
Abstract
SF3B1 is a highly mutated gene in myelodysplastic syndrome (MDS) patients, related to a specific subtype and parameters of good prognosis in MDS without excess blasts. More than 40% of MDS patients carry at least two myeloid-related gene mutations but little is known about the impact of concurrent mutations on the outcome of MDS patients. In applying next-generation sequencing (NGS) with a 117 myeloid gene custom panel, we analyzed the co-occurrence of SF3B1 with other mutations to reveal their clinical, biological, and prognostic implications in very low/low- and intermediate-risk MDS patients. Mutations in addition to those of SF3B1 were present in 80.4% of patients (median of 2 additional mutations/patient, range 0–5). The most frequently mutated genes were as follows: TET2 (39.2%), DNMT3A (25.5%), SRSF2 (10.8%), CDH23 (5.9%), and ASXL1, CUX1, and KMT2D (4.9% each). The presence of at least two mutations concomitant with that of SF3B1 had an adverse impact on survival compared with those with the SF3B1 mutation and fewer than two additional mutations (median of 54 vs. 87 months, respectively: p = 0.007). The co-occurrence of SF3B1 mutations with specific genes is also linked to a dismal prognosis: SRSF2 mutations were associated with shorter overall survival (OS) than SRSF2wt (median, 27 vs. 75 months, respectively; p = 0.001), concomitant IDH2 mutations (median OS, 11 [mut] vs. 75 [wt] months; p = 0.001), BCOR mutations (median OS, 11 [mut] vs. 71 [wt] months; p = 0.036), and NUP98 and STAG2 mutations (median OS, 27 and 11 vs. 71 months, respectively; p = 0.008 and p = 0.002). Mutations in CHIP genes (TET2, DNMT3A) did not significantly affect the clinical features or outcome. Our results suggest that a more comprehensive NGS study in low-risk MDS SF3B1mut patients is essential for a better prognostic evaluation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Myelodysplastic syndromes (MDS) are a heterogeneous group of clonal hematopoietic stem cell disorders leading to abnormal blood production and that have a variable risk of progression to acute myeloid leukemia (AML) [1,2,3].
In recent years, large-scale analysis using next-generation sequencing (NGS) has made it possible to identify recurrent genetic alterations, thereby improving our knowledge of MDS pathogenesis [4,5,6,7,8]. More than 80% of MDS patients harbor at least one mutation, affecting genes from a variety of functional groups: splicing machinery (SF3B1, SRSF2), DNA methylation (DNMT3A, TET2), transcription factors (TP53, RUNX1), chromatin modification (ASXL1, EZH2), RAS pathway (KRAS, NRAS), cohesin complex (STAG2, RAD21), kinases (JAK2, FLT3), and/or DNA repair (ATM, BRCC3) [4, 9,10,11].
Several clinical and biological implications of specific mutations have been demonstrated. Some of these gene mutations have been associated with morphological and clinical features such as complex karyotypes (TP53), excess proportions of bone marrow blast (RUNX1, NRAS) or ring sideroblasts (SF3B1), and with the prognosis for leukemia-free and overall survival [12, 13]. Mutations in TP53, U2AF1, RUNX1, SRSF2, IDH2, CUX1, ASXL1, and BCOR genes are associated with significantly worse leukemia-free survival [5]. Those in TP53, EZH2, ETV6, RUNX1, and ASXL1 are predictors of poor overall survival (OS), while mutations in SF3B1 are associated with a better outcome in MDS patients [6, 14]. Most of these studies have analyzed single mutations, but little is known about the impact of concurrent mutations on the outcome of MDS patients [15].
It is common for more than one mutation to be present in MDS. Papaemmanuil and colleagues clearly demonstrated the variability of gene mutation frequencies in a large series of MDS, whereby 40% of cases had 2 or 3 mutated genes and up to 10% of patients presented 4 to 8 oncogenic point mutations. All these features are associated with a more complex disease and have an adverse effect on OS [4, 5]. SF3B1 encodes a core component of the RNA splicing machinery. NGS studies have revealed that approximately 30% of MDS cases have a mutation of the SF3B1 gene, with a particularly high prevalence (> 90%) in the MDS with ring sideroblasts subtype (MDS-RS), as reflected in the most recent 2017 WHO classification [3, 13, 14]. Furthermore, SF3B1 mutations in low-risk MDS patients were associated with good prognostic parameters [13]. Although no significant affinity of SF3B1 with common mutational genes other than DNMT3A and JAK2 has been found, various gene mutations co-existing with SF3B1 have been described [4, 5].
Nevertheless, and given that the majority of MDS patients carry multiple genetic alterations, the well-known, better OS and leukemia-free survival referred to SF3B1 mutations in low-risk MDS patients may be worse. Recent studies in low-risk MDS–RS patients have highlighted the adverse influence of coexisting DNMT3A and ASXL1-SF3B1 mutations on clinical outcome [15, 16].
However, information about the influence of other gene mutations co-existing with SF3B1mut in MDS patients is scarce. Detailed molecular characterization of these groups would allow a better stratification of patients within the low-risk MDS categories as well as a better choice of treatment for these patients. The aim of this study was to analyze, by means of NGS, the presence of mutations associated with SF3B1 mut and to evaluate the prognostic value in a large series of very low/low/intermediate-risk MDS patients.
Materials and methods
Patients
The mutational profiles of 324 MDS patients, diagnosed in our center between 1999 and 2017, were analyzed. Diagnosis was based on the World Health Organization (WHO) criteria [3, 17]. Patients with refractory anemia with excess of blasts (RAEB) and MDS associated with isolated del(5q) were excluded from the study.
SF3B1 mutations were detected in 135 patients (42%). Detailed analysis of clinical parameters and cytogenetic findings was performed to facilitate risk stratification, according to the International Prognostic Scoring System (IPSS) and the revised IPSS (IPSS-R) [18, 19]. Finally, a total of 102 MDS cases with SF3B1 mutations and very low, low, and intermediate IPSS-R scores were included. The study was approved by the Local Ethical Committee (Comité Ético de Investigación Clínica, Hospital Universitario de Salamanca), and written informed consent was obtained for each patient according to the guidelines of the Declaration of Helsinki.
Mutational analysis
Mutational screening of genomic non-amplified DNA from bone marrow (BM) or peripheral blood (PB) cells was performed. A customized myeloid panel of 117 MDS-related genes was applied (Supplementary Table S1). NGS was carried out on a NextSeq sequencing platform (Illumina, San Diego, CA, USA) following Illumina’s standard protocol for Nextera Rapid Capture Enrichment. Sequencing data were analyzed by applying an in-house informatic pipeline that uses different software tools to perform quality assessment, alignment, and variant calling (Trimmomatic, FastQC, NGSQCToolkit, BWA, GATK, VarScan, SAMTools, ANNOVAR). The Integrative Genomics Viewer (IGV, Broad Institute) was used to evaluate variants visually.
After analysis, only those variants of good quality (Q > 30), supported by ≥ 100 total and ≥ 10 mutated reads, with a variant allele frequency (VAF) ≥ 3%, located in exonic or splicing regions, and which generate an amino acid change were considered. In addition, already reported polymorphisms (SNPs) (dbSNP144, 1000-genomes Project, ExAC, ESP-6500, when MAF ≥ 1%) and sequencing artifacts (internal laboratory database where distortions in data processing caused by a measuring instrument, already seen in previous NGS analysis in our laboratory, are annotated and stocked as a reference for further analysis) were discarded. For variant interpretation and oncogenic potential evaluation, COSMIC and ClinVar databases, SIFT, PolyPhen-2, and Mutation Taster predictors were used (Supplementary Fig. 1).
Statistical analysis
Numerical variables were summarized as the median and range; categorical variables were summarized as the frequency and percentage of subjects in each category. Group differences were examined with Student’s t test or the Mann-Whitney U test, respectively, for normally and non-normally distributed continuous variables. Overall survival was measured from the time of diagnosis to the time of last follow-up or death from any cause. Survival curves were generated using the Kaplan-Meier method and differences were assessed with the log-rank test. For multivariate analysis, Cox proportional hazards models were constructed, adjusting for potential confounding covariates. Significance of all statistical tests was defined as a value of p < 0.050. Analyses were carried out using the IBM SPSS version 22.0 statistical package (IBM Corp., Armonk, NY).
Results
One hundred and two patients with a mutation in SF3B1 gene were included as lower-risk cases according to the IPSS-R: very low (34.4%), low (60.4%), or intermediate (5.2%). Regarding the WHO 2017 classification, 1% of patients had MDS with single-lineage dysplasia (MDS-SLD), 5.2% had MDS with multilineage dysplasia (MDS-MLD), 40.6% had MDS with ring sideroblasts (MDS-RS) with single-lineage dysplasia (MDS-RS-SLD), and the remaining 53.1% had MDS-RS with multilineage dysplasia (MDS-RS-MLD). The median age was 76 years (range: 41–90 years). More detailed clinical features of the cohort are summarized in Table 1.
Characterization of SF3B1 mutations in MDS patients
One hundred and seven SF3B1 mutations were found in 102 MDS patients. The median VAF was 33.6% (range 5.72–64.00%). All mutations were missense, heterozygous, and, except for three (K748E, Y623F, and L536V), had been previously reported. The most frequent mutation was K700E (47/107, 43.9%) followed by K666R (12/107, 11.2%) and E622D (9/107, 8.4%). All SF3B1 mutational variants detected are illustrated in Fig. 1. Notably, five of the 102 MDS patients presented two SF3B1 mutations. The double variants of four patients were located in exon 14 (E622D and Y623F; H662Q and K666T; and two cases with E622V and T663I), while the double SF3B1 mutation in the other patients affected exons 14 and 15 (R625H and K700E) (detailed information about all mutations is shown in Supplementary Table S2).

SF3B1 mutations distributed in patients included in the study and classified by WHO 2017 MDS subtype
Co-occurrence of SF3B1 mutations with other gene mutations
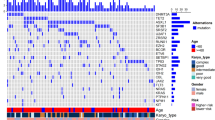
Co-occurrence of SF3B1 mutations with those of other genes was observed in 82 of 102 patients (80.4%). A total of 192 concomitant mutations, involving 51 genes, were found in the 102 SF3B1mut patients, with a median of two additional mutations per sample (range: 0–5), (Supplementary Table S2). Patients with one mutation in addition to that of SF3B1 were the most frequent type (n = 26, 25.5%), followed by two and three concurrent mutations (n = 23, 22.5%, and n = 19, 18.6%, respectively). Cases with four and five concomitant SF3B1 mutations were infrequent (n = 7, 6.9% and n = 8, 7.8%, respectively). The most frequently mutated genes co-occurring with SF3B1 were as follows: TET2 (n = 40, 39.2%), DNMT3A (n = 26, 25.5%), SRSF2 (n = 11, 10.8%), CDH23 (n = 6, 5.9%), and ASXL1, CUX1, KMT2D (n = 5, 4.9% each). Mutations were also present in the BCOR, NUP98, SMC3, SETBP1, and STAG2 genes, although at low frequencies (n = 3, 2.9% each) (Fig. 2).

Mutations in 51 genes co-occurring with SF3B1 in 102 very low, low, and intermediate-risk MDS patients
The great heterogeneity of the overall distribution of concomitant mutations is illustrated in Fig. 3. The addition of a single mutation to the SF3B1 mutation usually involves the TET2 or DNMT3A genes. However, the addition of two mutations concomitant with SF3B1 mutations did not show a clear pattern of distribution or evolution.

Mutational landscape of 102 MDS. Each column represents one patient. Patients are grouped by number of additional SF3B1 mutations. Each line represents one gene. Genes are grouped by function. Red circles indicate genes that reduce overall survival of MDS patients.VAF:  ≥ 3 < 5%
≥ 3 < 5%  ≥ 5 < 10%
≥ 5 < 10%  > 10%. Gene function:
> 10%. Gene function:  splicing,
splicing,  DNA methylation,
DNA methylation,  chromatin modification,
chromatin modification,  transcription factors,
transcription factors,  activated signaling,
activated signaling,  cohesin complex,
cohesin complex,  other. Cytogenetics:
other. Cytogenetics:  very good,
very good,  good,
good,  intermediate,
intermediate,  NA. IPSS-R:
NA. IPSS-R:  very low,
very low,  low,
low,  intermediate,
intermediate,  NA
NA
The effect of the frequency of co-occurring SF3B1 mutations
In the next step of the study, the influence of the number of concurrent mutations was analyzed, taking the clinical characteristics and overall survival (OS) into account. In general, no differences were observed in the clinical characteristic of the patients affected by between one and five of the additional SF3B1 mutations, (Supplementary Table S3). However, in the univariate analysis of the series, after a median follow-up of 54 months and a median OS of 70 months, we found that the presence of at least two mutations concomitant with SF3B1 had a more adverse effect on survival compared with those with SF3B1 + fewer than two additional mutations (median of 54 vs. 87 months, respectively, p = 0.007) (Fig. 4A). The negative impact of at least two additional SF3B1 mutations remained a significant factor in the multivariate analysis (Table 2).

Overall survival of MDS SF3B1mut patients by frequency of concomitant mutations (median 87 vs. 54 months) (A). Co-occurrence of SRSF2mut (median 75 vs. 27 months) (B). IDH2mut (median 75 vs. 11 months) (C). BCORmut (median 71 vs. 11 months) (D). NUP98mut (median 71 vs. 27 months) (E). STAG2mut (median 71 vs. 11 months) (F)
Several co-occurring SF3B1 gene mutations reduce OS of MDS patients
Frequently mutated genes overlapping with SF3B1 were analyzed (Fig. 2). There were no differences in the clinical features between groups of SF3B1mut patients with wild type or mutated forms of the most frequently mutated genes (data not provided). We observed that the presence of somatic mutations in other genes was able to modify the good prognosis of patients with isolated SF3B1mut. Thus, in the univariate analysis, co-occurrence of SF3B1 with SRSF2 mutations was associated with shorter OS than isolated SF3B1 mutations with SRSF2 wild type (median of 27 vs. 75 months, respectively; p = 0.001) (Fig. 4B). However, the negative impact of SRSF2 mutation was not retained as a significant term in the multivariate analysis (data not shown). Furthermore, although small groups of patients are involved, a similar adverse effect was observed with concomitant IDH2 mutations (median OS of 11 vs. 75 months, respectively; p = 0.001) (Fig. 4C) and BCOR mutations (median OS of 11 vs. 71 months, respectively; p = 0.036) (Fig. 4D). Interestingly, SF3B1 with NUP98, and SF3B1 with STAG2 also had a negative effect on patient prognosis (medians of 27 and 11 vs. 71 months, respectively; p = 0.008 and p = 0.002) (Fig. 4E and F). Very few patients bore these mutations, so they were not included in the multivariate analysis.
Co-occurrence of SF3B1 and clonal hematopoiesis indeterminate potential mutations
The presence of mutations in some genes considered CHIPs (clonal hematopoiesis indeterminate potentials) co-occurring with SF3B1 mutations was analyzed. More than half of MDS patients (57.8%, N = 59) displayed mutations in TET2, DNMT3A, and/or ASXL1 (Fig. 3). The comparison of the most relevant clinical and biological charactersitics, such as age, levels of hemoglobin, platelets and neutrophils, bone marrow percentage of blast and ring sideroblast, and OS, did not differ between the low-risk MDS patients with TET2 or DNMT3A as unique mutations associated with SF3B1 and SF3B1 mutations as the only abnormality (median OS of 70 and 87 months, respectively). Therefore, CHIP mutations had no effect on SF3B1-mutated cases (Table 3 and Fig. 5).

Overall survival of MDS SF3B1mut patients with respect to co-occurrence with mutations in CHIP genes (TET2 or DNMT3A) (median 87 vs. 70 months)
Discussion
SF3B1 is one of the most frequently mutated genes in MDS and the presence of mutations in this gene is associated with a favorable outcome in MDS patients without an excess of blasts [4, 13]. However, information about the appearance and influence of other concurrent mutations in prognosis is scarce, although they are important topics since at least 40% of patients with MDS have at least two mutations [4, 5, 20]. In the present study, we analyzed the co-occurrence of mutations in SF3B1 and other frequent mutations in myeloid-related genes, the consequences of these concomitant mutations for the clinical and biological phenotype, and the impact on prognosis in a cohort of 102 MDS low-risk patients without an excess of blasts. Our study confirmed that a complex mutational SF3B1 status (at least two associated mutations) is associated with a dismal prognosis in low-risk MDS patients.
To ensure the consistency of our analyses, we included only those low-risk patients who were categorized as very low, low, and intermediate risk according to the IPSS-R. Clinical features did not differ from those typical of low-risk MDS patients with respect to age, gender, and WHO classification [19, 21, 22]. Patients with excess blasts were excluded from the study because the frequency of SF3B1 mutations in this population was low and, due to an adverse outcome, predefined in this subset of patients [4, 5, 23]. In addition, and based on another recognized entity, with a clear genotype-phenotype relationship and prognosis, patients with MDS with an isolated 5q deletion were excluded from the study [24, 25].
The MDS subtype most frequently found in our cohort was MDS-RS (93.7%) according to the WHO’s 2017 classification. The percentage of RS ranged between 0 and 95%, with the majority of cases (83.3%) containing more than 15% of RS [26, 27]. The 2017 WHO classification expanded the MDS-RS group to include cases with an RS level between 5 and 15% and confirmed the SF3B1 mutation, which involved 4.9% of the cases in our cohort (Table 1) [3]. Six patients could not be classified because their RS frequency was not known. The increase in the proportion of patients diagnosed with MDS-RS following the 2017 WHO reclassification has been noted in other studies [28].
The extent of SF3B1 mutations among MDS-RS patients was similar to that reported in previous studies. All the mutations found were missense and heterozygous, with a change from lysine to glutamic acid in codon 700 (K700E) being the most frequent [14, 29]. Apart from three variants (K748E, Y623F, and L536V), all were already known to occur in hematological cancer, and at similar frequencies (Fig. 1) [30,31,32]. Interestingly, five of 102 patients had double SF3B1 mutations (E622D and Y623F; H662Q and K666T; two cases with E622V and T663I and R625H and K700E). Double mutations occur rarely in the SF3B1, although they have been reported [14, 20].
Co-occurrence analyses of the SF3B1 gene with other myeloid-related genes revealed that only 20 patients (19.3%) carried a mutation of SF3B1 as the only mutation. Previous studies had reported that more than 40% of cases of MDS had at least two mutations, which underlines the need for a detailed and complete molecular analysis to gain a better knowledge of the molecular pattern and risk to our patients, especially those with low-risk MDS [4, 5]. The isolated SF3B1 mutation appeared as a minority group in our cohort and in other studies, with a tendency to be higher (40%) among the MDS-RS cases [4, 20]. Those discrepancies may have arisen because the studies used different NGS methods and VAF cut-offs. Martin et al. used the amplicon-based technique to analyze regions of 39 genes, while we used a custom-capture strategy to interrogate exonic regions of 117 genes. Martin et al. chose a VAF cut-off of 5% whereas the threshold was 3% in the present study. Nevertheless, the majority group of patients in both series bore additional mutations that coexisted with those of SF3B1.
The TET2 and DNMT3A genes were the most frequently mutated, co-occurring with SF3B1 in 39.2% and 25.5% of cases, respectively, similar to what was noted in previous series [4, 5, 16]. Mutations in SRSF2 were the next most frequently observed. Although these are quite frequent in MDS or MDS-RS patients, they rarely co-occur with SF3B1 [8, 33]. In our previous study of splicing genes in MDS-RS patients, we identified double splicing gene mutations and noted their possible influence on the outcome of patients. This prompted us to investigate in more detail the group of patients in which SF3B1 and SRSF2 occur jointly [14]. As a consequence, we found that patients with the SRSF2 + SF3B1 mutation were another of the most frequently occurring types (10.8%) in our series. This is a novel observation; double splicing gene mutations have previosuly been considered to be mutually exclusive [4, 5]. The other gene mutations known in MDS were quite similar to those previously reported in the MDS and MDS-RS subtypes, as shown in Fig. 2 [4, 5, 20].
Previous studies suggested that a higher frequency of driver mutations worsens the clinical outcome in these patients [5, 34]. Our study confirms that SF3B1 with at least two additional mutations is an independent prognostic factor associated with shorter survival, even in these low-risk MDS patients. This could be interpreted as meaning that a highly heterogeneous mutational status gives rise to a more complex disease.
We now consider the role of the co-occurrence of particular gene mutations with SF3B1. Previous studies suggested that the presence of some gene mutations in low-risk SF3B1mut MDS patients could modify clinical features and prognosis. Malcovati et al. found that in SF3B1mut patients, RUNX1 mutations were significantly associated with worse OS and a higher cumulative incidence of disease progression [13]. Similary, Martin et al. demonstrated the same pattern in RARS patients with SF3B1 and DNMT3A mutations [15]. As far as we know, these findings have not been confirmed in other cohorts. Our own inability to corroborate these findings could be due to an insufficient number of SF3B1mut with RUNX1mut patients and the greater heterogeneity of our study cohort compared with other series [15]. Nevertheless, we found a negative impact on prognosis when there was co-occurrence of SF3B1 with SRSF2 and/or BCOR and/or IDH2 and/or NUP98 and/or STAG2 mutations. With the exception of SRSF2, the small number of affected cases available to us means that these findings need to be confirmed by the analysis of a larger cohort of patients.
In 2014, NGS studies in healthy individuals revealed that around of 10–20% of older people (≥ 65 years of age) carried somatic mutations in hematopoietic disorder–related genes, making hematopoiesis clonal. The majority of the variants occurred in three genes: DNMT3A, TET2, and ASXL1. The clonal hematopoiesis of indeterminate potential (CHIP) of healthy people helped create an increased risk of subsequent hematological cancer and of the appearance of cardiovascular diseases [35, 36]. In the current study, DNMT3A, TET2, and ASXL1 were among the most frequently mutated genes. No effect of co-occurrence with SF3B1 was seen in relation to clinical features and prognosis, although, according to previous findings, their existence could be crucial for initiating the clonal hematopoiesis that evolves to become MDS disease [35,36,37]. These results demonstrate the importance and utility of the NGS technique not only for monitoring patients’ disease course but also for making it possible to detect molecular changes (clonal hematopoiesis) early on in otherwise healthy people, thereby increasing the opportunity to improve the follow-up of receptors and immediately determine whether the disease has evolved.
In summary, our study demonstrated that the co-ocurrence of additional myeloid mutations is very frequent (80.4%) in very low/low/intermediate-risk SF3B1-mutated MDS patients. The presence of at least two additional mutations is associated with shorter survival and the presence of specific mutations concomitant with those of SF3B1 (SRSF2, IDH2, BCOR, NUP98, and/or STAG2). The comprehensive mutational study of low-risk MDS patients with SF3B1 mutation is essential if they are to receive a more accurate prognosis.
Data availability
Not applicable
References
Gangat N, Patnaik MM, Tefferi A 2016 Myelodysplastic syndromes: Contemporary review and how we treat. Am J Hematol 91(1):76–89
Bannon SA, DiNardo CD 2016 Hereditary Predispositions to Myelodysplastic Syndrome. Int J Mol Sci 17(6):838
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Vardiman JW (2016) The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127(20):2391–2405
Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, Schnittger S, Sanada M, Kon A, Alpermann T, Yoshida K, Roller A, Nadarajah N, Shiraishi Y, Shiozawa Y, Chiba K, Tanaka H, Koeffler HP, Klein HU, Dugas M, Aburatani H, Kohlmann A, Miyano S, Haferlach C, Kern W, Ogawa S (2014) Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 28(2):241–247
Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, Yoon CJ, Ellis P, Wedge DC, Pellagatti A et al (2013) Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 122(22):3616–3627 quiz 3699
Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, Kantarjian H, Raza A, Levine RL, Neuberg D, Ebert BL (2011) Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med 364(26):2496–2506
Bejar R, Greenberg PL (2017) The impact of somatic and germline mutations in myelodysplastic syndromes and related disorders. J Natl Compr Cancer Netw 15(1):131–135
Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, Chalkidis G, Suzuki Y, Shiosaka M, Kawahata R, Yamaguchi T, Otsu M, Obara N, Sakata-Yanagimoto M, Ishiyama K, Mori H, Nolte F, Hofmann WK, Miyawaki S, Sugano S, Haferlach C, Koeffler HP, Shih LY, Haferlach T, Chiba S, Nakauchi H, Miyano S, Ogawa S (2011) Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478(7367):64–69
Ganguly BB, Kadam NN (2016) Mutations of myelodysplastic syndromes (MDS): an update. Mutat Res Rev Mutat Res 769:47–62
Harada H, Harada Y (2015) Recent advances in myelodysplastic syndromes: molecular pathogenesis and its implications for targeted therapies. Cancer Sci 106(4):329–336
Itzykson R, Kosmider O, Fenaux P (2013) Somatic mutations and epigenetic abnormalities in myelodysplastic syndromes. Best Pract Res Clin Haematol 26(4):355–364
Greenberg PL, Stone RM, Al-Kali A, Barta SK, Bejar R, Bennett JM, Carraway H, De Castro CM, Deeg HJ, DeZern AE et al (2017) Myelodysplastic syndromes, version 2.2017, NCCN clinical practice guidelines in oncology. J Natl Compr Cancer Netw 15(1):60–87
Malcovati L, Karimi M, Papaemmanuil E, Ambaglio I, Jadersten M, Jansson M, Elena C, Galli A, Walldin G, Della Porta MG et al (2015) SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood 126(2):233–241
Janusz K, Del Rey M, Abaigar M, Collado R, Ivars D, Hernandez-Sanchez M, Valiente A, Robledo C, Benito R, Diez-Campelo M et al (2017) A two-step approach for sequencing spliceosome-related genes as a complementary diagnostic assay in MDS patients with ringed sideroblasts. Leuk Res 56:82–87
Martin I, Such E, Navarro B, Vicente A, Lopez-Pavia M, Ibanez M, Tormo M, Villamon E, Gomez-Segui I, Luna I et al (2017) Negative impact on clinical outcome of the mutational co-occurrence of SF3B1 and DNMT3A in refractory anemia with ring sideroblasts (RARS). Leuk Lymphoma 58(7):1686–1693
Mangaonkar AA, Lasho TL, Finke CM, Gangat N, Al-Kali A, Elliott MA, Begna KH, Alkhateeb H, Wolanskyj-Spinner AP, Hanson CA et al (2018) Prognostic interaction between bone marrow morphology and SF3B1 and ASXL1 mutations in myelodysplastic syndromes with ring sideroblasts. Blood Cancer J 8(2):18
Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, Harris NL, Le Beau MM, Hellstrom-Lindberg E, Tefferi A et al (2009) The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 114(5):937–951
Greenberg P, Cox C, LeBeau MM, Fenaux P, Morel P, Sanz G, Sanz M, Vallespi T, Hamblin T, Oscier D, Ohyashiki K, Toyama K, Aul C, Mufti G, Bennett J (1997) International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 89(6):2079–2088
Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Sole F, Bennett JM, Bowen D, Fenaux P, Dreyfus F et al (2012) Revised international prognostic scoring system for myelodysplastic syndromes. Blood 120(12):2454–2465
Martin I, Such E, Navarro B, Villamon E, Vicente A, Mora E, Pedrola L, Ibanez M, Lopez-Pavia M, Tormo M et al (2017) Prognostic impact of gene mutations in myelodysplastic syndromes with ring sideroblasts. Blood Cancer J 7(12):630
Schanz J, Tuchler H, Sole F, Mallo M, Luno E, Cervera J, Granada I, Hildebrandt B, Slovak ML, Ohyashiki K et al (2012) New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J Clin Oncol 30(8):820–829
Montalban-Bravo G, Garcia-Manero G (2018) Myelodysplastic syndromes: 2018 update on diagnosis, risk-stratification and management. Am J Hematol 93(1):129–147
Triantafyllidis I, Ciobanu A, Stanca O, Lupu AR (2012) Prognostic factors in myelodysplastic syndromes. Maedica (Buchar) 7(4):295–302
Boultwood J, Pellagatti A, McKenzie AN, Wainscoat JS (2010) Advances in the 5q- syndrome. Blood 116(26):5803–5811
Pellagatti A, Boultwood J (2015) Recent advances in the 5q- syndrome. Mediterr J Hematol Infect Dis 7(1):e2015037
Vardiman JW, Harris NL, Brunning RD (2002) The World Health Organization (WHO) classification of the myeloid neoplasms. Blood 100(7):2292–2302
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW (2008) WHO classification of tumours of haematopoietic and lymphoid tissues. IARC, Lyon
Strupp C, Nachtkamp K, Hildebrandt B, Giagounidis A, Haas R, Gattermann N, Bennett JM, Aul C, Germing U (2017) New proposals of the WHO working group (2016) for the diagnosis of myelodysplastic syndromes (MDS): characteristics of refined MDS types. Leuk Res 57:78–84
Malcovati L, Papaemmanuil E, Bowen DT, Boultwood J, Della Porta MG, Pascutto C, Travaglino E, Groves MJ, Godfrey AL, Ambaglio I, Gallì A, da Vià MC, Conte S, Tauro S, Keenan N, Hyslop A, Hinton J, Mudie LJ, Wainscoat JS, Futreal PA, Stratton MR, Campbell PJ, Hellström-Lindberg E, Cazzola M, on behalf of the Chronic Myeloid Disorders Working Group of the International Cancer Genome Consortium and of the Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie Mieloproliferative (2011) Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms. Blood 118(24):6239–6246
Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, Pellagatti A, Wainscoat JS, Hellstrom-Lindberg E, Gambacorti-Passerini C, Godfrey AL, Rapado I, Cvejic A, Rance R, McGee C, Ellis P, Mudie LJ, Stephens PJ, McLaren S, Massie CE, Tarpey PS, Varela I, Nik-Zainal S, Davies HR, Shlien A, Jones D, Raine K, Hinton J, Butler AP, Teague JW, Baxter EJ, Score J, Galli A, Della Porta MG, Travaglino E, Groves M, Tauro S, Munshi NC, Anderson KC, el-Naggar A, Fischer A, Mustonen V, Warren AJ, Cross NCP, Green AR, Futreal PA, Stratton MR, Campbell PJ (2011) Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med 365(15):1384–1395
Patnaik MM, Lasho TL, Hodnefield JM, Knudson RA, Ketterling RP, Garcia-Manero G, Steensma DP, Pardanani A, Hanson CA, Tefferi A (2012) SF3B1 mutations are prevalent in myelodysplastic syndromes with ring sideroblasts but do not hold independent prognostic value. Blood 119(2):569–572
Cui R, Gale RP, Xu Z, Qin T, Fang L, Zhang H, Pan L, Zhang Y, Xiao Z (2012) Clinical importance of SF3B1 mutations in Chinese with myelodysplastic syndromes with ring sideroblasts. Leuk Res 36(11):1428–1433
Thol F, Kade S, Schlarmann C, Loffeld P, Morgan M, Krauter J, Wlodarski MW, Kolking B, Wichmann M, Gorlich K et al (2012) Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood 119(15):3578–3584
Montalban-Bravo G, Takahashi K, Patel K, Wang F, Xingzhi S, Nogueras GM, Huang X, Pierola AA, Jabbour E, Colla S, Gañan-Gomez I, Borthakur G, Daver N, Estrov Z, Kadia T, Pemmaraju N, Ravandi F, Bueso-Ramos C, Chamseddine A, Konopleva M, Zhang J, Kantarjian H, Futreal A, Garcia-Manero G (2018) Impact of the number of mutations in survival and response outcomes to hypomethylating agents in patients with myelodysplastic syndromes or myelodysplastic/myeloproliferative neoplasms. Oncotarget 9(11):9714–9727
Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M et al (2014) Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 371(26):2477–2487
Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, Higgins JM, Moltchanov V, Kuo FC, Kluk MJ, Henderson B, Kinnunen L, Koistinen HA, Ladenvall C, Getz G, Correa A, Banahan BF, Gabriel S, Kathiresan S, Stringham HM, McCarthy MI, Boehnke M, Tuomilehto J, Haiman C, Groop L, Atzmon G, Wilson JG, Neuberg D, Altshuler D, Ebert BL (2014) Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 371(26):2488–2498
Chan SM, Majeti R (2013) Role of DNMT3A, TET2, and IDH1/2 mutations in pre-leukemic stem cells in acute myeloid leukemia. Int J Hematol 98(6):648–657
Acknowledgments
The authors would like to thank to Irene Rodríguez, Sara González, Teresa Prieto, María Ángeles Ramos, Filomena Corral, Almudena Martín, Ana Díaz, Ana Simón, María del Pozo, Isabel M Isidro, Vanesa Gutiérrez, Sandra Pujante, and María Angeles Hernández of the Cancer Research Center of Salamanca, Spain, for their technical assistance.
Funding
This work was supported by grants from the following: Contrato Rio Hortega, CM17/00171; Gerencia Regional de Salud (Castilla y León) para proyectos de investigación año 2018, 1850/A/18; Spanish Fondo de Investigaciones Sanitarias, PI15/01471, PI18/01500; Instituto de Salud Carlos III (ISCIII); European Regional Development Fund (ERDF) “Una manera de hacer Europa”; Consejería de Educación, Junta de Castilla y León (SA271P18); Proyectos de Investigación del SACYL, Spain, GRS1847/A/18, GRS1653/A17; SYNtherapy, Synthetic Lethality for Personalized Therapy-based Stratification In Acute Leukemia (ERAPERMED2018–275); ISCIII (AC18/00093), co-funded by ERDF/ESF, “Investing in your future”, by grants from Red Temática de Investigación Cooperativa en Cáncer (RTICC) (RD12/0036/0069) and Centro de Investigación Biomédica en Red de Cáncer (CIBERONC CB16/12/00233). JMHS is supported by a research grant from Fundación Española de Hematología y Hemoterapia. MM is currently supported by an Ayuda predoctoral de la Junta de Castilla y León from the Fondo Social Europeo (JCYL- EDU/556/2019 PhD scholarship).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethics approval
The study was approved by the Local Ethical Committee (Comité Ético de Investigación Clínica, Hospital Universitario de Salamanca) and written informed consent was obtained from each patient, in accordance with the guidelines of the Declaration of Helsinki.
Consent to participate
Informed consent was obtained from all individual participants included in the study.
Consent for publications
Not applicable
Code availability
Not applicable
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Janusz, K., Izquierdo, M.M., Cadenas, F.L. et al. Clinical, biological, and prognostic implications of SF3B1 co-occurrence mutations in very low/low- and intermediate-risk MDS patients. Ann Hematol 100, 1995–2004 (2021). https://doi.org/10.1007/s00277-020-04360-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-020-04360-4