicity of GC-derived DLBCL as demonstrated by increased acetylation of BCL6 and p53, and modulation of downstream targets of p21 and Blimp1 [50]. Extrapolating from these pre-clinical studies, a phase I clinical study utilizing combination therapy of vorinostat and niacinamide in heavily treated patients with relapsed/refractory lymphoma demonstrated an ORR of 24 %, with two patients achieving CRs and three patients with partial responses (PRs), suggesting a potential role of combination HDAC inhibitor therapy in DLBCL.
Since this development, other HDAC inhibitors have also been approved for the treatment of T cell lymphomas (Table 1). Romidepsin has been FDA approved for both relapsed CTCL and PTCL, and most recently, belinostat for PTCL [38–42]. Romidepsin is a cyclic peptide that has activity against class I–III HDACs. The activity of romidepsin in CTCL was first observed during a NCI Phase I clinical trial [51]. Given the promising results in CTCL, romidepsin was studied in two separate phase II multi-institutional clinical trials, one sponsored by the NCI and the other by Gloucester Pharmaceuticals (GPI) [38, 39]. Both studies demonstrated an ORR of approximately 35 %. Time to progression (TTP) in the GPI trial varied from 5.9 months, for patients who achieved stable disease, to 15.1 months, for those who achieved a major response (complete or partial responses), while the NCI study demonstrated a TTP of 8 months.
The NCI 1312 study analyzed the use of romidepsin in both CTCL and PTCL-NOS showing a CR rate of 18 % and PR rate of 20 % in patients with relapsed PTCL-NOS [40]. A separate phase II trial sponsored by GPI confirmed the activity observed in the NCI trial [41]. These studies led to accelerated FDA approval of romidepsin in PTCL.
Like vorinostat, belinostat is a derivative of hydroxyamic acid and inhibits both class I and class II HDACs. In the BELIEF trial, patients with relapsed/refractory PTCL received belinostat at 1,000 mg/m2 D1–5 on a 21-day cycle. The ORR was 26 %, with a median duration of response of >12 months [42]. Notably, patients with baseline thrombocytopenia tolerated belinostat with 98 % dose intensity and a low incidence of myelosuppression. Given the low incidence of myelosuppression, possible combination therapy with belinostat and traditional cytotoxic chemotherapy may prove to be more feasible compared to romidepsin and could serve a role in combination with regimens like CHOP for patients with PTCL.
The efficacy of the three approved HDAC inhibitors in TCL suggests that class I HDAC dysfunction plays a crucial role in pathogenesis of this disease. Additionally, the use of HDAC inhibitors has also been studied in other lymphoma subtypes and other hematological malignancies, leading to the recent FDA approval of panobinostat for Multiple Myeloma [52]. Given the success of single-agent HDAC inhibitors in relapsed/refractory TCL, further clinical investigation of HDAC inhibitors in combination therapy as well as in the first-line setting is warranted in not only TCL, but other hematological malignancies.
DNA Methylation Inhibitors
Azacitidine and decitabine are nucleoside analogues that are incorporated into DNA and act as hypomethylating agents by inhibiting DNA methyltransferases. Additionally, azacitidine is also integrated during RNA synthesis causing the disassembly of polyribosomes, and in turn, inhibition of translation [53]. The actions of azacitidine and decitabine are dose-dependent. At low doses, the desired effect of DNA methyltransferase inhibition occurs, whereas at higher concentrations cytotoxic effects including DNA damaging properties predominate often leading to myelosuppression [54]. Both drugs are FDA approved for the treatment of myelodysplastic syndromes [55, 56] and are currently under investigation in acute myeloid leukemia (AML) and lymphoma.
Several phase I studies with decitabine have been performed in lymphoma patients. In a study of patients with relapse/refractory small lymphocytic lymphoma (SLL), DLBCL, and MCL, dose-limiting myelosuppression and infectious complications prevented any significant change in DNA methylation levels and gene expression in tumor samples [57]. Among 20 patients, 8 patients experienced stable disease after one to two cycles of decitabine, with the remaining 12 patients progressing after a median of two to three cycles. However, in a study including both solid tumors and relapsed CTCL, treatment with decitabine decreased tumor global DNA methylation (percent of methylated CpG islands) by a median relative reduction of 6 %, with a median 12 % decrease in methylation at the highest dose tested (100 mg/m2/cycle). Reduction in tumor DNA methylation was seen at all decitabine dose levels, with relative methylation decreases of 6, 3, and 2.5 %, with 25, 50, and 75 mg/m2/cycle, respectively. To put into context, patients who had relapsed in <3 months were noted to have 55.2 % methylation of CpG islands as compared to 39.6 % methylation in patients who relapsed > 3 months (16 % difference). The reduction of global DNA methylation observed by treatment with decitabine may hypothetically reduce the risk of drug resistance [58]. At the first planned evaluation, 17 of 28 patients had stable disease, with one PR (thymoma) and three minor responses including one patient with CTCL. The median TTP was 7.1 months (range 1–7 months). Although a small study, these data support the notion that lower doses of DNMT inhibitors promote DNA hypomethylation without significant myelosuppression, and may be most useful in future combination strategies.
The combination of DNMT and HDAC inhibitors represents an area of increasing attention in the development of epigenetic therapies. Pre-clinical studies have demonstrated that histone deacetylation and DNA methylation are linked, contributing to the transcriptional inactivation of tumor-suppressor genes, such as hMLH-1, p57, CDKN2A, and CDKN2B [59, 60]. Moreover, the in vitro combination of a DNMT inhibitor with a HDAC inhibitor in hematologic and solid tumor cell lines have shown synergistic effects resulting in increased anti-tumor activity [59, 60]. Compared to their single-agent activity, combination therapy with decitabine and valproic acid, a HDAC inhibitor, enhanced apoptotic activity in leukemia cells [61]. In a phase I trial involving patients with heavily treated solid tumors and NHL, the use of decitabine and vorinostat together led to stable disease that lasted a median time of 4 months [62]. Notably, in this study, the majority of patients had a diagnosis of a solid tumor malignancy, while only four patients had a diagnosis of NHL (MCL, DLBCL, SLL, and anaplastic large cell lymphoma). Several other phase I studies performed in solid tumor and myeloid malignancies have demonstrated reversal of epigenetic markers when using a combination of DNMT inhibitors and HDAC inhibitors [63–65], and as discussed below, recent pre-clinical data also suggests a potential role in DLBCL and TCL [66••, 67].
EZH2 Inhibitors
Gain of function mutations in EZH2 have been linked to the downregulation of tumor-suppressor genes, which in turn, allows for the emergence of genetic mutations and lymphomagenesis [15, 17, 68–72]. Pre-clinical studies have shown that EZH2 is down-regulated in AML cells following treatment with panobinostat. This inhibition is enhanced in conjunction with 3-deazaneplanocin A, an EZH2 inhibitor [73]. Given the fact that GC-derived lymphomas, such as DLBCL and FL, often possess EZH2 mutations, EZH2 may represent a useful treatment target. At this time, EZH2 inhibitors are in development and are actively being incorporated into various clinical trials (NCT02395601) for the treatment of lymphoma [74].
Drug Resistance
As our understanding about the complexities of epigenetics grows, it is now known that epigenetic changes, such as methylation of CpG islands and mutations in HATs, may be occurring at a higher rate and influencing a greater set of pathways than the direct mutations of tumor-suppressor genes themselves. These mutations in epigenetic modulators have a global influence on both lymphomagenesis and drug sensitivity [75]. This likely occurs through modulation of a large scale of genes as opposed to the single gene rearrangement or mutation altering a single oncogene or tumor suppressor. The ability to silence multiple genes simultaneously leads to “polygenic drug resistance,” [76]. These epigenetic alterations, such as aberrant CpG methylation, are non-random, and have a tendency to occur repeatedly at specific promoter regions after repeated exposure to chemotherapy.
There are several examples of tumor-suppressor genes that have been identified as having hypermethylated CpG islands (Table 2) [6, 77–82]. Some well-characterized hypermethylated tumor suppressors include hMLH1, Apaf-1, p16, p73, Caspase 8, and SMAD1. As an example, Apaf-1 is a co-factor for Caspase-9 and leads to p53 induced apoptosis. Soengas et al. found that p53 is infrequently mutated in melanoma, but when apparent leads to a disease state that is refractory to chemotherapeutic agents [83]. Downstream targets of p53 were evaluated and Apaf-1 was discovered to have decreased expression by mRNA and protein levels. It was found that Apaf-1 was commonly mutated, but that this occurred exclusively in a mono-allelic fashion. After further investigation, it was elucidated that epigenetic inactivation occurred through methylation of enhancer elements of Apaf-1, and that expression of Apaf-1 could be restored following treatment with azacitidine. Similar findings were demonstrated in Burkitts lymphoma where Apaf-1 levels were found to be very low despite normal genetic structures [84]. It was found that methylation of CpG islands between +87 and +128 of the promoter regions was found exclusively in the cell lines with low Apaf-1 expression and correlated to over-expression of DNMT1. Treatment with azacitidine led to a reversal of these findings and restoration of Apaf-1 expression rendering these cells more sensitive to chemotherapeutic agents.
Additionally, genes may be silenced at the histone level. Tri-methylation of histone tails via histone methyltransferases (HMTs) leads to enforcement of chromatin in the condensed state. Interestingly, di-methylation can lead to both a permissive and repressed chromatin state. This dynamic process is altered not only by the “number” of methylation marks but also by other post-translational modifications such as acetylation, phosphorylation, ubiquitination, and sumolyation. Competitive acetylation at identical histone moieties such as H3K9 leads to a permissive chromatin state, displacing the possibility of methylation of a specific moiety, a modification leading to a repressed state [85]. Most lysines cannot accommodate both acetylation and methylation simultaneously as acetylation and methylation often lead to opposing effects. Enzymes leading to the acetylation and methylation of histones are often found in complexes creating a direct cross-talk between these states.
Given our understanding that hypermethylated CpG islands and methylated histone tails lead to a repressed chromatin state and that acetylation of these same sites in a competitive manner leads to a permissive chromatin state, treatment with a combination of HMTs, DNMT inhibitors, and HDAC inhibitors may have synergistic effects on re-sensitizing drug resistant malignancies.
Kalac and colleagues treated a panel of DLBCL cell lines with decitabine in combination with four separate pan-class HDAC inhibitors (panobinostat, belinostat, romidepsin, and vorinostat) and demonstrated synergy between the two classes of agents across six different cell lines [66••]. These findings were validated with a xenograft mouse model of DLBCL where the combination of decitabine plus panobinostat led to marked tumor growth delay compared to either agent alone. The combination also led to unique effects on gene expression and gene-specific CpG methylation as measured by bisulfite sequencing. In particular, it was found that panobinostat led to an increase in the gene expression of SMAD1 and DNMT3A. SMAD1 plays a role in differentiation, proliferation, and apoptosis, as well as chemotherapy-induced senescence. The finding of increased DNMT3A expression may explain the synergy demonstrated by adding a DNMT inhibitor. Verifying these findings, Clozel et al. demonstrated that DNA hypermethylation of SMAD1 contributed to doxorubicin resistance in DLBCL cell lines. However, resistance was overcome with a 5-day pre-treatment with decitabine. Extrapolating from this information, a small phase I study using azacitidine pre-treatment followed by standard R-CHOP demonstrated increased induction of apoptosis, a decrease in methylation markers, and 11/12 patients with adverse features achieved CRs [86••]. This collective data suggests a new a role for DNMT inhibitors in the treatment of DLBCL.
Similarly, the combination of HDAC and DNMT inhibition has been studied in the context of T cell lymphomas. O’Connor et al. evaluated the interaction of decitabine and romidepsin on modulation of gene expression and methylation array [67]. It was demonstrated that the combination led to increased number of modulated genes from 138 when treated with romidepsin alone, to 390 genes in the combination. In particular, there was significant up-regulation of cell cycle check points and a down-regulation of genes involved in biosynthetic pathways. Surprisingly, there was a decrease in the number of demethylated genes in the combination compared to cells treated with decitabine alone. The pattern of repressed to expressed genes in the T cell lymphoma cell line was completely reversed following treatment with both decitabine and romidepsin.
Based on these findings, a study of oral azacitidine in combination with romidepsin for relapsed refractory lymphoma is currently underway (NCT01998035). Although the primary objective of this phase I study is to assess safety, strong correlative studies will help to determine the effects of this combination on modulating gene expression and methylation pattern changes in these patients with chemotherapy resistant lymphomas. Different mechanisms for overcoming chemotherapy resistance may emerge for B cell lymphomas and TCL using this strategy.
Although PTCLs are heterogeneous, as a group, they are consistently very chemotherapy resistant with only 10–15 % of patients experiencing long-term survival after treatment with standard CHOP-based therapy. As treatment for this group of diseases evolves, HDAC inhibitors have emerged as one of the most active classes of drugs. Of the few other classes of drugs approved for this disease, pralatrexate also offers benefit leading to nearly 30 % response rate in heavily treated, relapsed PTCL patients. Jain et al. studied the combination of these two drugs in preclinical models of this disease and found striking synergy [87••]. Although mechanisms for synergy were evaluated, no change in RFC (reduced folate carrier) or FPGS (folylpolyglutamate synthase) was seen which are known to influence internalization of pralatrexate into cells. Treatment of a novel TCL mouse model with the combination led to complete responses in all mice in the combination arm, but none in the mice treated with romidepsin or pralatrexate alone. The combination was well tolerated and also led to a marked increased and durable survival compared to mice treated with single agents. These findings have been translated into a phase I/II clinical trial (NCT01947140) where early signals of response have been very promising in patients with refractory T cell lymphoma.
In keeping with this strategy, romidepsin has been combined with a multitude of agents, ranging from combination chemotherapy such as CHOP and ICE to other novel agents such as alisertib, a novel aurora A kinase inhibitor known to have activity in TCL with a response rate nearing 30 %. Zullo et al. analyzed the combination of romidepsin and alisertib across a large panel of lymphoma cell lines and found synergy exclusively in TCL by induction of polyploidy and failure of cytokinesis [88]. Again, the findings of this study have contributed to the development of a phase I study evaluating romidepsin plus alisertib in patients with refractory lymphoma (NCT01897012).
This approach has been taken with solid organ malignancies as well. Sharma et al. found that NSCLC exposed to EGFR TKIs eventually became resistant to these drugs [89]. Although this finding is not surprising, the mechanism by which resistance was conferred was not through direct genetic mutation of the drug target or efflux pumps, but rather through an altered chromatin state that was modified by the histone demethylase RBP2/KDM5A/Jarid1A as identified by gene expression profiling. Interestingly, this resistant phenotype could be rescued by chromatin-modifying drugs such as the HDAC inhibitors TSA, SAHA, MS-275, and Scriptaid which when given together with an EGFR TKI led to rapid cell kill and prevented emergence of new resistant clones. These findings were translated into a phase I clinical trial of panobinostat in combination with erlotinib for patients with advanced aerodigestive tract tumors [90]. In this study, 42 patients with refractory NSCLC and head-and-neck cancer were enrolled leading to 9 % ORR and an additional 42 % of patients achieved stable disease. Although perhaps underwhelming, these results were demonstrated in a heavily pretreated population and in a disease state not known to be inherently sensitive to HDAC inhibition, suggesting a broader applicability of this strategy.
Conclusion
The identification of mutations affecting epigenetic and transcriptional modifiers appears to be a driving force in both B cell and T cell lymphomas. Disruptions in DNA methylation and histone modification have emerged as hallmarks of these diseases, and have served as a foundation for epigenetic targeted therapy. With the use of next-generation sequencing, individualized approaches to therapy may arise based on unique expression patterns leading to specific molecular phenotypes. Because epigenetic modifications are potentially reversible, the development of epigenetic therapy alone as well in combination with traditional treatment regimens is promising. This may ultimately lead to treatment with less toxicity and increased tolerability. Another exciting potential use of epigenetic modification is to sensitize malignant cells to traditional chemotherapy agents, which may lead to lower doses of chemotherapeutic agents with similar cytotoxic effect. Future and on-going trials are currently evaluating various combinations of epigenetic therapies together as well as in combination with traditional chemotherapy agents.
References
Papers of particular interest, published recently, have been highlighted as: •• Of major importance
Waddington CH. Preliminary notes on the development of the wings in normal and mutant strains of drosophila. Proc Natl Acad Sci U S A. 1939;25(7):299–307.
Feinberg AP, Cui H, Ohlsson R. DNA methylation and genomic imprinting: insights from cancer into epigenetic mechanisms. Semin Cancer Biol. 2002;12(5):389–98.
Reik W, Lewis A. Co-evolution of X-chromosome inactivation and imprinting in mammals. Nat Rev Genet. 2005;6(5):403–10.
Bestor TH. Transposons reanimated in mice. Cell. 2005;122(3):322–5.
Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150(1):12–27.
Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349(21):2042–54.
Mack GS. Epigenetic cancer therapy makes headway. J Natl Cancer Inst. 2006;98(20):1443–4.
Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128(4):669–81.
Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90(4):595–606.
Rothgiesser KM, Fey M, Hottiger MO. Acetylation of p65 at lysine 314 is important for late NF-kappaB-dependent gene expression. BMC Genomics. 2010;11:22.
Cerchietti LC, et al. BCL6 repression of EP300 in human diffuse large B cell lymphoma cells provides a basis for rational combinatorial therapy. J Clin Invest. 2010.
Smith BC, Hallows WC, Denu JM. Mechanisms and molecular probes of sirtuins. Chem Biol. 2008;15(10):1002–13.
Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6(1):38–51.
Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 2007;26(37):5310–8.
Velichutina I et al. EZH2-mediated epigenetic silencing in germinal center B cells contributes to proliferation and lymphomagenesis. Blood. 2010;116(24):5247–55.
Morin RD et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476(7360):298–303.
Morin RD et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42(2):181–5.
Bodor C et al. EZH2 Y641 mutations in follicular lymphoma. Leukemia. 2011;25(4):726–9.
Bracken AP, Helin K. Polycomb group proteins: navigators of lineage pathways led astray in cancer. Nat Rev Cancer. 2009;9(11):773–84.
Su IH et al. Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat Immunol. 2003;4(2):124–31.
Chi P, Allis CD, Wang GG. Covalent histone modifications—miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer. 2010;10(7):457–69.
Liu H et al. Phosphorylation of MLL by ATR is required for execution of mammalian S-phase checkpoint. Nature. 2010;467(7313):343–6.
Esteller M et al. A gene hypermethylation profile of human cancer. Cancer Res. 2001;61(8):3225–9.
Martinez-Delgado B et al. Hypermethylation of a 5′ CpG island of p16 is a frequent event in non-Hodgkin’s lymphoma. Leukemia. 1997;11(3):425–8.
Odejide O et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood. 2014;123(9):1293–6.
Cairns RA et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood. 2012;119(8):1901–3.
Lemonnier F et al. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood. 2012;120(7):1466–9.
Couronne L, Bastard C, Bernard OA. TET2 and DNMT3A mutations in human T-cell lymphoma. N Engl J Med. 2012;366(1):95–6.
Losman JA, Kaelin Jr WG. What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013;27(8):836–52.
Losman JA et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339(6127):1621–5.
Rohle D et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340(6132):626–30.
Davis MI et al. Biochemical, cellular, and biophysical characterization of a potent inhibitor of mutant isocitrate dehydrogenase IDH1. J Biol Chem. 2014;289(20):13717–25.
Friend C et al. Hemoglobin synthesis in murine virus-induced leukemic cells in vitro: stimulation of erythroid differentiation by dimethyl sulfoxide. Proc Natl Acad Sci U S A. 1971;68(2):378–82.
O’Connor OA. For disease in need, a friend indeed. Blood. 2011;117(22):5787–8.
Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science. 1996;272(5260):408–11.
Duvic M et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood. 2007;109(1):31–9.
Olsen EA et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25(21):3109–15.
Piekarz RL et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol. 2009;27(32):5410–7.
Whittaker SJ et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol. 2010;28(29):4485–91.
Piekarz RL et al. Phase 2 trial of romidepsin in patients with peripheral T-cell lymphoma. Blood. 2011;117(22):5827–34.
Coiffier B et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. 2012;30(6):631–6.
O’Connor OA MT, Savage KJ, Pinter-Brown LC, Foss FM, Popplewell L, et al. Belinostat, a novel pan-histone deacetylase inhibitor (HDACi), in relapsed or refractory peripheral T-cell lymphoma (R/RPTCL): results from the BELIEF trial. J Clin Oncol. 2013;31 (suppl 1145; abstr 8507).
Kelly WK et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J Clin Oncol. 2005;23(17):3923–31.
Kelly WK et al. Phase I clinical trial of histone deacetylase inhibitor: suberoylanilide hydroxamic acid administered intravenously. Clin Cancer Res. 2003;9(10 Pt 1):3578–88.
Bates SE et al. Laboratory correlates for a phase II trial of romidepsin in cutaneous and peripheral T-cell lymphoma. Br J Haematol. 2010;148(2):256–67.
Sandor V et al. P21-dependent g(1)arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br J Cancer. 2000;83(6):817–25.
Zhang C et al. Selective induction of apoptosis by histone deacetylase inhibitor SAHA in cutaneous T-cell lymphoma cells: relevance to mechanism of therapeutic action. J Invest Dermatol. 2005;125(5):1045–52.
Kirschbaum M et al. Phase II study of vorinostat for treatment of relapsed or refractory indolent non-Hodgkin’s lymphoma and mantle cell lymphoma. J Clin Oncol. 2011;29(9):1198–203.
Crump M et al. Phase II trial of oral vorinostat (suberoylanilide hydroxamic acid) in relapsed diffuse large-B-cell lymphoma. Ann Oncol. 2008;19(5):964–9.
Amengual JE et al. Sirtuin and pan-class I/II deacetylase (DAC) inhibition is synergistic in preclinical models and clinical studies of lymphoma. Blood. 2013;122(12):2104–13.
Sandor V et al. Phase I trial of the histone deacetylase inhibitor, depsipeptide (FR901228, NSC 630176), in patients with refractory neoplasms. Clin Cancer Res. 2002;8(3):718–28.
San-Miguel JF et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014;15(11):1195–206.
Lee TT, Karon MR. Inhibition of protein synthesis in 5-azacytidine-treated HeLa cells. Biochem Pharmacol. 1976;25(15):1737–42.
Santi DV, Garrett CE, Barr PJ. On the mechanism of inhibition of DNA-cytosine methyltransferases by cytosine analogs. Cell. 1983;33(1):9–10.
Fenaux P et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10(3):223–32.
Lubbert M et al. Low-dose decitabine versus best supportive care in elderly patients with intermediate- or high-risk myelodysplastic syndrome (MDS) ineligible for intensive chemotherapy: final results of the randomized phase III study of the European Organisation for Research and Treatment of Cancer Leukemia Group and the German MDS Study Group. J Clin Oncol. 2011;29(15):1987–96.
Blum KA et al. Phase I trial of low dose decitabine targeting DNA hypermethylation in patients with chronic lymphocytic leukaemia and non-Hodgkin lymphoma: dose-limiting myelosuppression without evidence of DNA hypomethylation. Br J Haematol. 2010;150(2):189–95.
Stewart DJ et al. Decitabine effect on tumor global DNA methylation and other parameters in a phase I trial in refractory solid tumors and lymphomas. Clin Cancer Res. 2009;15(11):3881–8.
Cameron EE et al. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21(1):103–7.
Kikuchi T et al. Inactivation of p57KIP2 by regional promoter hypermethylation and histone deacetylation in human tumors. Oncogene. 2002;21(17):2741–9.
Yang H et al. Antileukemia activity of the combination of 5-aza-2′-deoxycytidine with valproic acid. Leuk Res. 2005;29(7):739–48.
Stathis A et al. Phase I study of decitabine in combination with vorinostat in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Clin Cancer Res. 2011;17(6):1582–90.
Garcia-Manero G et al. Phase 1/2 study of the combination of 5-aza-2′-deoxycytidine with valproic acid in patients with leukemia. Blood. 2006;108(10):3271–9.
Braiteh F et al. Phase I study of epigenetic modulation with 5-azacytidine and valproic acid in patients with advanced cancers. Clin Cancer Res. 2008;14(19):6296–301.
Gore SD et al. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer Res. 2006;66(12):6361–9.
Kalac M et al. HDAC inhibitors and decitabine are highly synergistic and associated with unique gene-expression and epigenetic profiles in models of DLBCL. Blood. 2011;118(20):5506–16. Compared to single agent activity, the use of hypomethylating agents in combination with HDAC inhibitors in DLBCL models demonstrated significant tumor growth inhibition and apoptosis with a unique gene expression after treatment. This data suggests a potential role of combinational epigenetic therapy in DLBCL.
O’Connor OA et al. The combination of hypomethylating agents and histone deacetylase inhibitors (HDACi) are synergistically cytotoxic and reverse the malignant phenotype in preclinical models of T-Cell lymphoma. Clin Adv Hematol Oncol. 2013;122:646.
Love C et al. The genetic landscape of mutations in Burkitt lymphoma. Nat Genet. 2012;44(12):1321–5.
Nikoloski G et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet. 2010;42(8):665–7.
Visser HP et al. The Polycomb group protein EZH2 is upregulated in proliferating, cultured human mantle cell lymphoma. Br J Haematol. 2001;112(4):950–8.
Eckerle S et al. Gene expression profiling of isolated tumour cells from anaplastic large cell lymphomas: insights into its cellular origin, pathogenesis and relation to Hodgkin lymphoma. Leukemia. 2009;23(11):2129–38.
Sasaki D et al. Overexpression of enhancer of zeste homolog 2 with trimethylation of lysine 27 on histone H3 in adult T-cell leukemia/lymphoma as a target for epigenetic therapy. Haematologica. 2011;96(5):712–9.
Fiskus W et al. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood. 2009;114(13):2733–43.
McCabe MT et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492(7427):108–12.
Fojo T. Multiple paths to a drug resistance phenotype: mutations, translocations, deletions and amplification of coding genes or promoter regions, epigenetic changes and microRNAs. Drug Resist Updat. 2007;10(1-2):59–67.
Glasspool RM, Teodoridis JM, Brown R. Epigenetics as a mechanism driving polygenic clinical drug resistance. Br J Cancer. 2006;94(8):1087–92.
Herman JG et al. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci U S A. 1994;91(21):9700–4.
Merlo A et al. 5′ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med. 1995;1(7):686–92.
Herman JG et al. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res. 1995;55(20):4525–30.
Gonzalez-Zulueta M et al. Methylation of the 5′ CpG island of the p16/CDKN2 tumor suppressor gene in normal and transformed human tissues correlates with gene silencing. Cancer Res. 1995;55(20):4531–5.
Kucuk C, et al. Global promoter methylation analysis reveals novel candidate tumor suppressor genes in natural killer cell lymphoma. Clin Cancer Res. 2015.
Hayslip J, Montero A. Tumor suppressor gene methylation in follicular lymphoma: a comprehensive review. Mol Cancer. 2006;5:44.
Soengas MS et al. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature. 2001;409(6817):207–11.
Furukawa Y et al. Methylation silencing of the Apaf-1 gene in acute leukemia. Mol Cancer Res. 2005;3(6):325–34.
Fischle W, Wang Y, Allis CD. Histone and chromatin cross-talk. Curr Opin Cell Biol. 2003;15(2):172–83.
Clozel T et al. Mechanism-based epigenetic chemosensitization therapy of diffuse large B-cell lymphoma. Cancer Discov. 2013;3(9):1002–19. DNA hypermethylation of SMAD1 contributes to doxorubicin resistance in DLBCL cell lines. This resistance can be reversed with pre-treatment using decitabine prior to standard R-CHOP therapy.
Jain S, et al. Preclinical pharmacologic evaluation of pralatrexate and romidepsin confirms potent synergy of the combination in a murine model of human t-cell lymphoma. Clin Cancer Res. 2015. Treatment of T-cell lymphoma mouse models with pralatrexate and romidepsin led to complete responses in all models tested. This drug combination is currently under investigation in a Phase I/II clinical trial.
Zullo K et al. The aurora a kinase inhibitor, alisertib, has broad activity in nonclinical models Of T-Cell lymphoma and is highly synergistic with romidepsin, but not with pralatrexate or the proteasome inhibitor, ixazomib. Blood. 2013;122:5141.
Sharma SV et al. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7(3):169–81.
Gray JE et al. A phase I, pharmacokinetic, and pharmacodynamic study of panobinostat, an HDAC inhibitor, combined with erlotinib in patients with advanced aerodigestive tract tumors. Clin Cancer Res. 2014;20(6):1644–55.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Jennifer K. Lue declares that she has no conflict of interest. Jennifer E. Amengual declares that she has no conflict of interest. Owen A. O’Connor has received research support through grants from Celgene and Spectrum Pharmaceuticals.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Lymphomas
Rights and permissions
About this article

Cite this article
Lue, J.K., Amengual, J.E. & O’Connor, O.A. Epigenetics and Lymphoma: Can We Use Epigenetics to Prime or Reset Chemoresistant Lymphoma Programs?. Curr Oncol Rep 17, 40 (2015). https://doi.org/10.1007/s11912-015-0464-y
Published:
DOI: https://doi.org/10.1007/s11912-015-0464-y