
Abstract
Aplastic anemia (AA) is the most serious non-malignant blood disorder in Pakistan, ranked second in prevalence, after thalassemia. We investigated various epidemiological, clinical, and genetic factors of AA in a Pakistani cohort of 214 patients reporting at our hospital between June 2014 and December 2015. A control group of 214 healthy subjects was included for comparison of epidemiological and clinical features. Epidemiological data revealed 2.75-fold higher frequency of AA among males. A single peak of disease onset was observed between ages 10 and 29 years followed by a steady decline. AA was strongly associated with lower socioeconomic profile, rural residence, and high rate of consanguineous marriages. Serum granulocyte colony-stimulating factor and thrombopoietin levels were significantly elevated in AA patients, compared to healthy controls (P < 0.0001), while there was no statistical significance in other nine cytokine levels screened. Allele frequencies of DRB1*15 (56.8%) and DQB1*06 (70.3%) were predominantly high in AA patients. Ten mutations were found in TERT and TERC genes, including two novel mutations (Val526Ala and Val777Met) in exons 3 and 7 of TERT gene. Despite specific features of the AA cohort, this study suggests that epidemiologic and etiologic factors as well as host genetic predisposition exclusively or cooperatively trigger AA in Pakistan.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Aplastic anemia (AA) is a rare and life-threatening hematopoietic disease characterized by peripheral blood pancytopenia accompanied with trilineage bone marrow (BM) aplasia. AA may develop at any stage of life [46, 48]. Most of the cases are caused by immune-mediated destruction of hematopoietic stem and progenitor cells by activated cytotoxic T lymphocytes (CTLs) inducing apoptosis in these cells. A significant proportion of AA patients respond to immunosuppressive treatment (IST) quite well, providing the best evidence for underlying immune pathophysiology of the disease. However, the majority of the cases are labeled as idiopathic because of inability to find a direct cause of disease. AA can transform to myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML) in some patients due to clonal evolution.
In AA, CD8+ CTLs with restricted TCR diversity (oligoclonal T cells) are expanded which secrete proinflammatory cytokines, such as IFN-γ and TNF-α. This results in elevated cytokine levels in the bone marrow and peripheral blood, inducing apoptosis of CD34+ cells [43]. The signature cytokine profile of AA also includes high levels of thrombopoietin (TPO) and granulocyte colony-stimulating factor (G-CSF) and low levels of some cytokines, such as CD40 ligand (CD40L), chemokine (C-X-C motif) ligand 5 (CXCL5), and epidermal growth factor (EGF) [10].
Association of some human leukocyte antigen (HLA) alleles with AA has also been reported by multiple studies, indicating influence of HLA polymorphism on AA pathophysiology [28, 32]. The HLA system is a gene complex encoding the major histocompatibility complex proteins in human. HLA genes are located at chromosome 6p2.13 and play crucial roles for immune regulation.
A subgroup of AA patients displays abnormal telomere shortening, due to mutations of telomerase and other telomere components [6]. Telomeres (TTAGGG in all vertebrates) are terminal cap structures of eukaryotic chromosomes and essential for chromosome integrity by preventing DNA degradation, end-to-end fusions, and rearrangements. A ribonucleoprotein enzyme, telomerase, maintains telomeres by synthesizing de novo repeats. The catalytic core of telomerase consists of two major components: a catalytic subunit of telomerase reverse transcriptase (TERT) and an RNA template (TERC). Telomerase activity is rate-limiting by transcriptional regulation of TERT [7].
According to epidemiological studies, the estimated annual incidence of AA is two per million in Western countries. The incidence rate is two to three times higher in Asian population, but the cause for this is still not clear [19]. In Pakistan, AA is the second most common serious non-malignant blood disorder after thalassemia [12]. AA has been reported predominantly in young adult male population [39]. According to a report in 2008, half of the patients undergoing bone marrow transplant (BMT) in Pakistan had AA [37].
To gain insight into the genetics and pathophysiology of AA in the Pakistani population, we investigated samples from AA patients reporting at the Armed Forces Bone Marrow Transplant Centre, Rawalpindi, Pakistan. This is the first study where examination of socioetiological data, cytokine profiling, HLA allelic association, and mutations in TERT and TERC genes was simultaneously studied to dissect a high incidence of AA cases in Pakistani population.
Methods
Study setting
Our center is the biggest center of clinical hematology and bone marrow transplantation in Pakistan, a country with population of 207 million according to 2017 census. It is a referral center for whole of the northern part and major area from central part of the country. The center has carried out more than 800 allogeneic HSCT that includes 330 cases of AA. The center is a 54-bedded facility with a team of 10 bone marrow transplant physicians.
Patients
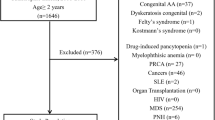
This study was approved by institutional review boards of Armed Forces Bone Marrow Transplant Center (AFBMTC), Pakistan and National Heart Lungs and Blood Institute (NHLBI) at National Institutes of Health (NIH), USA. Patients’ rights were protected in accordance with the Helsinki declaration. Informed written consent was taken from all patients before drawing blood sample for research. A detailed questionnaire was also filled by trained experts through direct interview detailing the patient and disease demographics. Blood samples were collected from 214 consecutive patients with AA reporting at AFBMTC between June 2014 and December 2015. The diagnosis of AA was based on the BM and blood count criteria of the International Agranulocytosis and Aplastic Anemia Study, and patients were classified as moderate, severe, and very severe AA using Camitta criteria.
Conventional karyotyping was carried out to identify chromosomal breakages. For Fanconi Anemia screening, Giemsa–trypsin banding technique was used on blood samples. Cells were cultured for 72 h with PHA at 37 °C with and without Mitomycin C. Twenty metaphases were examined with clastogenic stress and 20 without any stress. Microscopy was done for chromosomal aberrations such as chromatid breaks and exchanges. Paroxysmal nocturnal hemoglobinuria (PNH) screening was carried out as per guidelines proposed by Parker et al. [30]. Briefly, we carried out staining of two glycosylphosphatidylinositol-anchored proteins (GPI-AP) named CD59 (FITC) and CD55 (PE) antibodies (Becton Dickinson, USA) on BD FACS Calibur flow cytometer. Whole blood was stained using stain and wash technique. Red blood cells (RBCs) were selected for analysis using Cell Quest software. A total of 50,000 RBCs were acquired for analysis. Among these, 50 RBCs found deficient for CD55/59 were considered as a deficient clone, making sensitivity 0.1%.
HLA test was done in 74 patients to search for HLA-matched sibling donor and was offered HSCT if case matched donor was available. Not all patients with AA referred to our center are registered for BMT for various reasons like lack of entitlement, health insurance, inadequate funding, long waiting list, and lost to follow-up. HLA typing is done only for patients registered for treatment in the center. In patients with no HLA matched donor or comorbidities and those with non-severe AA were offered antithymocyte globulin (ATG) plus cyclosporin A (CsA) or CsA alone. CsA alone was given to patients who could not afford cost of ATG. Standard conditioning using ATG with cyclophosphamide with or without ATG was used as conditioning regimen for HSCT. Post-transplant GVHD prophylaxis consisted of CsA with or without short methotrexate. CsA was gradually tapered off after 9 months and stopped at 12 months post HSCT. Rabbit ATG (Fresenius) was given as 10–12 h infusion in a dose of 5 mg/kg/day for 5 days. CsA was added on day + 1 post ATG in a dose of 5–7 mg/kg to aim plasma trough level around 200 ng/mL with monitoring of renal functions. CsA was continued and stopped if there was no response after 6 months. In case of response, it was continued and dose was adjusted according to blood counts and side effects. In case of complete remission, CsAwas gradually reduced every 8–12 weeks and was either stopped or kept at as minimum dose as possible with regular monitoring of blood counts and side effects.
Controls
A control group of age and sex-matched healthy blood and marrow donors (n = 214) was also included in the study for comparison of sociodemographic variables. Blood samples were collected from 42 of these healthy volunteers for comparison of serum cytokine profile with that of AA patients.
Mutation analysis
Genomic DNA (gDNA) was extracted from whole blood and peripheral blood mononuclear cells (PBMCs) of AA patients using the DNeasy Blood and Tissue kit (Qiagen, Valencia, CA, USA), according to the manufacturer’s protocol. DNA was successfully extracted from 190 out of 214 patients. For mutation analysis, polymerase chain reaction (PCR) amplification of TERT and TERC genes was performed with appropriate primer sets and conditions as described previously [5, 44]. Purified PCR products were subjected to direct sequencing with the BigDye Terminator v3.1 Cycle Sequencing kit (Thermo Fisher Scientific, Waltham, MA, USA) and the 3130xl Genetic Analyzer (Thermo Fisher Scientific).
Cytokine expression profiling
Serum specimens were separated from clotted blood of 214 AA patients and 42 controls within 1 hour of collection, aliquoted, and stored at − 80 °C. Serum cytokine levels were measured by Magnetic Luminex Screening Assay using the Human Premixed Multi-Analyte kit composed of panels of capture antibody coated beads and labeled detection antibodies. The premixed kit used in this study contained 11 analytes (TNF-α, IL-1b, IL-17A, G-CSF, IL-6, INF-γ, IL-2, Tpo, IL-10, IL-4, and IL-17F) (R&D Systems, Minneapolis, MN, USA). In brief, Luminex assay was performed with 50 μl of 2-fold diluted serum/well in duplicate using the Premixed Multi-Analyte kit, according to manufacturer’s instructions, followed by analysis using Bio-Plex 200 System (Bio-Rad, Hercules, CA, USA). Assay sensitivity varied from 0.1 to 23.4 pg/ml depending on the analyte.
HLA typing
HLA typing was carried out in 74 of our patients by PCR with sets of sequence specific primers (SSP) [2, 3] or serological technique [24]. The frequencies of HLA class I, II, DRB, and DQB antigens expressed in >15% of patients are given in Supplemental Table 3. For comparison with normal healthy population, historical controls (approximately 2300) were searched from previously published literature reporting Pakistani HLA data [18, 25, 27, 31, 33].
Statistical analyses
The descriptive statistics were performed using SPSS version 17.0. It included frequency distribution of different patients and disease related factors. Variables like patient age, hematological parameters, and number of transfusions are shown as median with range, whereas serum cytokine levels were represented as means ± standard deviation (SD). GraphPad PRISM software version 6 was used for analysis of variance using Kruskal–Wallis test to compare serum levels of cytokines, growth factors, and hormones between different patient groups and control subjects. A P value < 0.05 was considered statistically significant.
Results
Epidemiological and clinico-hematological characteristics
Patients and disease characteristics are shown in Table 1. Out of 214 patients, 157 (73.4%) were male and 57 (26.6%) were female, showing a 2.75-fold higher frequency of AA among males. A single peak of disease onset was observed between ages 10 and 29 years in both males (67.5%) and females (50.8%) followed by a steady decline. Male and female patients exhibited similar hematological parameters (hemoglobin, platelet count, absolute neutrophil counts, and reticulocytes). Ten out of 214 patients (4.7%) were tested positive for presence of PNH clones. Cytogenetic studies were available in 195 out of 214 patients, all of them carrying normal findings. Conventional metaphase karyotyping was carried out in 184 cases. For rest of the cases, the test could not be done for various reasons. No structural abnormalities were observed on routine cytogenetics. Severity of disease varied slightly between the groups with more male patients labeled with severe AA (51%) compared to females (40.4%). In our cohort, 57 male and 9 female patients underwent HSCT with a complete response of 86% and 100%, respectively. Response to IST was rather poor with 12 out of 55 male and 7 out of 25 female patients achieving sustained CR beyond 6 months. Sixty-six of our patients (44 males and 22 females) who were undergoing pretransplant assessment or had no HLA matched donor were put on supportive treatment, comprising of oral hematinics, growth factors, and blood component support. Four of them recovered spontaneously with sustained increase in blood counts.
Epidemiological features of patients and controls are shown in Table 2. Among different occupations, students of both genders displayed high incidence of disease (52.2% in males and 33.3% in females, P < 0.0001). Regardless of gender, AA was strongly associated with a low socioeconomic profile (56.54% in patients compared to 16.35% in controls, P < 0.0001) and rural habitat (59.3% in patients and 36.9% in controls respectively, P < 0.0001). Another factor significantly associated with the incidence of AA in the country was high degree of consanguinity especially first or second cousins (87.9% male and 78.9% female). The sociodemographic variables found significantly associated with AA in univariate analysis were entered into a multivariate logistic regression model, and odd ratios with 95% confidence interval are shown in the form of a forest plot (Supplemental Figure 2).
Serum cytokine expression profiles in AA patients
To measure cytokine levels of our AA patients, we selected 11 cytokines described previously in AA. Cytokine measurement was performed on serum samples of AA patients divided into various treatment groups and one control group as indicated in Fig. 1. Compared to 42 healthy individuals of control group, significantly higher serum G-CSF levels were observed in three patient groups; pretreatment group (61 males and 23 females, P < 0.0001), cyclosporin A group (22 males and 7 females, P < 0.0001), and supportive therapy group (25 males and 18 females, P < 0.0001). Among post-treatment groups, G-CSF levels were significantly lower in partial (9 males and 9 females, P < 0.0001) and complete responder (53 males and 12 females, P < 0.0001) groups than pretreatment group. Compared to G-CSF, TPO levels were extremely variable between the groups. All seven AA groups showed significantly elevated TPO levels than control group (Fig. 1). Similarly, the pretreated group had significantly higher (P < 0.0001) TPO levels than those of complete responders. Modestly higher INF-γ levels were detected in the pretreated, CsA, ATG, and BMT groups. The levels however returned to within normal range in post-treatment groups. IL-2 was also found to be higher in certain groups but the difference was statistically non-significant compared to controls. No significant differences were observed for other cytokine levels among various patient groups and controls (TNF-α, IL-4, IL-1β, IL-6, IL-17A, IL-17F, and IL-10).

Serum cytokine levels among different groups
Next, we classified AA patients into three broad groups (pretreatment, during treatment, and post-treatment) and their serum cytokine levels were subjected to multiple group comparisons (Supplemental Figure 1). A similar trend in serum G-CSF and TPO levels was observed in these groups with highest levels among pretreatment and lowest among post-treatment group. No significant difference was observed in other nine cytokines between any of the four groups.
HLA typing
Multiple studies have demonstrated that specific HLA alleles confer susceptibility to AA. In this study, we examined HLA allele frequency in 74 AA patients whose HLA typing was carried out in order to find HLA matched donor. Based on the data, we selected HLA alleles with more than 15% frequencies in AA patients and compared it with that of corresponding alleles in healthy Pakistani population by extracting data from previously published reports (Supplemental Figure 2; Supplemental Table 1). DRB1*15 (56.8%) and sum of DQB1*06 (70.3%), which comprised of DQB1*06 (35.1%), DQB1*0601 (25.7%), and DQB1*0603 (12.2%), were dominant alleles in the patient group. These HLA frequencies were significantly higher than those of the corresponding healthy individuals (Fig. 2).

Frequency of HLA alleles among AA patients and healthy controls
Mutation analysis of TERT and TERC genes in AA patients
TERT and TERC mutations are well documented as risk factors for developing AA. We therefore screened mutations of these genes in Pakistani AA patients with various clinical characteristics (Table 1) using the direct sequencing method. DNA was successfully extracted from 190 out of 214 patients. Of the190 patients sequenced, nine patients carried heterozygous non-synonymous variants of TERT and one young female patient (12 years old) had a homozygous 36 C>T TERC mutation (Table 3). Among seven different TERT mutations, Val526Ala present in the N-terminus was a novel mutation while Val777Met located in the TERT-reverse transcriptase (RT) region was previously found during our routine TERT mutation screening of AA patients (data not published). One female patient (21 years old) carried two TERT mutations (Val777Met and Arg972Cys). Three patients had the same Ala279Thr while two had Ala1062Thr. Clinical and epidemiological features of the ten patients with TERT and TERC mutations are shown in Supplemental Table 2 and Supplemental Table 3, respectively. Two patients with TERT Val777Met and TERC 36C>T mutations respectively, had siblings with AA, strongly indicating a genetic predisposition in these patients.
Discussion
AA is a condition of bone marrow hypoplasia resulting from increased destruction of hematopoietic progenitor cells. Etiology of AA is poorly understood and involves a complex interplay of environmental, clinical, genetic, and immunological risk factors. In this study, simultaneous assessment of these risk factors was carried out for the first time in a cohort of 214 Pakistani AA patients enrolled at our center during a period of 18 months. It is a tertiary care facility providing comprehensive care including bone marrow transplant to patients suffering from various hematological diseases. There are only four centers providing bone marrow transplant facility in the country, and ours being the biggest and the only public sector hospital gets maximum number of patients referred from all over the country. A large study on epidemiology of AA in 1324 patients from our center is already in its final stages. Incidence of AA is high in Pakistan with current estimates being 4–5/million (unpublished data from our center).
AA incidence is more common in Asia than in the West with a 2–3-fold higher ratio. Even in Asia, AA frequency varies among different countries, such as 7.4/million in China and 3/million in Thailand [13, 14]. Till date, no study has been reported to establish exact reasons of increased incidence of AA in Asian countries. However, it is plausible that variable AA rates might reflect variations in environmental exposures in addition to underlying genetic factors, since a wide range of disease prevalence is observed among geographically different regions in the world [22, 47]. From the fact that the frequency of AA in Asian residents in Hawaii is at par with that of Americans (unpublished data), the environmental factors appeared to be more influential than the genetic predisposition (unpublished data).
In our study, the age and sex distribution of AA was comparable to data from other Asian regions, such as Bangkok, China, and India [15, 23, 42]. This and other studies from Pakistan have shown a single patient age peak of AA incidence [8]. This is in contrast to the bimodal patient age peak (one in younger adults and a second in the elderly) observed in Thailand [13] and Barcelona [26]. The overrepresentation of students (age between 10 and 29 years) among our patients might be due to enrollment of cases in a specialized BMT facility. This phenomenon may also be attributed to lower average life expectancy in Pakistani population (66.3 years). Further, a high rate of consanguinity was seen in our AA cohort, speculating that accumulation of some genetic factors might have increased susceptibility to AA in these subjects. A recent study in Pakistan has demonstrated significant association of AA with a lower socioeconomic profile and high consanguinity among parents [39]. Most of the other epidemiological features of our patients were also consistent with those previously reported from the country. Collectively, though, certain environmental risk factors were found in our AA patients, but no definitive association of these etiological agents could be asserted in most of the patients in our study.
Different growth factors, cytokines, and/or chemokines regulate hematopoiesis in an autocrine/paracrine manner. In this study, we measured 11 cytokines in serum samples on Pakistani AA patients as these cytokines are thought to play crucial roles in the disease pathogenesis. Among them, we found significantly elevated levels of G-CSF and particularly TPO in AA patients, compared to controls, with decreasing tendency of these cytokine levels after immunosuppressive therapies. These findings are consistent with our previous work using plasma samples of AA patients at NIH [10]. Elevated levels of G-CSF and TPO have also been reported previously in patients with AA [20, 35], thus providing strong evidence of involvement of both the cytokines in disease pathogenesis. It is also hypothesized that high levels of TPO and G-CSF might be a surrogate of blood counts as the highest median values of TPO (3787 pg/mL) and G-CSF (168 pg/mL) were observed in treatment naïve group, having lowest values for hemoglobin (Hb) [78 g/L], white blood cells (WBC) [2.9 × 109/L], platelets (7 × 109/L), and absolute neutrophil count (ANC) [0.76 × 109/L] respectively, as compared to other groups. On the other hand, patients in complete remission (CR) had highest counts of Hb (120 g/L), WBC (6.2 × 109/L), platelets (164 × 109/L), and ANC (3.1 × 109/L) respectively.
Sera of patients with AA contain high level of IFN-γ which is a direct mediator of hematopoietic suppression [51]. Schultz and Shahidi have reported increased TNF-α levels contributing to progressive hematopoietic suppression in AA [36]. Peripheral blood cells in AA also display abnormally high IL-2 levels that is followed by its reduction after high-dose immunosuppression [29]. Despite these reports, we could not find significant differences of IFN-γ and IL-2 levels between AA patients and the control group, though a slightly higher tendency was observed in some patient groups compared to the controls. As for TNF-α and other cytokine (TNF-α, IL-4, IL-1β, IL-6, IL-17A, IL-17F, and IL-10) levels, we did not detect any significant variation among the designated groups. This observation was different from the previously reported results for these cytokine levels and could be attributed to the qualitative and quantitative differences between patient groups. Besides, previously reported results of some cytokines in AA patients are still inconsistent [9, 10].
A variety of diseases including autoimmune disorders, cancer, and infections have been reported to be associated with particular HLA alleles. Multiple studies have demonstrated association of a significantly increased incidence of AA with particular HLA alleles. AA patients display overrepresentation of HLA-DR2 (HLA-DR15) especially DRB1*1501 and 1502 alleles and class I HLA-A*02:01, A*02:06, A*31:01, and B*40:02 [17, 21]. HLA typing was carried out in 74 of our patients. There was a significantly high frequency of HLA-DRB1*15 (56.8%) and sum of DQB1*06 (70.3%) in these patients. We compared our HLA allelic frequency data with historical controls (approximately 2300 subjects) pooled from previously published work in Pakistan which showed allelic frequencies ranging between 8.5–39% for HLA-DR15 and 13.5–48.4% for DQB1*06 respectively. According to a previous study in Pakistani AA patients, DRB1*15 and DRB1*03 were reported as susceptible and protective alleles respectively [31]. Another study in Pakistani AA patients has described HLA B5 [40, 49] as the highest allele (27%) in AA. To our best knowledge, there were no reports on association of the DQB1*06 allele with AA. DQB1*0601 and DQB1*0603 are known to be protective alleles; DQB1*0601 against narcolepsy, juvenile diabetes, and multiple sclerosis and DQB1*0603 against many autoimmune diseases. DQB1*0602 increases risks of cervical cancer and MS but protects against juvenile diabetes. Since there were no complete subtype allele data for DQB1*06 in our cases, we were unable to conclude which subtype allele of DQB1*06 was associated to AA. Nonetheless, it was curious to observe markedly high DQB1*06 allele frequency among our cases. This emphasizes on need of further studies in large cohorts with detailed DQB1*06 subtype allele analysis to establish a possible association. Since some HLA alleles are common in the general population, and only a fraction of individuals with these alleles actually develop AA, therefore it might be speculated that some HLA alleles trigger AA development by interacting with other genetic and environmental factors. Currently, it is difficult to draw definitive conclusions about HLA alleles predisposing to the development of AA as HLA alleles reported for association with AA are divergent among studies using different populations and ethnicities.
High penetrance of TERT and TERC mutations is observed in various diseases in addition to bone marrow failure (BMF) syndromes [6], modifying the host susceptibility to environmental risks. In our study, we were able to isolate good quality genomic DNA from 190 out of 214 enrolled patients. Among these, 10 patients (4.6%) carried TERT and TERC mutations. Heterozygous non-synonymous variants of TERT and homozygous variant of TERC were present in nine and one patient, respectively. Among them, a Val526Ala mutation present in the TERT-N-terminus was novel, while Val777Met was previously found during our routine mutation screening (data not published). Other mutations identified have been reported previously as pathogenic: Arg972Cys in a DKC patient [41] and two TERT mutations (Ala202Thr and Arg672Cys) in AA [16, 34] and in inherited BMF syndromes [45]. There is controversy regarding significance of Ala279Thr and Arg1062Thr TERT variants found in our current cohort work. However, in a recent study, Ala279Thr was reported to be pathogenic as it decreases telomere length and inhibits non-canonical telomerase activity in esophageal carcinomas [50].
As for the Arg1062Thr TERT variant, we have previously demonstrated that the allele frequency was 3-fold higher in patients than in controls in our three cohorts [4]. In addition, it has been reported that Arg1062Thr mutation is an independent negative prognostic factor in AML patients [1]. Therefore, it seems likely that Arg1062Thr TERT allele may cause telomere shortening over many years or that the Arg1062Thr mutation may perturb other functions, such as the recruitment of telomerase to telomeres, which were not revealed by simple enzyme assays. In this work, we found the homozygous 36 C>T TERC mutation although its heterozygous mutation has been shown in the congenital AA patients [38]. The 36 C>T TERC mutation site is in the P1b region of the Psuedoknot domain where many mutations related to various diseases, such as AA, myelodysplasia, dyskeratosis congenital (DKC), and pulmonary fibrosis, have been reported. Furthermore, our two patients carrying TERT (Arg672Cys) and TERC (36 C>T) mutation had one and two affected siblings, respectively, strongly indicating that these patients carried germline mutations. The patients were rigorously screened for DKC through absence of clinical findings, physical features, and family history. Similar findings were documented by other groups [11, 44]. However, this does not rule out DKC as (1) newer telomeropathy genes (others than TERT and TERC) have been identified and (2) a patient can heritage from short telomeres of one mutated parent without transmission of the mutation (pheno-copy phenomenon).
Collectively, we found TERT and TERC mutations in only 5% of our AA patients, suggesting addition of other candidate genes in the mutational screening panels, particularly those involved in DNA methylation and tumor suppression. Further, larger cohort studies should be carried out combining epidemiological, clinical, and laboratory findings to dissect the interplay of genetic predisposition, environmental triggers, and clinic-pathological features that might contribute to etiology of AA.
References
Aref S, El-Ghonemy MS, Abouzeid TE, El-Sabbagh AM, El-Baiomy MA (2014) Telomerase reverse transcriptase (TERT) A1062T mutation as a prognostic factor in Egyptian patients with acute myeloid leukemia (AML). Med Oncol 31(9):158. https://doi.org/10.1007/s12032-014-0158-6
Bunce M (2003) PCR-sequence-specific primer typing of HLA class I and class II alleles. In: Powis SH, Vaughan RW (eds) MHC protocols. Humana Press, Totowa, pp 143–171
Bunce M, Passey B (2013) HLA typing by sequence-specific primers. Methods Mol Biol 1034:147–159. https://doi.org/10.1007/978-1-62703-493-7_8
Calado RT, Regal JA, Hills M, Yewdell WT, Dalmazzo LF, Zago MA, … Young NS (2009a) Constitutional hypomorphic telomerase mutations in patients with acute myeloid leukemia. Proc Natl Acad Sci U S A 106(4):1187–1192. doi:https://doi.org/10.1073/pnas.0807057106
Calado RT, Yewdell WT, Wilkerson KL, Regal JA, Kajigaya S, Stratakis CA, Young NS (2009b) Sex hormones, acting on the TERT gene, increase telomerase activity in human primary hematopoietic cells. Blood 114(11):2236–2243. https://doi.org/10.1182/blood-2008-09-178871
Calado RT, Young NS (2009) Telomere diseases. N Engl J Med 361(24):2353–2365. https://doi.org/10.1056/NEJMra0903373
Ducrest AL, Szutorisz H, Lingner J, Nabholz M (2002) Regulation of the human telomerase reverse transcriptase gene. Oncogene 21(4):541–552. https://doi.org/10.1038/sj.onc.1205081
Ehsan A, Shah SA, Ibrahim T (2011) Epidemiology of acquired aplastic anaemia in Pakistan. J Ayub Med Coll Abbottabad 23(1):102–105
Feng X, Scheinberg P, Samsel L, Rios O, Chen J, McCoy JP Jr, … Young NS (2012) Decreased plasma cytokines are associated with low platelet counts in aplastic anemia and immune thrombocytopenic purpura. J Thromb Haemost 10(8):1616–1623. doi:https://doi.org/10.1111/j.1538-7836.2012.04757.x
Feng X, Scheinberg P, Wu CO, Samsel L, Nunez O, Prince C, … Young NS (2011) Cytokine signature profiles in acquired aplastic anemia and myelodysplastic syndromes. Haematologica 96(4):602–606. doi:https://doi.org/10.3324/haematol.2010.030536
Fogarty PF, Yamaguchi H, Wiestner A, Baerlocher GM, Sloand E, Zeng WS, Read EJ, Lansdorp PM, Young NS (2003) Late presentation of dyskeratosis congenita as apparently acquired aplastic anaemia due to mutations in telomerase RNA. Lancet 362(9396):1628–1630. https://doi.org/10.1016/S0140-6736(03)14797-6
Hafeez M, Aslam M, Ali A, Rashid Y, Jafri H (2007) Regional and ethnic distribution of beta thalassemia mutations and effect of consanguinity in patients referred for prenatal diagnosis. J Coll Physicians Surg Pak 17(3):144–147
Issaragrisil S, Chansung K, Kaufman DW, Sirijirachai J, Thamprasit T, Young NS (1997) Aplastic anemia in rural Thailand: its association with grain farming and agricultural pesticide exposure. Aplastic Anemia Study Group. Am J Public Health 87(9):1551–1554
Issaragrisil S, Kaufman DW, Anderson T, Chansung K, Leaverton PE, Shapiro S, Young NS (2006) The epidemiology of aplastic anemia in Thailand. Blood 107(4):1299–1307. https://doi.org/10.1182/blood-2005-01-0161
Issaragrisil S, Sriratanasatavorn C, Piankijagum A, Vannasaeng S, Porapakkham Y, Leaverton PE, … Young NS (1991) Incidence of aplastic anemia in Bangkok. The Aplastic Anemia Study Group. Blood 77(10):2166–2168
Jung M, Dunbar CE, Winkler T (2015) Modeling human bone marrow failure syndromes using pluripotent stem cells and genome engineering. Mol Ther 23(12):1832–1842. https://doi.org/10.1038/mt.2015.180
Katagiri T, Sato-Otsubo A, Kashiwase K, Morishima S, Sato Y, Mori Y et al (2011) Frequent loss of HLA alleles associated with copy number-neutral 6pLOH in acquired aplastic anemia. Blood 118(25):6601–6609. https://doi.org/10.1182/blood-2011-07-365189
Khan SW, Iftikhar N, Ahmed TA, Bashir M (2015) HLA-DR alleles in Pakistani patients of pemphigus vulgaris. J Coll Physicians Surg Pak 25(4):233–236
Kojima S (2002) Aplastic anemia in the Orient. Int J Hematol 76(Suppl 2):173–174
Kojima S, Matsuyama T, Kodera Y, Nishihira H, Ueda K, Shimbo T, Nakahata T (1996) Measurement of endogenous plasma granulocyte colony-stimulating factor in patients with acquired aplastic anemia by a sensitive chemiluminescent immunoassay. Blood 87(4):1303–1308
Maciejewski JP, Follmann D, Nakamura R, Saunthararajah Y, Rivera CE, Simonis T, … Young NS (2001) Increased frequency of HLA-DR2 in patients with paroxysmal nocturnal hemoglobinuria and the PNH/aplastic anemia syndrome. Blood 98(13):3513–3519
Maluf E, Hamerschlak N, Cavalcanti AB, Junior AA, Eluf-Neto J, Falcao RP, … Pasquini R (2009) Incidence and risk factors of aplastic anemia in Latin American countries: the LATIN case-control study. Haematologica 94(9):1220–1226. doi:https://doi.org/10.3324/haematol.2008.002642
Melinkeri SR (2015) Epidemiology, pathogenesis and diagnosis of aplastic anaemia. J Assoc Physicians India 63(3 Suppl):8–12
Moalic V, Ferec C (2005) HLA typing, analysis methods, and clinical applications. Presse Med 34(15):1101–1108
Mohyuddin A, Ayub Q, Khaliq S, Mansoor A, Mazhar K, Rehman S, Mehdi SQ (2002) HLA polymorphism in six ethnic groups from Pakistan. Tissue Antigens 59(6):492–501
Montane E, Ibanez L, Vidal X, Ballarin E, Puig R, Garcia N, … Aplastic A (2008) Epidemiology of aplastic anemia: a prospective multicenter study. Haematologica 93(4):518–523. doi:https://doi.org/10.3324/haematol.12020
Muazzam AG, Mansoor A, Ali L, Siddiqi S, Hameed A, Ajmal M, Mazhar K (2013) Association of HLA-DRB1 and -DQB1alleles and haplotypes with rheumatoid arthritis in a Pakistani population. Arthritis Res Ther 15(4):R95. https://doi.org/10.1186/ar4275
Nakao S, Takami A, Takamatsu H, Zeng W, Sugimori N, Yamazaki H, … Matsuda T (1997) Isolation of a T-cell clone showing HLA-DRB1*0405-restricted cytotoxicity for hematopoietic cells in a patient with aplastic anemia. Blood 89(10):3691–3699
Nissen C, Moser Y, Weis J, Wursch A, Gratwohl A, Speck B (1986) The release of interleukin-2 (IL-2) and colony stimulating activity (CSA) in aplastic anemia patients: opposite behaviour with improvement of bone marrow function. Blut 52(4):221–230
Parker C, Omine M, Richards S, Nishimura J, Bessler M, Ware R, Hillmen P, Luzzatto L, Young N, Kinoshita T, Rosse W, Socié G; International PNH Interest Group (2005) Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood 106(12):3699–3709
Rehman S, Saba N, Khalilullah, Munir S, Ahmed P, Mehmood T (2009) The frequency of HLA class I and II alleles in Pakistani patients with aplastic anemia. Immunol Investig 38(8):812–819. https://doi.org/10.3109/08820130903271415
Risitano AM, Kook H, Zeng W, Chen G, Young NS, Maciejewski JP (2002) Oligoclonal and polyclonal CD4 and CD8 lymphocytes in aplastic anemia and paroxysmal nocturnal hemoglobinuria measured by V beta CDR3 spectratyping and flow cytometry. Blood 100(1):178–183. https://doi.org/10.1182/blood-2002-01-0236
Saleem N, Ali S, Ahmed TA, Iqbal M, Bashir M (2013) HLA-DR alleles among Pakistani patients of coeliac disease. J Pak Med Assoc 63(10):1271–1274
Sarek G, Marzec P, Margalef P, Boulton SJ (2015) Molecular basis of telomere dysfunction in human genetic diseases. Nat Struct Mol Biol 22(11):867–874. https://doi.org/10.1038/nsmb.3093
Schrezenmeier H, Griesshammer M, Hornkohl A, Nichol JL, Hecht T, Heimpel H, … Raghavachar A (1998) Thrombopoietin serum levels in patients with aplastic anaemia: correlation with platelet count and persistent elevation in remission. Br J Haematol 100(3):571–576
Schultz JC, Shahidi NT (1994) Detection of tumor necrosis factor-alpha in bone marrow plasma and peripheral blood plasma from patients with aplastic anemia. Am J Hematol 45(1):32–38
Shamsi T, Hashmi K, Adil S, Ahmad P, Irfan M, Raza S, … Ansari S (2008) The stem cell transplant program in Pakistan—the first decade. Bone Marrow Transplant 42 Suppl 1:S114-S117. doi:https://doi.org/10.1038/bmt.2008.137
Sugino S, Bortz BJ, Vaida S, Karamchandani K, Janicki PK (2015) Peripartum anesthetic management and genomic analysis of rare variants in a patient with familial pulmonary fibrosis. A A Case Rep 5(10):169–172. https://doi.org/10.1213/XAA.0000000000000198
Taj M, Shah T, Aslam SK, Zaheer S, Nawab F, Shaheen S, … Shamsi TS (2016) Environmental determinants of aplastic anemia in Pakistan: a case-control study. Z Gesundh Wiss 24(5):453–460. doi:https://doi.org/10.1007/s10389-016-0743-6
Taj M, Shamsi TS, Ansari SH, Farzana T, Nazi A, Nadeem M, … Kazmi JH (2014) Epidemiologic and HLA antigen profile in patients with aplastic anemia. J Coll Physicians Surg Pak 24(8):549–552
Vulliamy TJ, Kirwan MJ, Beswick R, Hossain U, Baqai C, Ratcliffe A, … Dokal I (2011) Differences in disease severity but similar telomere lengths in genetic subgroups of patients with telomerase and shelterin mutations. PLoS One 6(9):e24383. doi:https://doi.org/10.1371/journal.pone.0024383
Wang W, Wang XQ, Li P, Lin GW (2011) Incidence of adult aplastic anemia in Shanghai, China. Zhonghua Nei Ke Za Zhi 50(4):284–286
Welsh JP, Rutherford TR, Flynn J, Foukaneli T, Gordon-Smith EC, Gibson FM (2004) In vitro effects of interferon-gamma and tumor necrosis factor-alpha on CD34+ bone marrow progenitor cells from aplastic anemia patients and normal donors. Hematol J 5(1):39–46. https://doi.org/10.1038/sj.thj.6200340
Yamaguchi H, Calado RT, Ly H, Kajigaya S, Baerlocher GM, Chanock SJ, … Young NS (2005) Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N Engl J Med 352(14):1413–1424. doi:https://doi.org/10.1056/NEJMoa042980
Yamaguchi H, Sakaguchi H, Yoshida K, Yabe M, Yabe H, Okuno Y et al (2015) Clinical and genetic features of dyskeratosis congenita, cryptic dyskeratosis congenita, and Hoyeraal-Hreidarsson syndrome in Japan. Int J Hematol 102(5):544–552. https://doi.org/10.1007/s12185-015-1861-6
Young NS, Calado RT, Scheinberg P (2006) Current concepts in the pathophysiology and treatment of aplastic anemia. Blood 108(8):2509–2519. https://doi.org/10.1182/blood-2006-03-010777
Young NS, Kaufman DW (2008) The epidemiology of acquired aplastic anemia. Haematologica 93(4):489–492. https://doi.org/10.3324/haematol.12855
Young NS, Scheinberg P, Calado RT (2008) Aplastic anemia. Curr Opin Hematol 15(3):162–168. https://doi.org/10.1097/MOH.0b013e3282fa7470
Zafar N, Khan S, Qadir A, Raza Y, Naqvi A, Rizvi A (1996) HLA frequencies in Pakistani population groups. J Pak Med Assoc 46(1):12–13
Zhang Y, Calado R, Rao M, Hong JA, Meeker AK, Dumitriu B, … Schrump DS (2014) Telomerase variant A279T induces telomere dysfunction and inhibits non-canonical telomerase activity in esophageal carcinomas. PLoS One 9(7):e101010. doi:https://doi.org/10.1371/journal.pone.0101010
Zoumbos NC, Gascon P, Djeu JY, Young NS (1985) Interferon is a mediator of hematopoietic suppression in aplastic anemia in vitro and possibly in vivo. Proc Natl Acad Sci U S A 82(1):188–192
Acknowledgments
We thank Dr. Imtenan Shareef, Miss. Muqadas, and Miss. Sonia for assistance in obtaining AA patients’ data. This research was supported by the Intramural Research Program of the NIH, National Heart, Lung, and Blood Institute and International Research Support Initiative Program of Government of Pakistan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This study was approved by institutional review boards of Armed Forces Bone Marrow Transplant Center (AFBMTC), Pakistan and National Heart Lungs and Blood Institute (NHLBI) at National Institutes of Health (NIH), USA. Patients’ rights were protected in accordance with the Helsinki declaration. Informed written consent was taken from all patients before drawing blood sample for research.
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Akram, Z., Ahmed, P., Kajigaya, S. et al. Epidemiological, clinical and genetic characterization of aplastic anemia patients in Pakistan. Ann Hematol 98, 301–312 (2019). https://doi.org/10.1007/s00277-018-3542-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-018-3542-z