
Abstract
Sickle cell disease (SCD) is a hereditary condition characterized by homozygosis of the hemoglobin S (HbS) gene. Marked morbimortality is observed due to chronic hemolysis, endothelial injury, and episodes of vaso-occlusion, which leads to multi-organ damage. Renal impairment is common and may have different presentations, such as deficiency in urinary acidification or concentration, glomerulopathies, proteinuria, and hematuria, frequently resulting in end-stage renal disease (ESRD). Novel biomarkers of renal function, such as kidney injury molecule 1 (KIM-1), and neutrophil gelatinase-associated lipocalin (NGAL) and monocyte chemoattractant protein 1 (MCP-1) are being studied in order to enable early diagnosis of kidney damage in SCD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sickle cell disease (SCD) is a common autosomal recessive hematological condition. Its mortality has risen 42% between 1990 and 2013, reaching 100,000 annual deaths worldwide [1]. Chronic hemolysis and repeated episodes of vaso-occlusion result in insidious damage of vital organs, among which the kidney is frequently affected. Sickle cell nephropathy (SCN) is considered one of the severest complications of SCD and may have its onset in childhood2. This review discusses SCD pathophysiology, treatment, clinical features, and the mechanisms involved in kidney injury. Considering the burden of this issue, there is an interest in prevention and early detection of SCN and novel biomarkers are being studied for that purpose [2,3,4,5,6,7,8,9,10].
Pathogenesis
Sickle cell disease (SCD) was first reported by Herrick in 1910, who described the characteristic shape of the erythrocytes. It is caused by a point mutation in chromosome 11p15.5, replacing timine by adenine in the β-globin gene, which leads to an aminoacid change from a hydrophilic glutamic acid to a hydrophobic valine in the sixth position in the β-globin chain (Fig. 1). As a result, a mutated hemoglobin S (HbS) tetramer is formed and SCD occurs when homozygous mutation is present [11, 12].

Point mutation of the β-globin gene
Pathophysiology
There are four main processes involved in SCD pathophysiology: HbS polymerization, vaso-occlusion, hemolysis associated with endothelial dysfunction, and inflammation [12].
Once glutamic acid is replaced by valine, HbS polymerization is favored in lower oxygen concentrations. HbS becomes stretched and the β-globin S chains are pulled closer together. Such conformational change favors contact between areas of deoxyhemoglobin, which is not possible in the oxygenated state. With the binding of multiple HbS tetramers, long polymers of double fibers are formed, which in turn aggregate in bundles with low solubility, leading to the precipitation of HbS crystals inside the erythrocytes. This process results in altered rheology, increased cell rigidity, energetic stress, dehydration, lower deformability, premature hemolysis, and membrane distortion, which is responsible for the sickle-like shape of the erythrocytes [12, 13].
The formation of HbS polymers inside the red cells causes potassium efflux, high intracellular calcium concentration, HbS polymerization with membrane components such as band 3 proteins and exposure of phosphatidylserine (PS), and CD36. Such changes facilitate erythrocyte’s endothelial adhesion, white blood cells migration, endothelial injury of the microvasculature, and nitric oxide (NO) depletion, creating a pro-inflammatory and pro-thrombotic environment [13].
Apart from the deoxygenated state, other factors influence erythrocyte sickling, including pH, temperature and 2,3-diphosphoglicerate levels, slow blood flow in the spleen and kidneys, for instance. High levels of fetal hemoglobin (HbF) prevent polymerization and are an important determinant of SCD clinical manifestations [12, 14].
The increased viscosity slows blood flow and favors episodic and sustained vaso-occlusion. In addition, due to their marked rigidity, sickle cells are likely to get trapped in small vessels, which contribute to vaso-occlusive events and chronic anaemia, since the erythrocytes trapped in splenic microvasculature are prematurely cleared from the circulation [13, 14].
Intravascular hemolysis of sickle cells releases intracellular hemoglobin in the circulation, which consumes NO in a reaction which also produces methemoglobin and inert nitrate. NO is an endothelium-derived regulator of vascular function, which promotes vasodilation, platelet inhibition, and expression of adhesion molecules, such as VCAM-1. Hemoglobin, heme, and ion catalyze the production of reactive oxygen species (ROS), further limiting NO bioavailability and activating endothelial cells. Hemolysis also releases arginase, which consume L-arginine, a substrate for NO synthesis. Chronic NO depletion may contribute to vasoconstriction, proliferative angiopathy, pulmonary hypertension, and activation of adhesion molecules, such as VCAM-1, platelet activation, and production of endothelin-1. The latter promotes vasoconstriction, increases soluble VCAM-1 and ICAM-1 levels, and induces monocytes to secrete inflammatory cytokines (e.g., IL-1, IL-6, IL-8, TNF-α) and substances which increase superoxide production by neutrophils. The physiological balance of vascular tonus is shift towards vasoconstriction, as well as endothelial activation and cells proliferation [13, 15, 16].
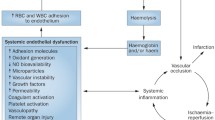
Therefore, SCD is characterized by decreased NO (which favors vascular occlusion), hemolysis, and endothelial activation, resulting in persistent inflammation. Long-term consequences of sustained hemolytic anaemia and episodes of vaso-occlusion include insidious development of damage to vital organs (Fig. 2) [17,18,19].

Pathophysiology of SCD. Deoxygenation induces HbS polymerization, which causes RBCs to take the characteristic sickle-like shape. Vaso-occlusion stems from the interaction between sickled RBCs and the endothelium. Vascular occlusion leads to infarction, hemolysis, and inflammation, which increases expression of adhesion molecules, worsening vaso-occlusion due to sickle cells adhesion to the endothelium. Reperfusion of the ischemic tissue originates free radicals and oxidative stress. Hemolysis releases free hemoglobin in the circulation causing functional NO deficiency, which contributes to the development of angiopathy
Such findings contribute to hypoperfusion in the microvasculature. In the macrovasculature, however, there is an increase in cardiac index (CI) and a decline in systemic vascular resistance (SVR), which is mediated by substances such as endothelium-derived hyperpolarizing factor (EDHF) and prostaglandins, but not NO, since it is decreased in SCD. Lower hemoglobin levels in SCD have also been associated with reduced SVR [20, 21]. Nath et al. have referred to this hemodinamical difference between macro and microcirculation as the “perfusion paradox,” for which the kidney is probably the best example. While there is glomerular hyperperfusion and hyperfiltration, the renal medulla shows a tendency to vaso-occlusion and hypoperfusion [20].
Clinical Manifestations of SCD
Patients with SCD present heterogenous and multifactorial clinical manifestations mainly related to hemolysis and vaso-occlusion. The former causes chronic anemia and elevation of serum markers, such as indirect bilirubin, lactate dehydrogenase (LDH), and reticulocyte count. Hemolysis is also associated with priapism, pulmonary hypertension, ulcers of inferior limbs, and stroke. Pain may be either an acute or chronic feature of SCD. In the acute setting, pain is related to tissular ischemia due to vaso-occlusion causing osteonecrosis or acute chest syndrome (ACS) [15, 16].
The natural history of SCD is marked by a wide range of complications, which promote multi-organ damage and may deeply compromise patient’s quality of life. Retinopathy, osteonecrosis (especially of the femur’s head), gallstones, recurrent infections, and renal failure are among the main complications of SCD [13].
Clinical determinants of SCD
Although all SCD patients possess the same β-globin mutation, different clinical phenotypes are observed. A known factor to significantly influence clinical manifestations in SCD is HbF serum concentration. It is predominantly produced during the fetal stages of life; thus, HbF is found in very low levels in adults, accounting for approximately 1% of total Hb, and is restricted to a small population of red blood cells (RBCs) called F cells. Levels of HbF vary among individuals and are genetically determined as a quantitative hereditary inheritance. High HbF reduces HbS concentration and hinders its polymerization, resulting in a milder SCD presentation [22, 23].
Apart from HbF levels, other genetic factors, such as coexistence of α-thalassemia and β-globin haplotypes are also able to influence SCD clinical course. Many single-nucleotide polymorphisms (located in the β-globin cluster in chromosome 11 or in other chromosomes) as well as epigenetic and environmental mechanisms are also being identified [14, 22].
Mechanisms of renal injury in SCD
The kidney may be affected by various pathophysiological pathways in SCD. Damage usually begins in childhood and progresses through adult life. Sickle cell nephropathy includes acidification and concentration disturbances, distal tubular dysfunction, renal papillary necrosis, and proteinuria, which may progress to ESRD. APOL1 genotypes G1 and G2 have been associated with worse kidney disease and progression to ESRD in SCD. A recent experimental study has demonstrated the upregulation of nephrin on podocytes of zebrafish injected with APOL1 G2 mRNA, which may be associated with the development of SCN [24,25,26].
The acidic, hyperosmolar, and hypoxic environment of the renal medulla dehydrates RBCs, increasing intracellular concentration of HbS, leading to polymerization and sickling inside the descending vasa recta. The slower blood flow through these small renal vessels also favors sickling and endothelial adhesion, increasing the probability of vaso-occlusion in the kidney, which results in renal infarction with tubular and endothelial lesion, reperfusion injury, oxidative stress, and inflammation with release of vasodilator substances, such as prostaglandins and NO, which in turn act on the glomeruli increasing the glomerular filtration rate (GFR). Prolonged hyperfiltration causes proteinuria, glomerulosclerosis, and eventually progression to CKD and ESRD. There is also a correlation between hyperfiltration and chronic hemolysis (Fig. 3) [27,28,29,30].

Pathophysiology of sickle cell nephropathy
Renal damage in SCD has been associated with chronic hemolysis and anemia [27, 31]. HbS is an unstable protein which undergoes auto-oxidation and denaturation, producing ROS and free heme radical. The passage of HbS through podocytes and tubular epithelium may cause oxidative damage to these cells. In addition, heme radical is ligand for type 4 Toll-like receptors, which is found on the endothelium, tubules, mesangium, and podocytes, leading to inflammatory responses of these cells. The oxidative effects of HbS and heme upregulate the transcription of pro-inflammatory and fibrogenic genes, leading to infiltration and fibrosis in the glomerular and tubulointerstitial compartments [27, 30].
Concentration defects
The destruction of the kidney’s medulla begins in childhood and is responsible for the urinary concentration deficit observed in many SCD patients. Hyposthenuria is the first renal manifestation of SCD and leads to polyuria and free water loss, which in turn may increase serum osmolarity if water intake does not balance the losses. Patients with higher HbF were able to concentrate the urine better, corroborating the hypothesis that hemoglobin polymerization causes renal medulla’s destruction [28, 30, 32, 33].
Hematuria
Hematuria is one of the most common renal manifestations of SCD, affecting up to 13––30% of patients. The majority is asymptomatic, but abdominal or back pain is occasionally reported by patients. Higher prevalence is associates with older age and male sex. Hematuria may be microscopic or macroscopic and occurs by capillary congestion, especially in the medulla, with RBCs leak to the lumen of the renal tubules. It may also be caused by papillary necrosis due to occlusion of the vasa recta, which might contribute to the development of CKD [30, 34, 35].
Hypertension
Patients with SCD present lower blood pressure (BP) in comparison with the overall population. This fact has been ascribed to the fluid loss secondary to concentration defects, but it is more likely to stem from the reduction in SVR, since lower BP is observed even in SCD patients with increased plasma volume [36, 37]. In SCD patients, not only overt hypertension is deleterious. Relative increments in systolic BP (resulting in elevated pulse pressure) have been associated with hemolysis and worse renal and cardiovascular outcomes, such as proteinuria, chronic kidney disease, pulmonary hypertension, diastolic heart dysfunction, stroke, and death [36, 38,39,40]. The term relative systemic hypertension (RSH) is currently being used to refer to SCD patients who do not present hypertension according to traditional criteria, but have increased risk for worse outcomes. Defined as systolic BP of 120–139 mmHg and diastolic BP of 70–89 mmHg, RSH is believed to affect approximately 45% of adult SCD patients, while overt hypertension has a prevalence of 19% in the same population [41].
Proteinuria and albuminuria
Moderately increased albuminuria (MIA; 30–300 mg/g Cr), formerly known as microalbuminuria progresses to severely increased albuminuria (SIA; > 300 mg/g Cr) as renal damage builds up. As a result, the intensity of proteinuria can be expected according to the patient’s age. Approximately 20–35% of patients present MIA in adolescence, whereas SIA is observed in 60% of adults with SCD [31]. Recent evidence has better defined the association between hyperfiltration in early childhood and the development of albuminuria later on [42].
Because of its early onset, albuminuria is currently considered a relevant biomarker of glomerular damage in SCD associated with hyperfiltration or not. Proteinuria is associated with a variety of factors, such as high blood pressure, low hemoglobin levels, hemolysis, leukocytosis, hematuria, previous episode of vaso-occlusion, β-globin S gene haplotype, pulmonary hypertension, stroke, and ACS [27, 30, 31, 43].
Chronic kidney disease and ESRD
Following the onset of proteinuria, some SCD patients develop CKD with GFR decrease due to interactions among multiple processes in glomerular, vascular, tubular, and interstitial compartments of the kidney. CKD may be caused either by repeated mild acute renal injuries or a single severe acute kidney injury. This concept is especially suitable for SCN, since it is characterized by intermittent episodes of vaso-occlusion and there is increased renal susceptibility to acute damage in SCD. Powars et al. [44] observed that renal failure in SCD was preceded by severe anemia, proteinuria, nephrotic syndrome, hypertension, and microscopic hematuria. In addition, patients with renal failure were three times more likely to develop chronic restrictive pulmonary disease, leg ulcers, and stroke than those without renal failure [28, 30, 44].
Although common, not all SCD patients will develop proteinuria and not all patients with proteinuria will develop SCN. The factors which influence progression to CKD are not completely understood, but older age is associated with development of renal failure in SCD. Also, it has been reported that male sex, hypertension, proteinuria, and worsening of anemia might be risk factors for ESRD [45]. The presence of comorbidities, such as chronic heart disease and diabetes, as well as the patient’s genetic background are related to progression of CKD in general. The intrarenal renin-angiotensin system has also been studied as a major contributor to SCN’s pathophysiology [2, 28].
Novel biomarkers of kidney injury
The traditional biomarkers of renal damage, such as serum creatinine, are not very sensitive, especially when hyperfiltration is present, since it increases GFR and creatinine clearance. As a result, a rise in serum creatinine is noticed in late stages of SCN, usually when GFR is below half of its normal value and marked proteinuria is present [2, 29, 34].
There is evidence of higher diagnostic accuracy using more than one biomarker; hence, the interest in validating new biomarkers for early diagnosis of SCN, before therapeutic interventions are no longer able to prevent the progression of renal damage [2, 34].
KIM-1
Kidney injury molecule 1 (KIM-1) is a transmembrane protein of the proximal tubule which is not found in normal conditions. It is specifically expressed by dedifferentiated tubular cells after ischemic or nephrotoxic damage, being associated with both acute and chronic tubular injury. Several studies have shown its usefulness in predicting adverse outcomes, but its role in CKD is still not well established. Sundaram et al. have demonstrated an association between KIM-1 and albuminuria in SCD; thus, it is a promising biomarker for SCN [2, 3, 10, 29].
Although undetectable in the normal kidney, mRNA for KIM-1 is rapidly produced after kidney injury and KIM-1 can be found in all three segments of the renal tubules, with especially high levels on the apical membrane of the proximal tubule. Soluble KIM-1 can be found in the urine of patients with acute tubular necrosis (ATN) and acts as an early biomarker of damage to the proximal tubule [4, 5].
The fact that KIM-1 is not present in normal kidneys but also increases its expression on the apical membrane of the injured proximal tubule and persists until it is fully recovered markedly increases its accuracy as a promising biomarker [4].
MCP-1
Monocyte chemotactic protein 1 (MCP-1) has been described as an important biomarker of mononuclear cell inflammation in ischemia-induced acute kidney injury (AKI). It is considered the most powerful chemotactic molecule for macrophages and monocytes recruitment and is expressed mainly by epithelial cells in the glomeruli and tubules during renal inflammation [4].
Patients with SCD presented higher MCP-1 levels than the control group and treatment with hydroxyurea was associated with a decrease in its measurement. Albuminuria was also associated with MCP-1 [6].
NGAL
Neutrophil gelatinase-associated lipocalin (NGAL) is a small protein specialized in the binding and transport of small hydrophobic molecules. Since the casual finding that NGAL is easily detected in the urine of animals with AKI, there is a growing interest in studying this molecule as a non-invasive AKI biomarker in humans [7].
Following ischemic renal damage in animal models, NGAL is sharply induced and its gene is one of the most up-regulated even in early stages. It has been detected in the urine in 2 h after the ischemic insult. In tubular injury, NGAL expression increases 1000 times in both humans and rodents and is promptly detected in the serum and urine [8–-10].
However, Sundaram et al. [2] have surprisingly found a subnormal NGAL concentration in the urine of SCD patients. It was ascribed to the increased reabsorption of NGAL by the proximal tubules, which are overfunctional in SCD. It was also hypothesized that the damage to the distal tubules in SCN would prevent these cells from increasing NGAL expression. Thus, it appears that the main role of NGAL in SCN is excluding other mechanisms of renal injury which are associated with increased urinary NGAL [2, 46].
Treatment
Despite the recent achievements in the comprehension of SCD, therapeutic options remain limited. Patients’ management is mostly supportive and includes control of symptoms with antibiotics, analgesic, and anti-inflammatory drugs. Patient who presented stroke, chronic refractory pain, or repeated splenic sequestration crises should receive blood transfusion regularly. Standard therapy also includes oral administration of hydroxyurea (HU) and bone marrow transplantation (BMT). The latter is the only treatment able to provide definite cure, but requires a compatible donor and may present complications, such as graft versus host disease (GVHD). Studies involving gene therapy are being carried out in order to develop a new curative treatment using the patient’s own stem cells without the risks of a BMT [47,48,49].
Hydroxyurea is a potent ribonucleotide reductase inhibitor and currently the main drug used to effectively treat SCD. It improves patient’s clinical course by progressively increasing HbF levels and lowering HbS concentration. In addition, studies have also demonstrated that HU is able to decrease RBC’s endothelial adhesion, endothelial activation and leukocytes and platelets counts. It also acts as a NO donor, reduces hemolysis, and effectively prevents acute vaso-occlusive events (Fig. 4) [50,51,52].

Effects of treatment with hydroxyurea on SCD. ACS, acute chest syndrome; HbF, fetal hemoglobin
In kidney disease, higher HbF was associated with lower incidence of MIA and with milder hyposthenuria. Treatment with HU decreases hyperfiltration and MIA. Although the mechanism of MIA reduction is still elusive, it has been proposed that the decrease in hemolysis and RBC sickling associated with hydroxyurea would reduce renal ischemic injury. Another study reported reduction of proteinuria in pediatric patients following hydroxyurea treatment. However, its use to specifically prevent the development or progression of SCN still lacks evidence from larger studies [31, 33, 34, 53,54,55]. Similarly, the benefit of angiotensin converting enzyme inhibitors (ACEI) in reducing proteinuria or preventing chronic kidney disease in normotensive SCD patients with MIA is uncertain [56].
Chronic transfusion prevents vaso-occlusive events, especially childhood strokes, but there is also evidence supporting a decline in ACS incidence [57]. Its main indication consists in abnormal transcranial Doppler velocity (> 200 cm/s) in children [58]. Long-term transfusion is capable of reducing erythrocyte deformability and improving the capacity of oxygen to be carried. When well-targeted, it increases hematocrit and hemoglobin levels without significant increments in blood viscosity in comparison to non-transfused individuals, because better oxygenation causes a decline in viscosity and compensates for the higher hematocrit [59]. However, this therapy should not aim for normal laboratory values, in order to avoid unnecessary increases in blood viscosity. Targeting hemoglobin at 10 g/dl (especially before surgical procedures) and hemoglobin S at < 30% seems to be effective and safe [58, 59].
Side effects of long-term transfusion may include alloimunization, transfusion reactions and, eventually, iron overload [60]. The spleen is the most commonly affected organ by iron deposits, followed by the liver, kidney, and pancreas [61]. Patients with renal hemosiderosis present higher urine albumin-creatinine ration and elevated markers of hemolysis [61]. The mechanism of iron deposit formation is determined by the filtered hemoglobin, which is endocyted into the proximal and distal tubules [62]. Although chronic transfusion is able to harm the kidneys in renal hemosiderosis, the overall impact of this therapy on kidney function and the new biomarkers are still elusive due to the lack of studies addressing this issue [56].
Conclusion
Nephropathy remains one of the most common chronic complications in patients with SCD and has its onset in childhood. It manifests by disturbances in both glomeruli and tubules, such as proteinuria, hematuria, and hyposthenuria, often progressing to CKD. The emergence of new non-invasive biomarkers of renal injury (such as KIM-1, MCP-1, and NGAL) and their assimilation into clinical practice is a promising area of improvement in SCD care. Their combined use in order to obtain a panel of biomarkers may contribute to the early diagnosis and the development of strategies to guide patients’ follow-up and tackle disease progression.
References
Reddy KS (2016) Global Burden of Disease Study 2015 provides GPS for global health 2030. Lancet 388(10053):1448–1449
Sundaram N, Bennett M, Wilhelm J, Kim MO, Atweh G, Devarajan P, Malik P (2011) Biomarkers for early detection of sickle nephropathy. Am J Hematol 86(7):559–566
Hamideh D, Raj V, Harrington T, Li H, Margolles E, Amole F, Garcia-Buitrago M, Ruiz P, Zilleruelo G, Alvarez O (2014) Albuminuria correlates with hemolysis and NAG and KIM-1 in patients with sickle cell anemia. Pediatr Nephrol 29(10):1997–2003
Peres LA, Cunha Júnior AD, Schäfer AJ, Silva AL, Gaspar AD, Scarpari DF, Alves JB, Girelli Neto R, Oliveira TF (2013) Biomarkers of acute kidney injury. J Bras Nefrol 35(3):229–236
Vaidya VS, Ferguson MA, Bonventre JV (2008) Biomarkers of acute kidney injury. Annu Rev Pharmacol Toxicol 48:463–493
Santos TE, Gonçalves RP, Barbosa MC et al (2015) Monocyte chemoatractant protein-1: a potential biomarker of renal lesion and its relation with oxidative status in sickle cell disease. Blood Cells Mol Dis 54(3):297–301
Sanjeevani S, Pruthi S, Kalra S, Goel A, Kalra OP (2014) Role of neutrophil gelatinase-associated lipocalin for early detection of acute kidney injury. Int J Crit Illn Inj Sci 4(3):223–228
Mishra J, Ma Q, Prada A, Mitsnefes M, Zahedi K, Yang J, Barasch J, Devarajan P (2003) Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J Am Soc Nephrol 14(10):2534–2543
Soni SS, Cruz D, Bobek I, Chionh CY, Nalesso F, Lentini P, de Cal M, Corradi V, Virzi G, Ronco C (2010) NGAL: a biomarker of acute kidney injury and other systemic conditions. Int Urol Nephrol 42(1):141–150
Devarajan P (2010) Review: Neutrophil gelatinase-associated lipocalin: a troponin-like biomarker for human acute kidney injury. Nephrology (Carlton) 15(4):419–428
Serjeant GR (2010) One hundred years of sickle cell disease. Br J Haematol 151(5):425–429
Sundd P, Gladwin MT, Novelli EM (2019) Pathophysiology of sickle cell disease. Annu Rev Pathol 14:263–292
Zago MA, Pinto ACS (2007) Fisiopatologia das doenças falciformes: da mutação genética à insuficiência de múltiplos órgãos. Rev. Bras Hematol. Hemoter. Set 29(3):207–214
Adekile AD (2013) What’s new in the pathophysiology of sickle cell disease? Med Princ Pract 22(4):311–312
Kato GJ, Gladwin MT, Steinberg MH (2007) Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev 21(1):37–47
Kato GJ, Hebbel RP, Steinberg MH, Gladwin MT (2009) Vasculopathy in sickle cell disease: Biology, pathophysiology, genetics, translational medicine, and new research directions. Am J Hematol 84(9):618–625
Rees DC, Gibson JS (2012) Biomarkers in sickle cell disease. Br J Haematol 156(4):433–445
Damanhouri GA, Jarullah J, Marouf S, Hindawi SI, Mushtaq G, Kamal MA (2015) Clinical biomarkers in sickle cell disease. Saudi J Biol Sci 22(1):24–31
Moreira JA, Laurentino MR, Machado RP, Barbosa MC, Gonçalves RP, Mota Ade M, Rocha LB, Martins AM, de Lima Arruda AB, de Souza IP, Gonçalves RP (2015) Pattern of hemolysis parameters and association with fetal hemoglobin in sickle cell anemia patients in steady state. Rev Bras Hematol Hemoter 37(3):167–171
Nath KA, Katusic ZS, Gladwin MT (2004) The perfusion paradox and vascular instability in sickle cell disease. Microcirculation 11(2):179–193
Lamarre Y, Hardy-Dessources MD, Romana M, Lalanne-Mistrih ML, Waltz X, Petras M, Doumdo L, Blanchet-Deverly A, Martino J, Tressières B, Maillard F, Tarer V, Etienne-Julan M, Connes P (2014) Relationships between systemic vascular resistance, blood rheology and nitric oxide in children with sickle cell anemia or sickle cell-hemoglobin C disease. Clin Hemorheol Microcirc 58(2):307–316
Steinberg MH (2009) Genetic etiologies for phenotypic diversity in sickle cell anemia. Sci World J 9:46–67
Orkin SH, Higgs DR (2010) Sickle cell disease at 100 years. Science. 329(5989):291–292
Maigne G, Ferlicot S, Galacteros F, Belenfant X, Ulinski T, Niaudet P, Ronco P, Godeau B, Durrbach A, Sahali S, Lang P, Lambotte O, Audard V (2010) Glomerular lesions in patients with sickle cell disease. Medicine (Baltimore) 89(1):18–27
Kormann R, Jannot AS, Narjoz C, Ribeil JA, Manceau S, Delville M, Joste V, Prié D, Pouchot J, Thervet E, Courbebaisse M, Arlet JB (2017) Roles of APOL1 G1 and G2 variants in sickle cell disease patients: kidney is the main target. Br J Haematol 179(2):323–335
Bundy JL, Anderson BR, Francescatto L, Garrett ME, Soldano KL, Telen MJ, Davis EE, Ashley-Koch AE (2019) RNA sequencing of isolated cell populations expressing human APOL1 G2 risk variant reveals molecular correlates of sickle cell nephropathy in zebrafish podocytes. PLoS One 14(6):e0217042
Haymann JP, Stankovic K, Levy P, Avellino V, Tharaux PL, Letavernier E, Grateau G, Baud L, Girot R, Lionnet F (2010) Glomerular hyperfiltration in adult sickle cell anemia: a frequent hemolysis associated feature. Clin J Am Soc Nephrol 5(5):756–761
Becker AM (2011) Sickle cell nephropathy: challenging the conventional wisdom. Pediatr Nephrol 26(12):2099–2109
Da Silva GB, Libório AB, Daher EF (2011) New insights on pathophysiology, clinical manifestations, diagnosis, and treatment of sickle cell nephropathy. Ann Hematol 90(12):1371–1379
Nath KA, Hebbel RP (2015) Sickle cell disease: renal manifestations and mechanisms. Nat Rev Nephrol 11(3):161–171
Aban I, Baddam S, Hilliard LM, Howard TH, Feig DI, Lebensburger JD (2017) Severe anemia early in life as a risk factor for sickle-cell kidney disease. Blood. 129(3):385–387
Tsaras G, Owusu-Ansah A, Boateng FO, Amoateng-Adjepong Y (2009) Complications associated with sickle cell trait: a brief narrative review. Am J Med 122(6):507–512
Miller ST, Wang WC, Iyer R, Rana S, Lane P, Ware RE, Li D, Rees RC, BABY-HUG Investigators (2010) Urine concentrating ability in infants with sickle cell disease: baseline data from the phase III trial of hydroxyurea (BABY HUG). Pediatr Blood Cancer 54(2):265–268
Hariri E, Mansour A, El Alam A et al (2018) Sickle cell nephropathy: an update on pathophysiology, diagnosis, and treatment. Int Urol Nephrol 50(6):1075–1083
Le Joncour A, Mesnard L, Hertig A, Robert T et al (2018) Red urine, updated for the nephrologist: a case report. BMC Nephrol 19(1):133
Pegelow CH, Colangelo L, Steinberg M, Wright EC, Smith J, Phillips G, Vichinsky E (1997) Natural history of blood pressure in sickle cell disease: risks for stroke and death associated with relative hypertension in sickle cell anemia. Am J Med 102(2):171–177
Nath KA, Katusic ZS, Gladwin MT (2004) The perfusion paradox and vascular instability in sickle cell disease. Microcirculation. 11(2):179–193
Gladwin MT (2016) Cardiovascular complications and risk of death in sickle-cell disease. Lancet. 387(10037):2565–2574
Novelli EM, Hildesheim M, Rosano C, Vanderpool R, Simon M, Kato GJ, Gladwin MT (2014) Elevated pulse pressure is associated with hemolysis, proteinuria and chronic kidney disease in sickle cell disease. PLoS One 9(12):e114309
Gordeuk VR, Sachdev V, Taylor JG, Gladwin MT, Kato G, Castro OL (2008) Relative systemic hypertension in patients with sickle cell disease is associated with risk of pulmonary hypertension and renal insufficiency. Am J Hematol 83(1):15–18
Benneh-Akwasi Kuma A, Owusu-Ansah AT, Ampomah MA, Sey F, Olayemi E, Nouraie M, Ofori-Acquah SF (2018) Prevalence of relative systemic hypertension in adults with sickle cell disease in Ghana. PLoS One 13(1):e0190347
Lebensburger JD, Aban I, Pernell B, Kasztan M, Feig DI, Hilliard LM, Askenazi DJ (2019) Hyperfiltration during early childhood precedes albuminuria in pediatric sickle cell nephropathy. Am J Hematol 94(4):417–423
Bartolucci P, Habibi A, Stehlé T, di Liberto G, Rakotoson MG, Gellen-Dautremer J, Loric S, Moutereau S, Sahali D, Wagner-Ballon O, Remy P, Lang P, Grimbert P, Audureau E, Godeau B, Galacteros F, Audard V (2016) Six months of hydroxyurea reduces albuminuria in patients with sickle cell disease. J Am Soc Nephrol 27(6):1847–1853
Powars DR, Elliott-Mills DD, Chan L, Niland J, Hiti AL, Opas LM, Johnson C (1991) Chronic renal failure in sickle cell disease: risk factors, clinical course, and mortality. Ann Intern Med 115(8):614–620
Sl Y, Paul Y, Oneal P, Nouraie M (2016) Renal failure in sickle cell disease: prevalence, predictors of disease, mortality and effect on length of hospital stay. Hemoglobin 40(5):295–299
Koyner JL, Vaidya VS, Bennett MR, Ma Q, Worcester E, Akhter SA, Raman J, Jeevanandam V, O'Connor MF, Devarajan P, Bonventre JV, Murray PT (2010) Urinary biomarkers in the clinical prognosis and early detection of acute kidney injury. Clin J Am Soc Nephrol 5(12):2154–2165 Epub 2010 Aug 26
Iannone R, Ohene-Frempong K, Fuchs EJ, Casella JF, Chen AR (2005) Bone marrow transplantation for sickle cell anemia: progress and prospects. Pediatr Blood Cancer 44(5):436–440
Kohne E (2011) Hemoglobinopathies: clinical manifestations, diagnosis, and treatment. Dtsch Arztebl Int 108(31-32):532–540
Orkin SH, Bauer DE (2019) Emerging genetic therapy for sickle cell disease. Annu Rev Med 70:257–271
Nahavandi M, Tavakkoli F, Wyche MQ, Perlin E, Winter WP, Castro O (2002) Nitric oxide and cyclic GMP levels in sickle cell patients receiving hydroxyurea. Br J Haematol 119(3):855–857
Agrawal RK, Patel RK, Shah V, Nainiwal L, Trivedi B (2014) Hydroxyurea in sickle cell disease: drug review. Indian J Hematol Blood Transfus 30(2):91–96
Nader E, Grau M, Fort R et al (2018) Hydroxyurea therapy modulates sickle cell anemia red blood cell physiology: impact on RBC deformability, oxidative stress, nitrite levels and nitric oxide synthase signalling pathway. Nitric Oxide 81:28–35
Mckie KT, Hanevold CD, Hernandez C et al (2007) Prevalence, prevention, and treatment of microalbuminuria and proteinuria in children with sickle cell disease. J Pediatr Hematol Oncol 29(3):140–144
Aygun B, Mortier NA, Smeltzer MP, Shulkin BL, Hankins JS, Ware RE (2013) Hydroxyurea treatment decreases glomerular hyperfiltration in children with sickle cell anemia. Am J Hematol 88(2):116–119
Laurin LP, Nachman PH, Desai PC et al (2014) Hydroxyurea is associated with lower prevalence of albuminuria in adults with sickle cell disease. Nephrol Dial Transplant 29(6):1211–1218
Roy NB, Fortin PM, Bull KR et al (2017) Interventions for chronic kidney disease in people with sickle cell disease. Cochrane Database Syst Rev 7:CD012380
Estcourt LJ, Fortin PM, Hopewell S et al (2017) Blood transfusion for preventing primary and secondary stroke in people with sickle cell disease. Cochrane Database Syst Rev 1:CD003146
Yawn BP, Buchanan GR, Afenyi-Annan AN et al (2014) Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. 312(10):1033–1048
Detterich JA (2018) Simple chronic transfusion therapy, a crucial therapeutic option for sickle cell disease, improves but does not normalize blood rheology: what should be our goals for transfusion therapy? Clin Hemorheol Microcirc 68(2-3):173–186
Ware RE, de Montalembert M, Tshilolo L, Abboud MR (2017) Sickle cell disease. Lancet 390(10091):311–323
Wood JC, Cohen AR, Pressel SL, Aygun B, Imran H, Luchtman-Jones L, Thompson AA, Fuh B, Schultz WH, Davis BR, Ware RE, TWiTCH Investigators (2016) Organ iron accumulation in chronically transfused children with sickle cell anaemia: baseline results from the TWiTCH trial. Br J Haematol 172(1):122–130
Gburek J, Verroust PJ, Willnow TE, Fyfe JC, Nowacki W, Jacobsen C, Moestrup SK, Christensen EI (2002) Megalin and cubilin are endocytic receptors involved in renal clearance of hemoglobin. J Am Soc Nephrol 13(2):423–430
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Laurentino, M.R., Parente Filho, S.L.A., Parente, L.L.C. et al. Non-invasive urinary biomarkers of renal function in sickle cell disease: an overview. Ann Hematol 98, 2653–2660 (2019). https://doi.org/10.1007/s00277-019-03813-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-019-03813-9