
Abstract
Autoimmune lymphoproliferative syndrome (ALPS) is a primary immune regulatory disorder characterized by benign or malignant lymphoproliferation and autoimmunity. Classically, ALPS is due to mutations in FAS and other related genes; however, recent research revealed that other genes could be responsible for similar clinical features. Therefore, ALPS classification and diagnostic criteria have changed over time, and several ALPS-like disorders have been recently identified. Moreover, mutations in FAS often show an incomplete penetrance, and certain genotypes have been associated to a dominant or recessive inheritance pattern. FAS mutations may also be acquired or could become pathogenic when associated to variants in other genes, delineating a possible digenic type of inheritance. Intriguingly, variants in FAS and increased TCR αβ double-negative T cells (DNTs, a hallmark of ALPS) have been identified in multifactorial autoimmune diseases, while FAS itself could play a potential role in carcinogenesis. These findings suggest that alterations of FAS-mediated apoptosis could trespass the universe of inborn errors of immunity and that somatic mutations leading to ALPS could only be the tip of the iceberg of acquired immunodeficiencies.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Autoimmune lymphoproliferative syndrome (ALPS) is a rare genetic disorder of immune regulation characterized by an impairment of lymphocyte homeostasis [1]. Clinically, ALPS has been known since the 60s [2], while its first genetic characterization dates back to 1995, when the first disease-causing mutations were identified in FAS gene [3,4,5].
ALPS clinical presentation is characterized by lymphoproliferation (lymphadenopathy and/or organomegaly), autoimmune phenomena (mainly cytopenias), and an increased incidence of lymphoma [6]. Therefore, clinical management aims at monitoring patients for the development of malignancies, while non-malignant lymphoproliferation and cytopenias benefit from several types of immunosuppressants (e.g., steroids and Sirolimus) [1, 7, 8]. Immunological tests typically show increased TCR α/β CD4-CD8- “double negative” T cells (DNTs, a hallmark of the disease) and other ALPS biomarkers, such as high levels of vitamin B12, IL-10, and sFASL and impaired FAS-mediated apoptosis [9, 10].
Clinical and laboratory features have been combined together creating diagnostic criteria for ALPS, first outlined in 1999 [11] and later revised in 2009 [9] and 2019 [12] (Tables 1, and 2). In parallel, research revealed that ALPS could also originate from germline mutations in FAS-Ligand (FASL) and caspase-10 (CASP10) genes — both involved in the extrinsic apoptosis pathway [13, 14] — and from somatic mutations in FAS [15]. On the other hand, a significant number of patients met the diagnostic criteria without evidence of mutations in the known causative genes. This complex genetic background brought to a classification of ALPS — first created in 1999 [11] and then revised in 2009 [9] (Table 3) — which denominated as ALPS-undetermined (ALPS-U) those cases where no known genetic defect was identified. Finally, in the last decade, alternative pathways of disease pathogenesis have been hypothesized, consistently with the identification of several genes whose mutations give rise to ALPS-like clinical phenotypes [16].
In addition to its genetic characterization, modalities of inheritance of ALPS are controversial. Both autosomal-dominant and autosomal-recessive types of inheritance have been described [17], while increasing reports of somatic mutations in FAS gene — detected in TCR αβ DNT cells — jeopardized the idea of ALPS as an exclusively inborn error of immunity [18]. Hence, a double-hit mechanism (i.e., predisposing germline mutations followed by disease-triggering somatic mutations) has been hypothesized [19]. Moreover, the variable penetrance seen in ALPS patients’ pedigrees [20] suggested that a digenic model of inheritance could be applicable, due to a possible role of disease-modifying genes [21].
Finally, variants in FAS and FASL have been reported in patients displaying multifactorial autoimmune diseases and cancer (e.g., systemic lupus erythematosus, SLE) [13, 22,23,24]. Such findings could indicate that genetic defects of lymphocyte apoptosis may be a possible underlying mechanism of autoimmunity and carcinogenesis [17, 22]. The crossroad between immune dysregulation and multifactorial autoimmunity is furtherly highlighted by reports of increased TCR αβ DNTs in patients with SLE and other autoimmune disorders [25], as well as in rare genetic disorders of immune regulation, such as STAT3 gain of function (STAT3-GoF), CTLA-4 haploinsufficiency (CHAI), and others [16, 26,27,28,29].
Herein, we review ALPS genetic background and possible relationships between disease-associated mutations and its type of inheritance. We will also describe current knowledge about somatic mutations in FAS, together with their possible underestimation and a potential disease-contributing role in multifactorial autoimmune disorders. Finally, we will outline current treatment options for ALPS and ALPS-related diseases.
ALPS: one disease, many genes
ALPS classification
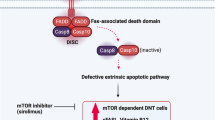
Genes included in ALPS classification (Table 3) retrace those implicated in the extrinsic, FAS-mediated, pathway of apoptosis [9]. Briefly, FAS (also known as CD95 or tumor necrosis factor receptor superfamily member 6 — TNFRSF6) is a homotrimeric receptor whose binding to FASL homotrimers activates the intracellular death domains of FAS molecules. These domains recruit the adaptor protein FADD (FAS-associated death domain) and ultimately bind caspase-8 and caspase-10 (CASP8-10), leading to the generation of a death-inducing signaling complex (DISC). Finally, DISC activates downstream effector caspases, paving the way for the occurrence of programmed cell death [30] (Fig. 1B).

FAS structure, biology, and molecular mechanism of FAS mutations. A Intron-exon structure of FAS gene delineating the exons coding for the extracellular, transmembrane, and intracellular portions of FAS. Cysteine-rich domain 1 (CRD1) is also known as pre-ligand assembly domain (PLAD) and allows homotrimerization of FAS monomers. UTR, untranslated region. B Biological mechanism of extrinsic, FAS-mediated, pathway of apoptosis. FAS is a homotrimeric receptor expressed at the surface of many cell types, whose correct trimerization is mediated by PLAD. Binding of cognate ligand FASL allows the recruitment of FAS-associated death domain (FADD), an adaptor protein that bridges the death domain of FAS to pro-caspases-8 and caspase-10, leading to the generation of death-inducing stimulating complex (DISC). DISC formation allows the downstream activation of the caspases, resulting in a biochemical cascade that ultimately leads to apoptosis. C FAS mutations exerting a dominant-negative effect do not impair the expression of mutated, PLAD-sparing, monomers (in gray) at the cell surface. Such monomers may interact with wild-type ones in a 1:2 or 2:1 ratio, resulting in non-functional trimers that impair downstream activation of apoptosis. These mutations are clinically penetrant and usually all affected individuals develop ALPS. D FAS haploinsufficient mutations usually involve PLAD. These mutations completely impair FAS expression at the cell surface only when both alleles are affected (either because of germline homozygosity or somatic events on the second allele). In heterozygous individuals, the wild-type allele preserves FAS expression and its correct functionality, leading to apoptosis
Germline mutations in FASL and CASP10, together with germline or somatic mutations in FAS, give rise to typical ALPS clinical and laboratory features [18, 20, 31, 32] and have therefore been included in ALPS classification [9]. On the other hand, mutations in FADD and CASP8 lead both to an impairment in FAS-mediated apoptosis and a profound susceptibility to infections [33, 34]. Indeed, these genes underlie other immunological pathways leading to pleiotropic effects that trespass ALPS phenotype. Therefore, mutations in CASP8 — formerly labeled as ALPS IIb [34] — have been removed from current ALPS classification and termed caspase eight deficiency state (CEDS), while FADD deficiency has never been actually defined as ALPS.
Regardless of the genes involved, specific criteria need to be satisfied in order to achieve a diagnosis of ALPS. These have been recently updated by the European Society for Immunodeficiencies (ESID) (Table 2) [12]. If compared to the previous ones [9], increased TCR αβ DNTs are not anymore mandatory, while the TCR αβ DNT ratio threshold has been raised from 2.5 to 6% of total TCR αβ CD3+ T cells. Moreover, immunohistological findings have been excluded, and the identification of pathogenic mutations is not needed to define ALPS, though of course necessary in order to attain a molecular diagnosis [12]. Growing relevance has been given to biomarkers, that — once combined — demonstrated to bear elevated positive and negative predictive values for ALPS [35, 36]. In particular, the combination of normal TCR αβ DNTs and in vitro apoptosis assay can essentially rule out ALPS [36, 37].
ALPS-U: determining the undetermined
Since its first definition, 20–30% of ALPS cases lack a molecular diagnosis (ALPS-U) [9, 37]. Recent studies showed that patients with ALPS-U display a different biomarker profile, compared to those affected by ALPS-FAS [35, 36]. In detail, Molnar et al. demonstrated that ALPS-FAS is characterized by significantly higher proportions of TCR αβ DNTs, in vitro apoptosis alterations, and soluble FAS-Ligand (sFASL) levels [36]. The biological background of ALPS-U might therefore be different from classical ALPS. In accordance with this, next-generation sequencing analyses in these patients identified pathogenic or likely pathogenic mutations in signal transducer and activator of transcription 3 (STAT3), inhibitor of nuclear factor kappa B kinase regulatory subunit gamma/NF-kappa-B essential modulator (IKBKG/NEMO), perforin 1 (PRF1), and recombination activating 1 (RAG1). Such findings are consistent with ALPS-like features previously described in patients carrying defects in the same genes, even though the overall clinical phenotypes of these disorders are extremely heterogeneous and different from ALPS [38,39,40,41,42,43,44].
Nevertheless, in the vast majority of ALPS-U cases, an underlying genetic defect is unknown. Many genes have been recalled as hypothetic players in ALPS-U pathogenesis [16]. In order to be called ALPS-U, however, the patient’s phenotype must respond to the above-mentioned diagnostic criteria. Therefore, together with clinical features, either increased TCR αβ DNTs or two biomarkers (including impaired FAS-mediated apoptosis) must be displayed. In line with this, several candidates standing behind an ALPS-U phenotype have been described in the past decades. First of all, multiple cases of lymphoproliferation, autoimmunity, and apoptosis defect but normal TCR αβ DNTs without a genetic explanation have formerly been described as Dianzani autoimmune lymphoproliferative disease (DALD) [45, 46]. Later, germline gain-of-function (GoF) mutations in NRAS or somatic GoF variants in KRAS were found, depicting a phenotype called RAS-associated autoimmune lymphoproliferative disease (RALD), characterized by intrinsic apoptosis defect and normal or slightly elevated TCR αβ DNTs [47, 48]. Increased TCR αβ DNTs and autoimmune features were also found in STAT3-GoF, CHAI and X-linked immunodeficiency with magnesium defect, Epstein-Barr virus (EBV) infection, and neoplasia (XMEN), demonstrating that ALPS-U may comprise a wide variety of responsible genes [26, 28, 29].
Finally, we can speculate that somatic mutations in FAS [18] or in other related genes could be responsible for a significant portion of ALPS-U cases. These could potentially be underestimated, given that somatic mutation analysis is not easily feasible, since patients’ samples are often not enough to perform TCR αβ DNTs sorting [35]. Future developments in this field will hopefully clarify the impact of somatic mutations on ALPS-U pathogenesis.
ALPS-FAS: one gene, many inheritance patterns
FAS was the first gene associated with ALPS [3, 4]. Since its identification, hundreds of mutations in it have been described, accounting for roughly 70% of ALPS cases [49]. FAS gene consists of an extracellular portion (exons 1–5), a transmembrane domain (exon 6), and an intracellular part (exons 7–9) [50]. The extracellular portion contains 3 cysteine-rich domains (CRDs): while CRDs 2–3 are paramount to bind FASL, CRD1 allows homotrimerization of FAS molecules and is therefore named pre-ligand assembly domain (PLAD). On the intracellular side, exon 9 encodes for the death domain (DD), which is critical for FADD binding and downstream activation of apoptosis [8] (Fig. 1A).
In the last two decades, mutations have been described throughout any site of FAS: the majority (70%) of ALPS-FAS cases are due intracellular FAS mutations, while 50% involve the DD [8]. Increasing reports speculated the existence of a relationship between genotype and inheritance pattern. This implies that different mutations exert distinct molecular effects, leading to either dominant or recessive types of inheritance [17]. Moreover, the discovery of somatic FAS mutations in TCR αβ DNTs furtherly complicated this model [15]. Below, we attempt to clarify these important aspects.
Dominant inheritance in ALPS-FAS
Studies on mice strains Ipr [51] and Iprcg [52] initially allowed to understand the role of Fas gene, the murine analog of FAS [21]. Iprcg strains beared mutations in the DD of Fas: importantly, their inheritance pattern proved to be not fully recessive as in Ipr mice [52]. Similarly, a dominant-interfering effect has been described since the first human reports of ALPS-FAS [4]. Later, mutations in the intracellular portion of FAS that did not abolish its surface expression were considered to be responsible for a dominant type of inheritance [53]. Finally, Siegel et al. shed more light on this aspect, establishing that PLAD preservation was always correlated with the presence of a dominant-negative mechanism [54].
According to this model, transmembrane wild-type FAS combines with the mutated form (via PLAD-PLAD interactions) in a 2:1 or 1:2 ratio. This results in a trimer which is unable to recruit FADD after binding of cognate ligand FASL. Hence, the majority of FAS trimers display defective intracellular signaling, even though 50% of total FAS in the cell is in a wild-type form [55] (Fig. 1C).
Recently, a large retrospective study demonstrated that a dominant-negative effect stands behind the majority of ALPS-FAS cases [8], justifying a dominant type of inheritance (i.e., one mutation leads to ALPS) [17]. Moreover, reports of single-allele somatic dominant mutations in FAS confirmed that this important molecular mechanism is also conserved in ALPS-sFAS [18, 56].
Recessive inheritance in ALPS-FAS
In contrast to Iprcg mice, murine Ipr strains bore mutations in intron 3, leading to reduced Fas expression and behaving as a recessive defect [21, 51]. Later on, several examples of autosomal recessive ALPS-FAS have been reported, though less frequently than cases with a dominant type of inheritance [53, 57, 58].
These patients displayed either mutations in the extracellular portion of FAS (which particularly involved PLAD) [8, 54] or in the transmembrane domain (i.e., exon 6) [55]. In any case, surface expression of FAS was abolished, due to impaired trimerization. This implies that mutated monomers cannot anymore bind their wild-type (wt) homologue, which may be undisturbedly displayed on the cell surface. This situation jeopardizes a dominant-interfering effect, while the presence of two mutated alleles gives rise to haploinsufficiency [59, 60] (Fig. 1D). In line with this, FAS-wt transfection in FAS-mutated cell cultures both corrected the apoptosis defect and increased surface FAS expression, demonstrating the hypothesis of a haploinsufficiency mechanism [59].
Nevertheless, germline haploinsufficient mutations were found both in patients and asymptomatic relatives, and this fact did not find an immediate explanation. Initially, a reduced penetrance due to this genotype was hypothesized [61]. The conundrum was partially solved by the identification of somatic events in patients, while not in asymptomatic carriers, with familial haploinsufficient mutations [19]. Therefore, these defects are non-penetrant, and an additional event on the second allele is needed in order to determine clinical manifestations [17]. Such “second hit” may either be inherited [53, 57] or — more frequently — acquired [19, 62, 63], as furtherly described.
Somatic mutations in FAS (ALPS-sFAS)
Somatic heterozygous dominant mutations in FAS were initially reported in 2004 [15], depicting for the first time the role of somatic mutations in a non-malignant disease [17]. Such variants were recognized in patients’ TCR αβ DNTs, which were found to be increased, just as in germline ALPS-FAS. Clinical features were also not distinguishable but — compared to germline ALPS-FAS — patients with somatic mutations did not display a severe impairment in FAS-mediated apoptosis [18]. Such finding has been repeatedly confirmed [1] and bears a possible explanation in the fact that TCR αβ DNTs do not survive in cell culture, with only a fraction of mononuclear cells being actually mutated. This fraction is apparently sufficient to give rise to clinical features, but not enough to significantly impair apoptosis [18].
Further studies revealed that somatic FAS mutations are not rare and represent up to 15% of total ALPS cases [63]. Indeed, these variants respond to the same molecular mechanisms described above. Therefore, dominant-interfering somatic mutations alone are sufficient to induce the disease [15, 56], while patients displaying germline haploinsufficient mutations require a somatic event in order to develop ALPS clinical features [19, 56]. The latter, moreover, exhibit symptoms at a later stage [56], coherently with the fact that somatic mutations initially occur in hematopoietic stem cells (HSCs) and furtherly provide a selective advantage to mutated cells. These lymphocytes, however, require several years of proliferation in order to reach sufficient numbers to have a clinical impact [17].
We may speculate that somatic mutations in FAS are probably underdiagnosed, for a couple of reasons. First, TCR αβ DNTs sorting is often challenging to perform and such difficulty discourages single-cell DNA sequencing [21, 35]. Moreover, somatic mutations are potentially responsible for disease penetrance in many patients displaying heterozygous haploinsufficient germline FAS mutations. However, TCR αβ DNTs sorting and sequencing is seldom performed in these cases, since clinicians have already achieved a genetic diagnosis. Finally, somatic variants may potentially lie at the base of more common, multifactorial, autoimmune disorders [17].
Double-hit hypothesis in ALPS: one disease, two mutations needed
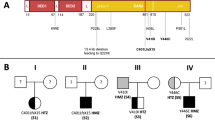
ALPS patients’ pedigrees often show an incomplete disease penetrance [20, 63]. However, additional genetic mechanisms may come into play and ultimately determine the expression of an ALPS disease phenotype [1, 17]. These predisposing mutations act as a “second hit” and include somatic FAS mutations (leading to loss of heterozygosity, LoH), a second germline mutation (leading to homozygous/compound heterozygous genotypes), or variants in other disease-modifying genes (Fig. 2).

Double-hit hypothesis in ALPS. Germline heterozygous haploinsufficient mutations in FAS are not sufficient to determine the disease: a second hit is needed in order to develop ALPS. Different mechanisms may account for the second hit. First, a somatic event in the second FAS allele could be possible (e.g., a new acquired mutation or uniparental disomy). Second, germline homozygous or compound heterozygous mutations in FAS or FASL can determine ALPS. Finally, a second mutation in other disease-modifying genes could induce the disease, depicting a digenic inheritance pattern. These genes may involve both the extrinsic and intrinsic pathway of apoptosis [47, 48, 71, 73, 74, 77], perforin secretion [38, 76], or other possible mechanisms [78, 79]
Somatic loss of heterozygosity in FAS
ALPS has been recently summoned as a paradigm of inborn errors of immunity due to a somatic mosaicism [64, 65]. Indeed, evidence suggests that ALPS may respect a “two-hit” mechanism similar to the one postulated in the 70s for retinoblastoma’s pathogenesis [66]. In line with this, ALPS was described in 7 patients bearing germline mutations in the extracellular domain (ECD) of FAS, together with a somatic event detected in the second FAS allele in TCR αβ DNTs [19]. Such somatic event is not necessarily a new mutation, but often corresponds to uniparental disomy that makes the cell homozygous for the original germline variant [17, 56].
Not surprisingly, ALPS-causing somatic events are almost exclusively identified in individuals bearing haploinsufficient mutations, which ideally behave in a recessive fashion [63]. Among these subjects, only those displaying a “second hit” actually develop an ALPS phenotype [62], which typically manifests at an older age [56]. However, not every patient with ALPS and a haploinsufficient mutation was found to bear a somatic event. This could be potentially due to intronic FAS mutations or variants in other disease-modifying genes [56].
Germline double-hit
In rare instances, both hits on the FAS gene may be inherited. This often leads to severe phenotypes and may be due either to compound heterozygous or homozygous FAS mutations (formerly described as ALPS type 0) [1, 57, 58, 67]. The effect of the mutation (i.e., haploinsufficiency or dominant-negative) is trivial in these cases, since both alleles are involved.
Similarly, ALPS-FASL supposedly observes an autosomal recessive inheritance, giving rise to severe clinical phenotypes [31, 32, 68, 69]. However, ALPS-FASL inheritance may also behave outside the box, since a dominant-negative effect has been reported [70] and heterozygous mutations have been associated to SLE [13]. The precise mechanism hiding behind FASL mutations is still not clear, given that these reports are extremely rare [17].
Second hit in disease-modifying genes
Studies in mice originally noted that double heterozygous subjects (i.e., lpr/+, gld/+) for murine analogs of FAS and FASL could develop lymphoproliferation and autoimmunity [52]. In the last two decades, reports of ALPS and ALPS-like patients revealed that CASP10 and other genes may influence disease pathogenesis. These observations implicated that ALPS could be inherited in a digenic or oligogenic fashion, thus trespassing a classical Mendelian transmission model [17, 71].
CASP10
Only two pathogenic mutations in CASP10 have been identified thus far (i.e., p.I406L and p.L258F), both leading to a dominant-negative effect [14, 72, 73]. Conversely, several other variants or polymorphisms in CASP10, whose pathogenicity is still controversial, have been described [14, 73, 74]. Nevertheless, multiple reports of FAS-mutated individuals bearing a concomitant CASP10 variant and developing ALPS clinical features suggested the existence of an underlying digenic mechanism [71, 74, 75]. Both FAS and CASP10 belong to the same — extrinsic — apoptosis pathway; therefore, mutations in these genes would produce complementary effects, leading to impairment of programmed cell death. Similarly, some CASP10 variants may also display a protective effect towards the development of ALPS [72]. Hence, interactions between variants in genes belonging to the same pathway may be both synergic and antagonistic. Finally, ALPS or ALPS-like features may also arise from the complementary effect of variants in CASP10 and in another non-FAS gene, such as CASP8 or TNFRSF13C (i.e., BAFF receptor, whose mutations are typically associated with common variable immunodeficiency, CVID) [73].
Other disease-modifying genes
In the last two decades, interesting reports revealed that the “second hit” in ALPS pathogenesis may concern other genes, beyond those involved in the extrinsic apoptosis pathway. For instance, variants in UNC13D and PRF1 highlighted that genes involved in perforin secretion by natural killer (NK) and cytotoxic T lymphocytes (CTLs) may bestow clinical significance to certain heterozygous FAS mutations [38, 76]. A similar role could be played by genes belonging to the intrinsic apoptosis pathway, such as XIAP [77] or NRAS and KRAS. Mutations in these last two genes may alone give rise to RALD [47, 48], but we may speculate that variants in them could also determine ALPS in genetically predisposed individuals. Moreover, since specific osteopontin haplotypes have been formerly related to DALD [78], a possible role of OPN gene in ALPS could be contemplated. Finally, other biological mechanisms, such as microRNA overexpression (e.g., miR-146a), are involved in Fas downregulation in mice and could ideally be involved in ALPS pathogenesis [79].
ALPS, cancer, and autoimmunity: one mechanism, many diseases
Alterations in the extrinsic pathway of apoptosis are not prerogative of ALPS but may also hide behind more common conditions such as cancer and autoimmune diseases [21, 22]. Indeed, research in the last 10 years revealed that single-nucleotide polymorphisms (SNPs) in FAS or FASL may correlate with some types of cancer and multifactorial autoimmune diseases [23, 80, 81]. Similarly, an expansion of TCR αβ DNTs is not exclusive of ALPS but may also be found in other conditions [25], where these cells may play pathogenic roles [82]. Therefore, the genetic and immunological features of ALPS push beyond the limits of this disease and involve more common clinical situations (Fig. 3). In this section, we try to summarize this cutting-edge topic.

ALPS, cancer and autoimmune diseases. Venn diagram showing mutual relationships and shared clinical and pathophysiologic features connecting ALPS to more common conditions such as cancer and autoimmune diseases. TCR αβ DNTs, T-cell receptor αβ double-negative T cells
FAS and cancer
While initially only non-malignant lymphoproliferation was taken into account in ALPS definitions [9, 11], the latest diagnostic criteria comprise a history of lymphoma as a major clinical feature (Table 2) [12]. In parallel, recent research highlighted potential carcinogenic implications of FAS, whose role is still controversial [22].
Most cancer cells are resistant to FAS-mediated apoptosis [22]. Such an escape mechanism is required since the CD95/CD95L interaction may destroy cells by inducing DISC formation [83]. However, baseline FAS signaling seems crucial for cancer cell survival [84], since a complete elimination of CD95/CD95L interplay leads to an irreversible cell death called DICE (death induced by CD95R/L elimination) [22]. Such peculiar behavior might be explained by non-apoptotic functions of FAS, whose engagement also promotes inflammation and carcinogenesis [83]. In light of this, preclinical studies on therapies targeting the FAS/FASL pathway have been implemented [85], even though severe hepatotoxicity hampers their clinical applicability [22].
In a clinical setting, lymphoma may be a threatening part of the natural history of ALPS. Interestingly, this malignancy may also develop in healthy FAS-mutated relatives of ALPS patients [8]. In a wider perspective, a relationship between cancer risk and FAS polymorphisms is controversial [23, 86,87,88]. A meta-analysis argues for a reduction of cancer risk in individuals bearing FAS-1377 G/A SNP [86]. Similarly, a previous hypothetical association between FAS-670 A/G SNP and acute myeloid leukemia (AML) has been rejected [88]. On the other hand, SNPs in codons FAS-1377 and FASL-844 correlated with bladder cancer [81] and neuroblastoma [23]. Further research may shed more light on these aspects and allow to identify possible factors determining FAS-mediated carcinogenesis.
FAS, FASL, and autoimmunity
B cell differentiation is typically altered in ALPS-FAS [1]. Murine models showed that an impaired germinal center reaction inappropriately allows survival of autoreactive B cells [89]. Therefore, along with defective T cell apoptosis, a dysregulated B cell compartment is characteristic of ALPS.
Such immunological background is coherent with a clinical overlap among ALPS, CVID [90] and — most importantly — SLE [91, 92]. In particular, lupus and lupus-like features may characterize the clinical course of ALPS-FAS and ALPS-FASL [13, 92]. Moreover, a role for apoptosis in SLE susceptibility is plausible, since SNPs FAS-670 A/A and FASL-844 C/C (alone or combined in a digenic type of inheritance) are associated with an increased risk of lupus [24, 93]. On the other hand, variants in other genes involved in apoptosis (i.e., BAX) seem to be protective towards the development of SLE [93]. Other autoimmune diseases have been associated with SNPs in FAS and these include Hashimoto’s thyroiditis, systemic sclerosis, and multiple sclerosis [80, 94].
Apart from polymorphisms, the increasing discovery of somatic mutations in FAS opens an intriguing debate. Somatic variants in this and other genes could potentially account for the clinical discordance of monozygotic twins, a frequent phenomenon in autoimmunity whose explanation has been classically attributed to epigenetic modifications [95]. Future research could reveal if FAS somatic mosaicism hides behind those autoimmune conditions that interestingly display an ALPS-like expansion of TCR αβ DNTs [25].
DNTs in ALPS and autoimmune diseases
TCR αβ DNTs are a peculiar T cell subset whose origin and pathogenetic role has not been clearly elucidated [82, 96]. Initially, TCR αβ DNTs were thought to have a thymic origin [97]. However, the expression of both the senescent marker CD57 and CD45RA makes them more similar to terminally differentiated effector T cells re-expressing CD45RA (TEMRA) [98]. Such phenotype inclines towards the hypothesis that TCR αβ DNTs may peripherally derive from downregulation of CD8 in autoreactive CD8+ T cells [96, 99]. Their pathogenic role is controversial since TCR αβ DNTs are not only a hallmark of ALPS [100, 101], but have also been described in pro-inflammatory contexts (e.g., in SLE and other autoimmune diseases) or as mediators of immune regulation (e.g., in graft-versus-host disease — GVHD) [82, 102].
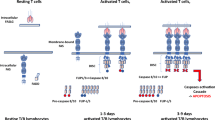
In ALPS, TCR αβ DNTs are not only a key feature for diagnostic purposes, but they also seem to play a relevant role in disease pathogenesis [100, 101]. An expansion of TCR αβ DNTs is tightly associated with disease development [82, 103], and their number often correlates with the presence of autoantibodies [87, 104]. Recently, an elegant study by Maccari et al. shed more light on this topic, revealing two main populations among TCR αβ DNTs: FAS-controlled DNTs (FC-DNTs, CD38+ CD45RA+) and conventional DNTs (cDNTs, CD45RA+/− CD38-) [101]. The former are IL-10 producers and are the real hallmark of ALPS-FAS, where they represent a significant proportion of total TCR αβ DNTs (usually > 25%). Moreover, these FC-DNTs share a transcriptional profile with certain CD28 + CD57+ single-positive T cells (SPTs, both CD4+ or CD8+), corroborating the hypothesis that TCR αβ DNTs derive from downregulation of CD4/CD8 coreceptor at a late differentiation step in T cell development. On the other hand, cDNTs are closely related to “canonical” CD8+ T cells, since they express both interferon-γ (IFNγ) and cytolytic molecules (granzyme B and perforin), oppositely to FC-DNTs. Unexpectedly, both TCR αβ DNT populations have been found also in healthy subjects, indicating that these cell types may also play a physiologic role. Moreover, treatment with Rapamycin led to a decrease in both ALPS and healthy subjects-derived FC-DNTs, highlighting that the maintenance of this lymphocyte subset tightly depends on mammalian target of rapamycin (mTOR) signaling [101].
Such recent findings have not yet been applicated to more common, multifactorial autoimmune diseases (e.g., SLE). Anyhow, in these disorders, TCR αβ DNTs have been identified as main actors of an increased production of the pro-inflammatory interleukin-17A (IL-17A). Such process seems to be mediated by the transcription factor cAMP-responsive element modulator (CREM) α [96]. CREMα also plays a significant role in the downregulation of CD8 from autoreactive CD8+ T cells [82, 96], which may take place in an inflammatory milieu, such as the one seen in spleens of SLE patients due to the lack of the tolerogenic splenic marginal zone macrophages (MZMs) [105]. In addition, MZMs express scavenger receptors that efficiently clear apoptotic and necrotic cellular fragments, avoiding the generation of autoimmunity against these debris [106]. Consistently, a lack of MZMs and an increase in TCR αβ DNTs are particularly displayed in SLE, where TCR αβ DNTs showed to promote the production of anti-dsDNA antibodies [96]. In a clinical context, association studies and pathologic specimens also revealed a possible role of TCR αβ DNTs in Sjögren’s syndrome [107], psoriasis [108], axial spondylarthritis [109], mixed connective tissue disease, juvenile idiopathic arthritis, juvenile dermatomyositis [110], and Behçet’s disease [111].
In contrast to the findings above, evidence suggests that at least a subset of TCR αβ DNTs may exhibit a regulatory activity [82, 112]. Even though specific markers allowing their identification are lacking, these TCR αβ DNTs have been called double-negative regulatory T cells (TCR αβ DN Tregs). Despite what the name suggests, DN Tregs are FOXP3- and must not be confused with CD4+ CD25+ FOXP3+ regulatory T cells [113]. A first interesting role of TCR αβ DN T regs was demonstrated in non-obese diabetic (NOD) mouse models, where this cell type lowered the risk of developing islet autoimmunity through the production of IL-10 [114]. In addition, TCR αβ DN T regs seem to play a significant role in the development of immune tolerance after hematopoietic stem cell transplantation (HSCT). Initially, murine models revealed that TCR αβ DN T regs were able to inhibit natural killer (NK) cell-mediated rejection of allogenic bone marrow in a perforin-dependent manner [115]. Moreover, clinical studies showed an inverse correlation between TCR αβ DNTs frequency and the risk of developing GVHD [116, 117]. Proposed mechanisms of TCR αβ DN T regs functioning are IL-10 production, the peculiar use of IFNγ to regulate immune responses, and the acquisition of alloantigens from dendritic cells (i.e., trogocytosis) in order to present them to antigen-specific CD8+ T cells and killing them through Fas-dependent apoptosis [113]. However, such immunological processes still need to be fully clarified by further studies [82].
ALPS and ALPS-like disorders: one phenotype, many treatments
Treatment options and management of ALPS
The most frequent presentation of ALPS is benign lymphoproliferation; however, despite potentially massive manifestations during childhood, lymphadenopathies and splenomegaly generally shrink spontaneously with age [21]. On the other hand, malignant lymphoproliferation may develop at any time during follow-up [118, 119]. Nonetheless, autoimmune cytopenias occur in more than 80% of ALPS-FAS patients and often represent a therapeutic challenge [8, 21]. Therefore, management of ALPS should cover all these aspects, and clinicians should choose treatment options taking into account that they might be administered for a lifelong span [7].
Treatment of isolated benign lymphoproliferation for cosmetic reasons is not usually indicated [7]. In case of symptoms or concomitant cytopenias, the only drug that demonstrated to significantly diminish lymphoproliferation is Sirolimus [120,121,122], consistently with the description of a hyperactive mTOR pathway in ALPS [123]. Splenomegaly should be carefully managed using thermoplastic spleen guards, in order to avoid splenic rupture, allowing children to participate to sport programs [7]. ALPS is characterized by poor anti-polysaccharide response and disorganized splenic marginal zone [124], correlated with a high risk of streptococcal sepsis [8]. Therefore, in order to preserve a minimal anti-polysaccharide response, splenectomy is contraindicated and should be taken into consideration if it is the only remaining therapeutic option. However, it is possible to deal with already splenectomized ALPS cases. Asplenic ALPS patients should receive long-term antibiotic prophylaxis (e.g., penicillin V) and periodic anti-pneumococcal reimmunization every 4–5 years [7].
Autoimmune cytopenias in ALPS have been classically treated as sporadic immune cytopenias, using corticosteroids and intravenous immunoglobulins (IVIG) as first-line options [7, 125,126,127]. Due to refractoriness to these treatments, cytopenias in ALPS often require the use of second-line agents. Rituximab and/or Mycophenolate mofetil (MMF) were the first studied, the former being particularly effective for thrombocytopenia, though not usually recommended due to a consistent increase in the risk of severe infections [128]. On the other hand, MMF proved to be an effective steroid-sparing agent, without evidence of significant toxicities or infections [7, 129]. However, further evidence revealed a dramatic effectiveness of Sirolimus [120, 121], which may be considered a targeted treatment for ALPS [123], consistently with the high degree of remission achieved and the significant reduction of ALPS biomarkers after 6 months of therapy [122]. For these reasons, Sirolimus may be considered as a first-line treatment option, and, once complete remission is achieved, its serum levels may be maintained at a lower therapeutic range (i.e., 2–5 ng/mL) [122].
ALPS patients exhibit an increased risk of lymphoma; therefore, periodic surveillance with CT and positron emission tomography (PET) scans should be carried out, and lymph node biopsies must be performed in case of clinical or radiological suspect of malignancy [7]. Conventional multiagent chemotherapy and radiation are usually effective, and no specific treatment protocols are available for ALPS-related lymphoma [7, 118, 119, 130]. Finally, life-threatening, early-onset lymphoproliferation may seldom represent ALPS clinical onset, requiring HSCT as the only therapeutic option. Sporadic cases have been described, often exhibiting severe GVHD even in favorable conditions [131, 132].
Targeted treatment in ALPS-related disorders
Clinical presentation of ALPS-related disorders is often challenging and characterized by ALPS clinical signs with or without other organ-specific involvement [133]. However, autoimmune cytopenias are a hallmark of these disorders [134, 135] and are often refractory to first-line treatments (i.e., corticosteroids and IVIG) [136]. Recent evidence suggests that a rapid switchover to disease-specific therapies targeting underlying pathomechanisms is recommended [134]. Therefore, promptly achieving a genetic diagnosis is paramount in order to select a specific targeted treatment [137].
Target therapies include Janus kinase (JAK) inhibitors (e.g., ruxolitinib) and anti-IL-6 (tocilizumab) for STAT3-GoF [138], CTLA4-Ig (abatacept) for CHAI and lipopolysaccharide (LPS)-responsive and beige-like anchor protein (LRBA) deficiency [139, 140], PI3Kδ inhibitor (leniolisib) for activated PI3Kδ syndrome (APDS) [141], and others under study [134]. These treatments showed to be very effective also for other organ involvements, apart from cytopenias [138,139,140]. However, no targeted molecules may be available for certain disorders, and a specific molecular pathway is unknown in patients that still lack a genetic diagnosis. In these circumstances, clinical signs, immunophenotype, and laboratory parameters should guide treatment decisions [134, 135]. For instance, increased TCR αβ DNTs could be an indication to start treatment with Sirolimus, postulating a pathogenic mechanism similar to ALPS [134].
Just as in ALPS, periodic surveillance for malignancies should be carried out also in ALPS-related disorders and must be life-long, since cancer may develop at decades from disease onset (e.g., Juvenile myelomonocytic leukemia in RALD) [142]. Due to the short follow-up, it is not known whether targeted treatments may actually determine a reduction of malignant degeneration in these diseases [122]. Finally, gene editing trials are currently not available for ALPS and related syndromes, though future research might introduce this definitive treatment option, similarly to other primary immune regulatory disorders (PIRDs) [143, 144].
Conclusions
Since its discovery 25 years ago, ALPS has become a model of monogenic autoimmunity with hundreds of disease-causing mutations in different genes being identified [8, 145]. ALPS diagnostic criteria evolved over time: the latest highlight the fact that malignant lymphoproliferation could be a warning sign of this disorder, while TCR αβ DNTs are only one of several possible disease biomarkers (Table 2) [12, 37]. Widespread use of genetic testing is revealing an increasing amount of genes hiding behind ALPS-U [36]. Moreover, an expanding number of ALPS-like syndromes are being identified, sharing clinical and immunological features with ALPS, though not completely satisfying its diagnostic criteria [16]. Concurrently, Sirolimus is becoming a first choice for treatment of ALPS [121], while targeted therapies for ALPS-related disorders are getting a foothold [134].
ALPS inheritance pattern is whatsoever complicated. According to the molecular effect of the mutation, both dominant-interference or haploinsufficiency mechanisms may develop, determining a dominant or recessive transmission, respectively [17]. The discovery of somatic FAS mutations as causative of ALPS furtherly complicated the plot [15]. Somatic mutations follow the same molecular mechanisms as germline ones and could present alone or in association with inherited mutations.
While heterozygous, dominant mutations in FAS may be sufficient to determine the disease [8], severe ALPS phenotypes often require “two hits,” which may arise in different fashions. Rarely, both of them are inherited in FAS or FAS-related genes [32, 57]. Scattered reports reveal that a “second hit” could develop in disease modifying genes that bestow pathogenic significance to heterozygous variants in FAS, demonstrating that a digenic inheritance model is applicable to ALPS [17, 71, 75]. Most importantly, a second somatic mutation in FAS — detected in TCR αβ DNTs — may often account as a “second hit,” potentially explaining the reduced penetrance of this disorder [17, 19, 56].
Due to the difficulties of performing TCR αβ DNT sorting and single-cell DNA sequencing on scant specimens [35], somatic mutations in FAS are probably underdiagnosed. Suspicion of somatic mutations should especially arise when facing heterozygous, non-penetrant, haploinsufficient mutations in FAS that inexplicably become pathogenic in an individual with healthy FAS-mutated family members. Moreover, somatic FAS mutations could potentially underlie several cases of multifactorial autoimmune diseases such as SLE, which may display clinical and immunological features that are superimposable to ALPS. Among these, an expansion of TCR αβ DNTs may occur in SLE and other autoimmune conditions: these cells could therefore represent a helpful screening tool for autoimmunity. Hence, ALPS and more common disorders may be significantly interconnected (Fig. 3), and future research may shed more light on the role of FAS signaling in both autoimmunity and cancer.
The number of discovered inborn errors of immunity (IEIs) is growing exponentially, and atypical presentations of formerly known immunodeficiencies are being progressively unmasked [145, 146]. In this context, ALPS is the prototypical example of how more genes can determine one univocal phenotype [145]. ALPS was the first example of IEI that could be also caused by somatic mosaicism. Therefore, both germline and acquired mutations can be responsible for IEIs, and research has already identified and may furtherly reveal other disorders behaving in a similar fashion [65]. ALPS could only be the tip of the iceberg.
References
Speckmann C, Borkhardt A, Gaspar HB et al (2017) Genetic disorders of immune regulation. In: Primary immunodeficiency diseases. Springer-Verlag Berlin Heidelberg 2017:295–338
Canale VC, Smith CH (1967) Chronic lymphadenopathy simulating malignant lymphoma. J Pediatr 70:891–899. https://doi.org/10.1016/S0022-3476(67)80262-2
Rieux-Laucat F, Le Deist F, Hivroz C et al (1995) Mutations in fas associated with human lymphoproliferative syndrome and autoimmunity. Science 268(80):1347–1349. https://doi.org/10.1126/science.7539157
Fisher GH, Rosenberg FJ, Straus SE et al (1995) Dominant interfering fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell 81:935–946. https://doi.org/10.1016/0092-8674(95)90013-6
Drappa J, Vaishnaw AK, Sullivan KE et al (1996) Fas gene mutations in the Canale-Smith syndrome, an inherited lymphoproliferative disorder associated with autoimmunity. N Engl J Med 335:1643–1649. https://doi.org/10.1056/NEJM199611283352204
Straus MSE, Sneller M, Lenardo MJ et al (1999) An inherited disorder of lymphocyte apoptosis: The autoimmune lymphoproliferative syndrome. Ann Intern Med 130:591–601. https://doi.org/10.7326/0003-4819-130-7-199904060-00020
Rao VK, Oliveira JB (2011) How I treat autoimmune lymphoproliferative syndrome. Blood 118:5741–5751. https://doi.org/10.1182/blood-2011-07-325217
Price S, Shaw PA, Seitz A et al (2014) Natural history of autoimmune lymphoproliferative syndrome associated with FAS gene mutations. Blood 123:1989–1999. https://doi.org/10.1182/blood-2013-10-535393
Oliveira JB, Bleesing JJ, Dianzani U et al (2010) Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): report from the 2009 NIH International Workshop. Blood 116:35–41. https://doi.org/10.1182/blood-2010-04-280347
Bowen RAR, Dowdell KC, Dale JK et al (2012) Elevated vitamin B 12 levels in autoimmune lymphoproliferative syndrome attributable to elevated haptocorrin in lymphocytes. Clin Biochem 45:490–492. https://doi.org/10.1016/j.clinbiochem.2012.01.016
Bleesing JJH, Straus SE, Fleisher TA (2000) Autoimmune lymphoproliferative syndrome: A human disorder of abnormal lymphocyte survival. Pediatr Clin North Am 47:1291–1310. https://doi.org/10.1016/S0031-3955(05)70272-8
Abinun M, Beaussant Cohen S, Bustamante J, et al (2019) ESID Registry – working definitions for clinical diagnosis of PID
Wu J, Wilson J, He J et al (1996) Fas ligand mutation in a patient with systemic lupus erythematosus and lymphoproliferative disease. J Clin Invest 98:1107–1113. https://doi.org/10.1172/JCI118892
Wang J, Zheng L, Lobito A et al (1999) Inherited human caspase 10 mutations underlie defective lymphocyte and dendritic cell apoptosis in autoimmune lymphoproliferative syndrome type II. Cell 98:47–58. https://doi.org/10.1016/S0092-8674(00)80605-4
Holzelova E, Vonarbourg C, Stolzenberg MC et al (2004) Autoimmune lymphoproliferative syndrome with somatic Fas mutations. N Engl J Med 351:1409–1418. https://doi.org/10.1056/NEJMoa040036
Teachey D, Bride K (2017) Autoimmune lymphoproliferative syndrome: more than a FAScinating disease. F1000Research 6:. https://doi.org/10.12688/f1000research.11545.1
Rieux-Laucat F (2017) What’s up in the ALPS. Curr Opin Immunol 49:79–86. https://doi.org/10.1016/j.coi.2017.10.001
Dowdell KC, Niemela JE, Price S et al (2010) Somatic FAS mutations are common in patients with genetically undefined autoimmune lymphoproliferative syndrome. Blood 115:5164–5169. https://doi.org/10.1182/blood-2010-01-263145
Magerus-Chatinet A, Neven B, Stolzenberg MC et al (2011) Onset of autoimmune lymphoproliferative syndrome (ALPS) in humans as a consequence of genetic defect accumulation. J Clin Invest 121:106–112. https://doi.org/10.1172/JCI43752
Gaefke CL, Metts J, Imanirad D et al (2021) Case report: a novel pathogenic missense mutation in FAS: a multi-generational case series of autoimmune lymphoproliferative syndrome. Front Pediatr 9:1–7. https://doi.org/10.3389/fped.2021.624116
Rieux-Laucat F, Magérus-Chatinet A, Neven B (2018) The autoimmune lymphoproliferative syndrome with defective FAS or FAS-ligand functions. J Clin Immunol 38:558–568. https://doi.org/10.1007/s10875-018-0523-x
Peter ME, Hadji A, Murmann AE et al (2015) The role of CD95 and CD95 ligand in cancer. Cell Death Differ 22:549–559. https://doi.org/10.1038/cdd.2015.3
Han W, Zhou Y, Zhong R et al (2013) Functional polymorphisms in FAS/FASL system increase the risk of neuroblastoma in Chinese population. PLoS One 8. https://doi.org/10.1371/journal.pone.0071656
Moudi B, Salimi S, Farajian Mashhadi F et al (2013) Association of FAS and FAS ligand genes polymorphism and risk of systemic lupus erythematosus. Sci World J. https://doi.org/10.1155/2013/176741
Tarbox JA, Keppel MP, Topcagic N et al (2014) Elevated double negative T cells in pediatric autoimmunity. J Clin Immunol 34:594–599. https://doi.org/10.1007/s10875-014-0038-z
Haapaniemi EM, Kaustio M, Rajala HLM et al (2015) Autoimmunity, hypogammaglobulinemia, lymphoproliferation and mycobacterial disease in patients with activating mutations in STAT3. Blood 125:639–649. https://doi.org/10.1182/blood-2014-04-570101.The
Consonni F, Dotta L, Todaro F et al (2017) Signal transducer and activator of transcription gain-of-function primary immunodeficiency/immunodysregulation disorders. Curr Opin Pediatr 29:1. https://doi.org/10.1097/MOP.0000000000000551
Kucuk ZY, Charbonnier LM, McMasters RL et al (2017) CTLA-4 haploinsufficiency in a patient with an autoimmune lymphoproliferative disorder. J Allergy Clin Immunol 140:862–864.e4. https://doi.org/10.1016/j.jaci.2017.02.032
Ravell JC, Matsuda-Lennikov M, Chauvin SD et al (2020) Defective glycosylation and multisystem abnormalities characterize the primary immunodeficiency XMEN disease. J Clin Invest 130:507–522. https://doi.org/10.1172/JCI131116
Rieux-Laucat F, Fischer A, Le Deist F (2003) Cell-death signaling and human disease. Curr Opin Immunol 15:325–331. https://doi.org/10.1016/S0952-7915(03)00042-6
Nabhani S, Hönscheid A, Oommen PT et al (2014) A novel homozygous Fas ligand mutation leads to early protein truncation, abrogation of death receptor and reverse signaling and a severe form of the autoimmune lymphoproliferative syndrome. Clin Immunol 155:231–237. https://doi.org/10.1016/j.clim.2014.10.006
Sobh A, Crestani E, Cangemi B et al (2016) Autoimmune lymphoproliferative syndrome caused by a homozygous FasL mutation that disrupts FasL assembly. J Allergy Clin Immunol 137:324–327.e2. https://doi.org/10.1016/j.jaci.2015.08.025
Bolze A, Byun M, McDonald D et al (2010) Whole-exome-sequencing-based discovery of human FADD deficiency. Am J Hum Genet 87:873–881. https://doi.org/10.1016/j.ajhg.2010.10.028
Chun HJ, Zheng L, Ahmad M et al (2002) Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature 419:395–399. https://doi.org/10.1038/nature01063
Rensing-Ehl A, Janda A, Lorenz MR et al (2013) Sequential decisions on FAS sequencing guided by biomarkers in patients with lymphoproliferation and autoimmune cytopenia. Haematologica 98:1948–1955. https://doi.org/10.3324/haematol.2012.081901
Molnár E, Radwan N, Kovács G et al (2020) Key diagnostic markers for autoimmune lymphoproliferative syndrome with molecular genetic diagnosis. Blood 136:1933–1945. https://doi.org/10.1182/BLOOD.2020005486
McClain KL, Gulati N (2020) ALPS or not? Blood 136:1895–1896. https://doi.org/10.1182/BLOOD.2020007418
Clementi R, Dagna L, Dianzani U et al (2004) Inherited perforin and Fas mutations in a patient with autoimmune lymphoproliferative syndrome and lymphoma. N Engl J Med 351:1419–1423. https://doi.org/10.1056/NEJMoa041432
Stepp SE, Dufourcq-lagelouse R, Le Deist F et al (1999) Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science 286(80):1957–1959. https://doi.org/10.1126/science.286.5446.1957
Hanson EP, Monaco-Shawver L, Solt LA et al (2008) Hypomorphic nuclear factor-κB essential modulator mutation database and reconstitution system identifies phenotypic and immunologic diversity. J Allergy Clin Immunol 122:1169–1193. https://doi.org/10.1016/j.jaci.2008.08.018
Niehues T, Reichenbach J, Neubert J et al (2004) Nuclear factor κB essential modulator-deficient child with immunodeficiency yet without anhidrotic ectodermal dysplasia. J Allergy Clin Immunol 114:1456–1462. https://doi.org/10.1016/j.jaci.2004.08.047
Todaro F, Tamassia N, Pinelli M et al (2019) Multisystem autoimmune disease caused by increased STAT3 phosphorylation and dysregulated gene expression. Haematologica 104:322–325. https://doi.org/10.3324/haematol.2018.202374
Milner JD, Vogel TP, Forbes L et al (2015) Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood 125:591–599. https://doi.org/10.1182/blood-2014-09-602763
Delmonte OM, Schuetz C, Notarangelo LD (2018) RAG deficiency: two genes, many diseases. J Clin Immunol 38:646–655. https://doi.org/10.1007/s10875-018-0537-4
Dianzani U, Bragardo M, DiFranco D et al (1997) Deficiency of the Fas apoptosis pathway without Fas gene mutations in pediatric patients with autoimmunity/lymphoproliferation. Blood 89:2871–2879. https://doi.org/10.1182/blood.v89.8.2871
Ramenghi U, Bonissoni S, Migliaretti G et al (2000) Deficiency of the Fas apoptosis pathway without Fas gene mutations is a familial trait predisposing to development of autoimmune diseases and cancer. Blood 95:3176–3182. https://doi.org/10.1182/blood.v95.10.3176
Oliveira JB, Bidère N, Niemela JE et al (2007) NRAS mutation causes a human autoimmune lymphoproliferative syndrome. Proc Natl Acad Sci USA 104:8953–8958. https://doi.org/10.1073/pnas.0702975104
Niemela JE, Lu L, Fleisher TA et al (2011) Somatic KRAS mutations associated with a human nonmalignant syndrome of autoimmunity and abnormal leukocyte homeostasis. Blood 117:2883–2886. https://doi.org/10.1182/blood-2010-07-295501
Shah S, Wu E, Rao VK, Tarrant TK (2014) Autoimmune lymphoproliferative syndrome: an update and review of the literature. Curr Allergy Asthma Rep 14:1–10. https://doi.org/10.1007/s11882-014-0462-4
Huang B, Eberstadt M, Olejniczak ET et al (1996) NMR structure and mutagenesis of the Fas (APO-1/CD95) death domain. Nature 384:356–358. https://doi.org/10.1038/384638a0
Watanabe-Fukunaga R, Brannan CI, Copeland NG et al (1992) Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature 356:314–317
Matsusawa A, Moriyama T, Kaneko T et al (1990) A new allele of the lpr locus, lprcg, that complements the gld gene in induction of lymphadenopathy in the mouse. J Exp Med 171:519–531. https://doi.org/10.1084/jem.171.2.519
Kasahara Y, Wada T, Niida Y et al (1998) Novel Fas (CD95/APO-1) mutations in infants with a lymphoproliferative disorder. Int Immunol 10:195–202. https://doi.org/10.1093/intimm/10.2.195
Siegel RM, Frederiksen JK, Zacharias DA et al (2000) Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science 288(80):2354–2357. https://doi.org/10.1126/science.288.5475.2354
Roesler J, Izquierdo JM, Ryser M et al (2005) Haploinsufficiency, rather than the effect of an excessive production of soluble CD95 (CD95ΔTM), is the basis for ALPS Ia in a family with duplicated 3′ splice site AG in CD95 intron 5 on one allele. Blood 106:1652–1659. https://doi.org/10.1182/blood-2004-08-3104
Lambotte O, Neven B, Galicier L et al (2013) Diagnosis of autoimmune lymphoproliferative syndrome caused by FAS deficiency in adults. Haematologica 98:389–392. https://doi.org/10.3324/haematol.2012.067488
Bettinardi A, Brugnoni D, Quiròs-Roldan E et al (1997) Missense mutations in the fas gene resulting in autoimmune lymphoproliferative syndrome: a molecular and immunological analysis. Blood 89:902–909. https://doi.org/10.1182/blood.v89.3.902
Van Der Burg M, De Groot R, Comans-Bitter WM et al (2000) Autoimmune lymphoproliferative syndrome (ALPS) in a child from consanguineous parents: a dominant or recessive disease? Pediatr Res 47:336–343. https://doi.org/10.1203/00006450-200003000-00009
Kuehn HS, Caminha I, Niemela JE et al (2011) FAS haploinsufficiency is a common disease mechanism in the human autoimmune lymphoproliferative syndrome. J Immunol 186:6035–6043. https://doi.org/10.4049/jimmunol.1100021
Simesen De Bielke MG, Yancoski J, Rocco C et al (2012) A missense mutation in the extracellular domain of Fas: the most common change in argentinean patients with autoimmune lymphoproliferative syndrome represents a founder effect. J Clin Immunol 32:1197–1203. https://doi.org/10.1007/s10875-012-9731-y
Hsu AP, Dowdell KC, Davis J et al (2012) Autoimmune lymphoproliferative syndrome due to FAS mutations outside the signal-transducing death domain: molecular mechanisms and clinical penetrance. Genet Med 14:81–89. https://doi.org/10.1038/gim.0b013e3182310b7d
Hauck F, Magerus-Chatinet A, Vicca S et al (2013) Somatic loss of heterozygosity, but not haploinsufficiency alone, leads to full-blown autoimmune lymphoproliferative syndrome in 1 of 12 family members with FAS start codon mutation. Clin Immunol 147:61–68. https://doi.org/10.1016/j.clim.2013.02.019
Neven B, Magerus-Chatinet A, Florkin B et al (2011) Asurvey of 90 patients with autoimmune lymphoproliferative syndrome related to TNFRSF6 mutation. Blood 118:4798–4807. https://doi.org/10.1182/blood-2011-04-347641
Chinn IK, Chan AY, Chen K et al (2020) Diagnostic interpretation of genetic studies in patients with primary immunodeficiency diseases: a working group report of the Primary Immunodeficiency Diseases Committee of the American Academy of Allergy, Asthma & Immunology. J Allergy Clin Immunol 145:46–69. https://doi.org/10.1016/j.jaci.2019.09.009
Aluri J, Cooper MA (2021) Genetic mosaicism as a cause of inborn errors of immunity. J Clin Immunol 41:718–728. https://doi.org/10.1007/s10875-021-01037-z
Knudson AG (1971) Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A 68:820–823. https://doi.org/10.1073/pnas.68.4.820
Le Deist F, Emile JF, Rieux-Laucat F et al (1996) Clinical, immunological, and pathological consequences of Fas-deficient conditions. Lancet 348:719–723. https://doi.org/10.1016/S0140-6736(96)02293-3
Del-Rey M, Ruiz-Contreras J, Bosque A et al (2006) A homozygous Fas ligand gene mutation in a patient causes a new type of autoimmune lymphoproliferative syndrome. Blood 108:1306–1312. https://doi.org/10.1182/blood-2006-04-015776
Magerus-Chatinet A, Stolzenberg MC, Lanzarotti N et al (2013) Autoimmune lymphoproliferative syndrome caused by a homozygous null FAS ligand (FASLG) mutation. J Allergy Clin Immunol 131:486–490. https://doi.org/10.1016/j.jaci.2012.06.011
Bi LL, Pan G, Atkinson TP et al (2007) Dominant inhibition of Fas ligand-mediated apoptosis due to a heterozygous mutation associated with autoimmune lymphoproliferative syndrome (ALPS) Type Ib. BMC Med Genet 8:1–14. https://doi.org/10.1186/1471-2350-8-41
Cerutti E, Campagnoli MF, Ferretti M et al (2007) Co-inherited mutations of Fas and caspase-10 in development of the autoimmune lymphoproliferative syndrome. BMC Immunol 8:1–9. https://doi.org/10.1186/1471-2172-8-28
Zhu S, Hsu AP, Vacek MM et al (2006) Genetic alterations in caspase-10 may be causative or protective in autoimmune lymphoproliferative syndrome. Hum Genet 119:284–294. https://doi.org/10.1007/s00439-006-0138-9
Miano M, Cappelli E, Pezzulla A et al (2019) FAS-mediated apoptosis impairment in patients with ALPS/ALPS-like phenotype carrying variants on CASP10 gene. Br J Haematol 187:502–508
Campagnoli MF, Garbarini L, Quarello P et al (2006) The broad spectrum of autoimmune lymphoproliferative disease: molecular bases, clinical features and long-term follow-up in 31 patients. Haematologica 91:538–541
Martinez-Feito A, Melero J, Mora-Diaz S et al (2016) Autoimmune lymphoproliferative syndrome due to somatic FAS mutation (ALPS-sFAS) combined with a germline caspase-10 (CASP10) variation. Immunobiology 221:40–47. https://doi.org/10.1016/j.imbio.2015.08.004
Aricò M, Boggio E, Cetica V et al (2013) Variations of the UNC13D gene in patients with autoimmune lymphoproliferative syndrome. PLoS One 8:1–9. https://doi.org/10.1371/journal.pone.0068045
Boggio E, Aricò M, Melensi M et al (2013) Mutation of FAS, XIAP, and UNC13D genes in a patient with a complex lymphoproliferative phenotype. Pediatrics 132. https://doi.org/10.1542/peds.2012-1838
Chiocchetti A, Indelicato M, Bensi T et al (2004) High levels of osteopontin associated with polymorphisms in its gene are a risk factor for development of autoimmunity/lymphoproliferation. Blood 103:1376–1382. https://doi.org/10.1182/blood-2003-05-1748
Guo Q, Zhang J, Li J et al (2013) Forced miR-146a expression causes autoimmune lymphoproliferative syndrome in mice via downregulation of Fas in germinal center B cells. Blood 121:4875–4883. https://doi.org/10.1182/blood-2012-08-452425
Yan H, Hong Y, Cai Y (2020) Association between FAS gene −670 A/G and −1377 G/A polymorphisms and the risk of autoimmune diseases: a meta-analysis. Biosci Rep 40:1–18. https://doi.org/10.1042/BSR20191197
Verim L, Timirci-Kahraman O, Akbulut H et al (2014) Functional genetic variants in apoptosis-associated FAS and FASL genes and risk of bladder cancer in a Turkish population. In Vivo (Brooklyn) 28:397–402
Li H, Tsokos GC (2021) Double-negative T cells in autoimmune diseases. Curr Opin Rheumatol 33:163–172. https://doi.org/10.1097/BOR.0000000000000778
Guegan JP, Legembre P (2018) Nonapoptotic functions of Fas/CD95 in the immune response. FEBS J 285:809–827. https://doi.org/10.1111/febs.14292
Chen L, Park SM, Tumanov AV et al (2010) CD95 promotes tumour growth. Nature 465:492–496. https://doi.org/10.1038/nature09075
Villa-Morales M, Fernandez-Piqueras J (2012) Targeting the Fas/FasL signaling pathway in cancer therapy. Expert Opin Ther Targets 16:85–101. https://doi.org/10.1517/14728222.2011.628937
Zhong-Xing Z, Yuan-Yuan M, Hai Zhen M et al (2013) FAS-1377 G/A (rs2234767) Polymorphism and cancer susceptibility: a meta-analysis of 17,858 cases and 24,311 controls. PLoS One 8:1–9. https://doi.org/10.1371/journal.pone.0073700
Li P, Huang P, Yang Y et al (2016) Updated understanding of autoimmune lymphoproliferative syndrome (ALPS). Clin Rev Allergy Immunol 50:55–63. https://doi.org/10.1007/s12016-015-8466-y
Huang Y, Deng D, Li H et al (2016) Fas-670A>G polymorphism is not associated with an increased risk of acute myeloid leukemia development. Biomed Reports 4:153–160. https://doi.org/10.3892/br.2015.564
Hao Z, Duncan GS, Seagal J et al (2008) Fas receptor expression in germinal-center B cells is essential for T and B lymphocyte homeostasis. Immunity 29:615–627. https://doi.org/10.1016/j.immuni.2008.07.016
Rensing-Ehl A, Warnatz K, Fuchs S et al (2010) Clinical and immunological overlap between autoimmune lymphoproliferative syndrome and common variable immunodeficiency. Clin Immunol 137:357–365. https://doi.org/10.1016/j.clim.2010.08.008
Ghodke-Puranik Y, Niewold TB (2015) Immunogenetics of systemic lupus erythematosus: a comprehensive review. J Autoimmun 64:125–136. https://doi.org/10.1016/j.jaut.2015.08.004
Chasset F, Richez C, Martin T et al (2019) Rare diseases that mimic systemic lupus erythematosus (lupus mimickers). Jt Bone Spine 86:165–171. https://doi.org/10.1016/j.jbspin.2018.10.007
Glesse N, Vianna P, Paim LMG et al (2017) Evaluation of polymorphic variants in apoptotic genes and their role in susceptibility and clinical progression to systemic lupus erythematosus. Lupus 26:746–755. https://doi.org/10.1177/0961203316678671
Erdogan M, Kulaksizoglu M, Ganidagli S, Berdeli A (2017) Fas/FasL gene polymorphism in patients with Hashimoto’s thyroiditis in Turkish population. J Endocrinol Invest 40:77–82. https://doi.org/10.1007/s40618-016-0534-5
Xiang Z, Yang Y, Chang C, Lu Q (2017) The epigenetic mechanism for discordance of autoimmunity in monozygotic twins. J Autoimmun 83:43–50. https://doi.org/10.1016/j.jaut.2017.04.003
Brandt D, Hedrich CM (2018) TCRαβ+CD3+CD4−CD8− (double negative) T cells in autoimmunity. Autoimmun Rev 17:422–430. https://doi.org/10.1016/j.autrev.2018.02.001
Mixter PF, Russell JQ, Morrissette GJ et al (1999) A model for the origin of TCR-alphabeta+ CD4-CD8- B220+ cells based on high affinity TCR signals. J Immunol 162:5747–5756
Tian Y, Babor M, Lane J et al (2017) Unique phenotypes and clonal expansions of human CD4 effector memory T cells re-expressing CD45RA. Nat Commun 8. https://doi.org/10.1038/s41467-017-01728-5
Crispín JC, Tsokos GC (2009) Human TCR-αβ + CD4 − CD8 − T cells can derive from CD8 + T cells and display an inflammatory effector phenotype. J Immunol 183:4675–4681. https://doi.org/10.4049/jimmunol.0901533
Rensing-Ehl A, Volkl S, Speckmann C et al (2014) Abnormally differentiated CD41 or CD81 T cells with phenotypic and genetic features of double negative T cells in human Fas deficienc. Blood 124:851–860. https://doi.org/10.1182/blood-2014-03-564286
Maccari ME, Fuchs S, Kury P et al (2020) A distinct CD38+ CD45RA+ population of CD4+,CD8+, and double-negative T cells is controlled by FAS. J Exp Med 218. https://doi.org/10.1084/jem.201921911of
D’Acquisto F, Crompton T (2011) CD3+CD4-CD8- (double negative) T cells: saviours or villains of the immune response? Biochem Pharmacol 82:333–340. https://doi.org/10.1016/j.bcp.2011.05.019
Bleesing JJH, Brown MR, Novicio C et al (2002) A composite picture of TcRα/β+ CD4-CD8- T cells (α/β-DNTCs) in humans with autoimmune lymphoproliferative syndrome. Clin Immunol 104:21–30. https://doi.org/10.1006/clim.2002.5225
Lisco A, Wong CS, Price S et al (2019) Paradoxical CD4 lymphopenia in autoimmune lymphoproliferative syndrome (ALPS). Front Immunol 10:1–13. https://doi.org/10.3389/fimmu.2019.01193
Li H, Adamopoulos IE, Moulton VR et al (2020) Systemic lupus erythematosus favors the generation of IL-17 producing double negative T cells. Nat Commun 11:21–27. https://doi.org/10.1038/s41467-020-16636-4
McGaha TL, Chen Y, Ravishankar B et al (2011) Marginal zone macrophages suppress innate and adaptive immunity to apoptotic cells in the spleen. Blood 117:5403–5412. https://doi.org/10.1182/blood-2010-11-320028
Alunno A, Bistoni O, Bartoloni E et al (2013) IL-17-producing CD4-CD8- T cells are expanded in the peripheral blood, infiltrate salivary glands and are resistant to corticosteroids in patients with primary Sjögren’s syndrome. Ann Rheum Dis 72:286–292. https://doi.org/10.1136/annrheumdis-2012-201511
Brandt D, Sergon M, Abraham S et al (2017) TCR+ CD3+ CD4− CD8− effector T cells in psoriasis. Clin Immunol 181:51–59. https://doi.org/10.1016/j.clim.2017.06.002
Sherlock JP, Joyce-Shaikh B, Turner SP et al (2012) IL-23 induces spondyloarthropathy by acting on ROR-γt+ CD3+ CD4- CD8- entheseal resident T cells. Nat Med 18:1069–1076. https://doi.org/10.1038/nm.2817
Tarbox JA, Keppel MP, Topcagic N et al (2014) Elevated double negative T cells in pediatric autoimmunity. J Clin Immunol 34:594–599. https://doi.org/10.1007/s10875-014-0038-z
Ling E, Shubinsky G, Press J (2007) Increased proportion of CD3+CD4-CD8- double-negative T cells in peripheral blood of children with Behcet’s disease. Autoimmun Rev 6:237–240. https://doi.org/10.1016/j.autrev.2006.08.008
Fischer K, Voelkl S, Heymann J et al (2005) Isolation and characterization of human antigen-specific TCRab+CD4- CD8- double-negative regulatory T cells. Blood 105:2828–2836. https://doi.org/10.1182/blood-2004-07-2583.Supported
Juvet SC, Zhang L (2012) Double negative regulatory T cells in transplantation and autoimmunity: Recent progress and future directions. J Mol Cell Biol 4:48–58. https://doi.org/10.1093/jmcb/mjr043
Duncan B, Nazarov-Stoica C, Surls J et al (2010) Double negative (CD3+4-8-) TCRαβ splenic cells from young NOD mice provide long-lasting protection against type 1 diabetes. PLoS One 5. https://doi.org/10.1371/journal.pone.0011427
Su Y, Huang X, Wang S et al (2012) Double negative Treg cells promote nonmyeloablative bone marrow chimerism by inducing T-cell clonal deletion and suppressing NK cell function. Eur J Immunol 42:1216–1225. https://doi.org/10.1002/eji.201141808
McIver Z, Serio B, Dunbar A et al (2008) Double-negative regulatory T cells induce allotolerance when expanded after allogeneic haematopoietic stem cell transplantation. Br J Haematol 141:170–178. https://doi.org/10.1111/j.1365-2141.2008.07021.x
Hillhouse EE, Thiant S, Moutuou MM et al (2019) Double-negative T cell levels correlate with chronic graft-versus-host disease severity. Biol Blood Marrow Transplant 25:19–25. https://doi.org/10.1016/j.bbmt.2018.09.008
Straus SE, Jaffe ES, Puck JM et al (2001) The development of lymphomas in families with autoimmune lymphoproliferative syndrome with germline Fas mutations and defective lymphocyte apoptosis. Blood 98:194–200. https://doi.org/10.1182/blood.V98.1.194
Poppema S, Maggio E, Van den Berg A (2004) Development of lymphoma in autoimmune lymphoproliferative syndrome (ALPS) and its relationship to Fas gene mutations. Leuk Lymphoma 45:423–431. https://doi.org/10.1080/10428190310001593166
Teachey DT, Greiner R, Seif A et al (2009) Treatment with sirolimus results in complete responses in patients with autoimmune lymphoproliferative syndrome. Br J Haematol 145:101–106. https://doi.org/10.1111/j.1365-2141.2009.07595.x
Bride KL, Vincent T, Smith-Whitley K et al (2016) Sirolimus is effective in relapsed/refractory autoimmune cytopenias: results of a prospective multi-Institutional trial. Blood 127:17–28. https://doi.org/10.1182/blood-2015-07-657981
Klemann C, Esquivel M, Magerus-Chatinet A et al (2017) Evolution of disease activity and biomarkers on and off rapamycin in 28 patients with autoimmune lymphoproliferative syndrome. Haematologica 102:e52–e56. https://doi.org/10.3324/haematol.2016.153411
Völkl S, Rensing-Ehl A, Allgäuer A et al (2016) Hyperactive mTOR pathway promotes lymphoproliferation and abnormal differentiation in autoimmune lymphoproliferative syndrome. Blood 128:227–238. https://doi.org/10.1182/blood-2015-11-685024
Neven B, Bruneau J, Stolzenberg MC et al (2014) Defective anti-polysaccharide response and splenic marginal zone disorganization in ALPS patients. Blood 124:1597–1609. https://doi.org/10.1182/blood-2014-02-553834
Go RS, Winters JL, Kay NE (2017) How I treat autoimmune hemolytic anemia. Blood 129:2971–2979. https://doi.org/10.1182/blood-2016-11-693689
Farruggia P, Dufour C (2015) Diagnosis and management of primary autoimmune neutropenia in children: Insights for clinicians. Ther Adv Hematol 6:15–24. https://doi.org/10.1177/2040620714556642
Cuker A, Neunert CE (2016) How I treat refractory immune thrombocytopenia. Blood 128:1547–1554. https://doi.org/10.1182/blood-2016-03-603365
Rao VK, Price S, Perkins K et al (2009) Use of rituximab for refractory cytopenias associated with autoimmune lymphoproliferative syndrome (ALPS). Pediatr Blood Cancer 52:847–852. https://doi.org/10.1002/pbc.21965
Rao VK, Dugan F, Dale JK et al (2005) Use of mycophenolate mofetil for chronic, refractory immune cytopenias in children with autoimmune lymphoproliferative syndrome. Br J Haematol 129:534–538. https://doi.org/10.1111/j.1365-2141.2005.05496.x
Boulanger E, Rieux-Laucat F, Picard C et al (2001) Diffuse large B-cell non-Hodgkin’s lymphoma in a patient with autoimmune lymphoproliferative syndrome. Br J Haematol 113:432–434. https://doi.org/10.1046/j.1365-2141.2001.02749.x
Sleight BJ, Prasad VS, DeLaat C et al (1998) Correction of autoimmune lymphoproliferative syndrome by bone marrow transplantation. Bone Marrow Transplant 22:375–380. https://doi.org/10.1038/sj.bmt.1701306
Benkerrou M, Le Deist F, De Villartay JP et al (1997) Correction of Fas (CD95) deficiency by haploidentical bone marrow transplantation. Eur J Immunol 27:2043–2047. https://doi.org/10.1002/eji.1830270831
Hafezi N, Zaki-Dizaji M, Nirouei M et al (2021) Clinical, immunological, and genetic features in 780 patients with autoimmune lymphoproliferative syndrome (ALPS) and ALPS-like diseases: a systematic review. Pediatr Allergy Immunol 32:1519–1532. https://doi.org/10.1111/pai.13535
Seidel MG (2020) Treatment of immune-mediated cytopenias in patients with primary immunodeficiencies and immune regulatory disorders (PIRDs). Hematol (United States) 20:673–679. https://doi.org/10.1182/HEMATOLOGY.2020000153
Schiavo E, Martini B, Attardi E et al (2022) Autoimmune cytopenias and dysregulated immunophenotype act as warning signs of inborn errors of immunity: results from a prospective study. Front Immunol 12:1–12. https://doi.org/10.3389/fimmu.2021.790455
Seidel MG (2014) Autoimmune and other cytopenias in primary immunodeficiencies: pathomechanisms, novel differential diagnoses, and treatment. Blood 124:2337–2344. https://doi.org/10.1182/blood-2014-06-583260
Delmonte OM, Castagnoli R, Calzoni E, Notarangelo LD (2019) Inborn errors of immunity with immune dysregulation: from bench to bedside. Front Pediatr 7:1–14. https://doi.org/10.3389/fped.2019.00353
Forbes LR, Vogel TP, Cooper MA et al (2018) Jakinibs for the treatment of immune dysregulation in patients with gain-of-function signal transducer and activator of transcription 1 (STAT1) or STAT3 mutations. J Allergy Clin Immunol 142:1665–1669. https://doi.org/10.1016/j.jaci.2018.07.020
Lee S, Moon JS, Lee CR et al (2016) Abatacept alleviates severe autoimmune symptoms in a patient carrying a de novo variant in CTLA-4. J Allergy Clin Immunol 137:327–330. https://doi.org/10.1016/j.jaci.2015.08.036
Lo B, Zhang K, Lu W et al (2015) Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science 349(80):436–440. https://doi.org/10.1126/science.aaa1663
Rao VK, Webster S, Dalm VASH et al (2017) Effective “activated PI3Kδ syndrome”–targeted therapy with the PI3Kδ inhibitor leniolisib. Blood 130:2307–2316. https://doi.org/10.1182/blood-2017-08-801191
Lanzarotti N, Bruneau J, Trinquand A et al (2014) RAS-associated lymphoproliferative disease evolves into severe juvenile myelo-monocytic leukemia. Blood 123:1960–1963. https://doi.org/10.1182/blood-2014-01-548958
Goodwin M, Lee E, Lakshmanan U et al (2020) CRISPR-based gene editing enables FOXP3 gene repair in IPEX patient cells. Sci Adv 6. https://doi.org/10.1126/sciadv.aaz0571
Consonni F, Favre C, Gambineri E (2021) IL-2 signaling axis defects: how many faces? Front Pediatr 9. https://doi.org/10.3389/fped.2021.669298
Notarangelo LD, Bacchetta R, Casanova JL, Su HC (2020) Human inborn errors of immunity: an expanding universe. Sci Immunol 5. https://doi.org/10.1126/sciimmunol.abb1662
Consonni F, Mannurita SC, Gambineri E (2021) Atypical presentations of IPEX : expect the unexpected. Front Pediatr 9:1–11. https://doi.org/10.3389/fped.2021.643094
Acknowledgements
Figures 1, 2, and 3 were created with BioRender.com and were exported under a paid subscription.
Author information
Authors and Affiliations
Contributions
CF and FC had the idea for the article, EG and FC performed literature search and data analysis, FC wrote the original draft, and EG and CF critically revised the work. All authors commented on the previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
This article does not contain any studies with human participants performed by any of the authors.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Consonni, F., Gambineri, E. & Favre, C. ALPS, FAS, and beyond: from inborn errors of immunity to acquired immunodeficiencies. Ann Hematol 101, 469–484 (2022). https://doi.org/10.1007/s00277-022-04761-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-022-04761-7