
Abstract
The elastic behavior and the structural evolution at high pressure of a natural phillipsite have been investigated by in situ single-crystal X-ray diffraction up to 9.44 GPa, using a diamond anvil cell and the nominally penetrating P-transmitting fluid methanol:ethanol:water (16:3:1) mix. Although no phase transition was observed within the P-range investigated, two different compressional regimes occur. Between 0.0001 and 2.0 GPa, the refined elastic parameters, calculated by a second-order Birch–Murnaghan equation of state (BM-EoS) fit, are V 0 = 1005(1) Å3, K 0 = 89(8) GPa for the unit-cell volume; a 0 = 9.914(7) Å, K a = 81(12) GPa for the a-axis; b 0 = 14.201(9) Å, K b = 50(5) GPa for the b-axis; and c 0 = 8.707(2) Å, K c = 107(8) GPa for the c-axis (K a :K b :K c ~1.62:1:2.14). Between 2.0 and 9.4 GPa, a P-induced change in the configuration of H2O molecules, coupled with a change in the tilting mechanisms of the framework tetrahedra, gives rise to a second compressional regime, in which the phillipsite structure is softer if compared to the first compressional range. In the second compressional regime, the refined elastic parameters, calculated by a second-order BM-EoS fit, are V 0 = 1098 (7) Å3, K 0 = 18.8(7) GPa for the unit-cell volume; a 0 = 10.07(3) Å, K a = 30(2) GPa for the a-axis; b 0 = 14.8(1) Å, K b = 11(1) GPa for the b-axis; and c 0 = 8.94(2) Å, K c = 21(1) GPa for the c-axis (K a :K b :K c ~2.72:1:1.90). The evolution of the monoclinic β angle with pressure shows two distinct trends in the two compressional regimes: with a negative slope between 0.0001 and 2.0 GPa, and a positive slope between 2.0 and 9.4 GPa. The mechanisms, at the atomic scale, that govern the two compressional regimes of the phillipsite structure are described.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Phillipsite is one of the most common natural zeolites with ideal composition K2(Na, Ca0.5)3[Al5Si11O32]·12H2O (Passaglia and Sheppard 2001), commonly found in altered sediments in “closed” or “open” hydrologic systems, such as saline lakes, deep-sea sediments, hot spring deposits, as well as in vugs of basalt and in tuffs (Galli and Loschi Ghittoni1972; Rinaldi et al. 1974; Gottardi and Galli 1985; Armbruster and Gunter 2001). In Langella et al. (2001), it is reported how the formation of phillipsite requires low silica activity, along with high pH.
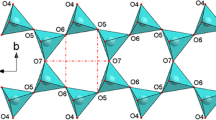
Phillipsite is monoclinic (space group P21 /m, a ~9.865 Å, b ~14.300 Å, c ~8.693 Å, β ~124.92°), with framework density: 15.8 T/1000 Å3 (Baerlocher et al. 2007). In nature, crystals are often found in spherical radiating aggregates, frequently twinned on [001], [021] and [110] (Rinaldi et al. 1974). Several examples of intergrowths with other zeolites (e.g., faujasite, offretite, gismondine, garrionite and gobbinsite) are reported in the literature (Rinaldi et al. 1975, Passaglia and Sheppard 2001). In a very recent paper, Danisi et al. (2015) reported coherent twinned intergrowths of merlinoite and phillipsite in a sample originated from Monte Somma, Vesuvius (Italy). The framework of phillipsite (PHI topology, Baerlocher et al. 2007) is built up by four corner-shearing Si/Al tetrahedra, which form four- and eight- membered rings. The four-membered rings (hereafter 4-mRs) and the eight-membered rings (hereafter 8-mRs) are also referred as the secondary building units of PHI framework type (SBU, codes 4 and 8, respectively, Baerlocher et al. 2007). In PHI framework, the 4-mRs are connected to form double-crankshaft chains running parallel to [100]. The PHI framework contains two independent channel systems, running along [100] and [010] (hereafter: 8-mRs[100] and 8-mRs[010] channels, respectively) (Fig. 1). The two sets of channels intersect each other (Gatta and Lee 2007; Gatta et al. 2009a). The channel running along [100] is confined by two different 8-mRs, hereafter 8-mRs[100]-1 and 8-mRs[100]-2. In the 8-mRs[100]-1, the distance O9–O8 < O7–O7, whereas, in the 8-mRs[100]-2, O9–O8 > O7–O7 (Fig. 1). The 4-mRs double-crankshaft chains form 8-mRs, here referred as 8-mRs(001) (Fig. 1). As pointed out by Rinaldi et al. (1974) and Gatta et al. (2009a, 2010, 2015), the Si/Al distribution between the tetrahedral sites is completely disordered in natural phillipsite.

Configuration of the 8-mR[100]-1, 8mR[100]-2, 8-mR[010] and 8-mR(001), and of the extra-framework population in phillipsite (as viewed down [100]), based on the structural refinements at 0.20 GPa (P1)
In phillipsite from Richmond, Victoria, Australia (i.e., the same natural species used in this study), the extra-framework population (Fig. 1) is represented by two main cation sites in which Ca (or Na) and K lie. Viewing the structure perpendicular to (010), the Ca site lies above and below the mirror plane, where the two independent systems of channels intersect. As reported by Gatta et al. (2009a), the coordination shell of Ca site is complex, with a maximum distance of ~3 Å, and at least nine mutually excluding configurations are possible. The coordination number (CN) of the Ca sites is 6–7: three oxygen atoms of the tetrahedral framework (O4, O3, O7) and three or four H2O molecules (among W1, W3, W4–W4′, W5, W6). The K site is actually split into two subsites, K1 and K2, only 0.5 Å apart. The two K sites lie close to the 8-mRs[100] channel wall. If a maximum bond length of 3.4 Å is considered, the CN of both sites is 9 (five oxygen atoms of the framework and four H2O molecules) (Gatta et al. 2009a). As shown in Gatta et al. (2009a), the H2O molecules are distributed over seven independent sites: Only W1 and W2 lie on the mirror plane, occupying the Wyckoff special position 2e; the others sites (i.e., W3, W5, W6, W4, W4′ in Gatta et al. 2009a) occupy the general Wyckoff position 4f. W4 and W4′ are two subsites only 0.4 Å apart, and mutually exclusive. The sites W4, W4′, W5, W6 occupy central positions in the 8-mRs[010] channel, whereas the W3 site lies close to the channel wall. Among them, W4, W4′ and W6 lie at the intersection of the two channels. The W1 and W2 sites lie approximately in the center of the 8-mRs[100] channel (Fig. 1).
The high-temperature behavior of natural and synthetic phillipsites has been the object of several investigations (e.g., Steinfink 1962; Stuckenschmidt et al. 1990; Gualtieri et al.1999a, b, 2000; Passaglia et al. 2000; Sani et al. 2002). The thermal behavior and, in particular, the dehydration mechanisms are connected to the nature of the extra-framework population. Due to the lack of single crystals of phillipsite suitable for non-ambient conditions experiments, Gatta and Lee (2007) investigated the high-pressure behavior of a natural phillipsite powder from Richmond, Victoria, Australia, up to 3.64 GPa, by in situ synchrotron X-ray powder diffraction with a diamond anvil cell, using the methanol:ethanol:water = 16:3:1 mix as P-transmitting fluid. Axial and volumetric bulk moduli were reported. No unambiguous evidence of phase transition was observed within the P-range investigated. Moreover, the quality of the high-P powder data, as well as the complex structure of phillipsite, prevented a fulfill discussion about the role played by the extra-framework population on the elastic behavior. Although the general aspects of the high-P behavior of the zeolite phillipsite were discussed by Gatta and Lee (2007), a comprehensive characterization of the mechanisms which involve framework and extra-framework population is still missing. In addition, the previous high-P study explored a modest P-range. In light of this, the aim of this work is to reinvestigate the P-induced structural evolution of a natural phillipsite (using the same natural sample previously used by Gatta and Lee 2007) by in situ single-crystal synchrotron X-ray diffraction, using a diamond anvil cell.
Experimental methods
The HP-synchrotron X-ray single-crystal diffraction experiments were conducted at the Extreme Conditions Beamline P02.2 at PETRA III/DESY, Hamburg, Germany. Data collections were performed with an incident beam of 42.7 keV in energy, and a focusing spot of ~8.5 (H) μm × 1.8 (V) μm originated from a compound refractive lenses system, consisting of 121 Be lenses with a radius of 50 μm (400 μm beam acceptance) and a focal length of 1221 mm. A single crystal of phillipsite (~50 × 50 × 20 μm), free of twinning under polarized-light microscope and with lamellar morphology, was selected for the experiment. The crystal was loaded in a symmetric DAC equipped with Boehler–Almax design diamonds/seats with a 70° opening and 300 μm culet size. A 250-μm-thick foil of stainless steel was used as gasket, which was pre-indented to ~60 μm and then drilled with a hole of ~200 μm in diameter, using a spark-erosion device. A few ruby spheres were added into the gasket hole for P measurement, by the ruby-fluorescence method (Mao et al. 1986; pressure uncertainty ±0.05 GPa). The mix of methanol:ethanol:water = 16:3:1 (hereafter m.e.w.) was used as hydrostatic P-transmitting fluid (Angel et al. 2007). This pressure medium is considered potentially “penetrating” (Gatta and Lee 2014), as at least the molecules of H2O (∅ ~ 2.65Å) and CH3OH (∅ ~ 3.76 Å) may theoretically be incorporated into the 8-mRs[100] channels of phillipsite (∅ ~ 3.8 Å, Baerlocher et al. 2007). Pressure was increased (up to P max = 9.4 GPa, Table 1) with an automated pressure-driven system from Sanchez Technology (Viarmes, France) and measured with the online ruby/alignment system powered by a 100 mW 458-nm laser. Diffraction images were collected with a PerkinElmer XRD 1621 flat-panel detector, using an in-house script for collecting step-scan diffraction images. The sample-to-detector distance (402.34 mm) was calibrated using a CeO2 standard (NIST 674a). A few data collections were performed in decompression (Table 1). The diffraction images were then converted to conform to the “Esperanto” format of the program CrysAlis (Agilent 2012; Rothkirch et al. 2013). At all pressure points, the adopted data collection strategy consisted in a pure ω-scan (−28° ≤ ω ≤ +28°), with a step size of 1° and an exposure time of 1 s/frame; then, Bragg peaks were indexed. Intensities of the diffraction peaks were integrated and corrected for Lorentz-polarization effects, using the CrysAlis package (Agilent 2012). Corrections for adsorption (due to the DAC components) were applied by the semiempirical ABSPACK routine implemented in CrysAlis. The refined unit-cell parameters are listed in Table 1.
Structure refinement protocol
All the structure refinements, at different pressures, were performed using the software JANA2006 (Petříček et al. 2014) in the space group P21 /m, as suggested by the reflection conditions. Reflections were “observed” down to an interplanar distance d ~0.8 Å. At P ≥ 2.57 GPa, an abrupt decrease in the number of observed reflections, with a consequent increase in the R 1 factor, was observed (Table 2). The isotropic structure refinements were possible up to 4.85 GPa (P11). For the first refinement (P1, 0.2 GPa), the input fractional coordinates of the framework sites were taken from Gatta et al. (2009a). The positions of the extra-framework sites were obtained by a careful analysis of the difference-Fourier maps of the electron density. At P1, one Ca site and two mutually independent K sites were identified, along with six independent sites assigned to the H2O molecules, according to the structure model of Gatta et al. (2009a); the same sites’ labeling scheme was used (i.e., Ca, K1, K2 and W1-6, Table 3). Briefly, W1 and W2 sites lie in the center of 8-mRs[100]-1 and 8-mRs[100]-2, respectively, whereas W3 and W5 occupy the 8-mRs[010] channel; the W4 and W6 sites lie at the intersection of the two channel systems. As the analysis of the difference-Fourier maps revealed only one strong peak lying on the mirror plane between the position of the sites W4–W4′ in Gatta et al. (2009a), in the structure model of this study, we refined only one site (labeled as W4, Table 3). No specific restraints were applied to the Ca site, whereas the K1–K2 sites were refined with the same displacing parameter (DP). The same restriction was applied to all the H2O sites. This protocol was used in all the refinements at different pressures, with the following exceptions:
-
The DPs of the H2O sites were forced to have an U iso value fixed to 0.075 Å2. This value is arbitrary, although very reasonable: The refined U iso of H2O sites at P1 was ~0.079(4) Å2 (Table 3).
-
The occupancy of the Ca site was fixed to the one obtained from the refinement of P1. The sum of the K1 and K2 site occupancies, in all the refinements at P > P1, was forced to assume the value found at P1.
-
The R 1 factors were all ≤10 % up to 2.56 GPa (P8). At P > P8, the R 1 values increased significantly; the last refinement which reached convergence is that at 4.85 GPa (P11). At P > P8, the occupancy of the H2O sites showed an anomalous increase. As the adsorption of H2O molecule at this pressure is not realistic (i.e., all the previous experimental findings available in the literature reported P-induced over-hydration effects at P < 1 GPa, Gatta and Lee 2014, for a review), we introduced a further restraint: The sum of the H2O site occupancies was fixed to the value obtained by the refinement at P8 (i.e., 14.36 ± 1.16 molecules per formula unit, m.p.f.u.). The sum at P8 is equal, within 1σ, to that obtained at P1 (i.e., 14.00 ± 0.84 m.p.f.u.).
For all the structure refinements, Table 2 lists the principal statistical parameters. Site coordinates and occupancies are given in Table 3. Refined bond distances are reported in Tables 4 and 5. Other relevant structural parameters are reported in Tables 6, 7, 8, and 9.
Results
High-pressure elastic behavior
The unit-cell parameters at eighteen P-points up to 9.44(5) GPa, and three points in decompression, are reported in Table 1. The P-induced evolution of the unit-cell parameters is shown in Fig. 2. The evolution of the unit-cell parameters shows at least two different compressional regimes. From P1 to P6 (1.82 GPa), phillipsite is stiffer than in the second regime [i.e., from P9 (3.13 GPa) to P19 (9.44 GPa)]. Up to P8 (2.56 GPa), the value of the β angle decreases, whereas at P ≥ P9, there is a clear inversion of the trend (Fig. 2). The evolution of the β angle is diagnostic for the change in the compressional regimes. For the two regimes, a Birch–Murnaghan equation of state truncated to the second order was fitted to the experimental P–V data using the EoSFit 7.0 software (Angel et al. 2000, 2014). We did not consider the unit-cell parameters of P7 (2.14 GPa) and P8 in the calculation of the elastic parameters, considering the P-range between P7 and P8 as a transitional interval between the two regimes. The refined elastic parameters are reported in Table 10. Bulk moduli, K V , K a , K b , K c of the first regime (hereafter named K 1 x ), are significantly higher than the ones obtained for the second regime (hereafter named K 2 x ), suggesting that, at low pressure, phillipsite is “stiffer.” In the second regime, the linear bulk moduli decrease drastically and anisotropically (e.g., K a decreases by about 2.5 times, whereas K b and K c decrease by about 5 times). The reasons of such changes in the elastic behavior, at the atomic level, will be discussed in the next sessions.

High-pressure evolution of the normalized (to P1) unit-cell volume and axial parameters of phillipsite compressed in m.e.w. V/V 0 in black squares, a/a 0 red circles, b/b 0 blue triangles, c/c 0 cyan triangles
Pressure-induced structural evolution: deformation of the Si/Al framework
The intra-tetrahedral T–O distances (reported in Table 4) do not show drastic changes within the P-range investigated. The most significant changes in the inter-tetrahedral angles are those observed for the T1–O7–T3, T2–O8–T2, T4–O6–T2 and T4–O1–T3 angles, reported in Table 11. Figure 3 shows the trends of these T–O–T angles (normalized to the value at P1). At P7, all trends change markedly. The inter-tetrahedral tilting gives rise to a pronounced increase in the ellipticity of all the 8-mRs. In order to explain the behavior of the 8-mRs, we introduce the ε parameter (i.e., the ellipticity ratio) as the b/a ratio, where b is the minor and a the major axis of any given ring (e.g., Gatta and Lee 2007; Gatta et al. 2009a). The ε ratio for the 8-mRs[100]-1 (hereafter ε8-mRs[100]-1) is defined as O9–O8/O7–O7 (with O7–O7 > O9–O8); for the 8-mRs[100]-2, the ε ratio (hereafter ε8-mRs[100]-2) is defined as O7–O7/O9–O8 (as O7–O7 < O9–O8). For the 8-mRs[010] and the 8-mRs(001) (hereafter ε8-mRs[010] and ε8-mRs(001)), the ε ratios are defined as O3–O3/O1–O1 and O9–O8/O5–O5, respectively. The trend of all the ε values of the 8-mRs is shown in Fig. 4. Every ring tends to increase its ellipticity with the pressure increase, as previously observed in several others zeolites (e.g., Gatta et al. 2005, 2009b; Lotti et al. 2016). Contrarily to what was observed by Gatta and Lee (2007), the ɛ ratio of the 8-mRs[100]-2 is more pronounced than that of the 8mRs[100]-1. This feature is maintained at high-P, and, interestingly, the trends of ε8-mRs[100]-1, ε8-mRs[100]-2 and ε8-mRs[010] are practically parallel (Fig. 4). However, at a careful analysis from P7 to P11, ε8-mRs[100]-2 decreases more pronouncedly than ε8-mRs[100]-1 and ε8-mRs[010]. In fact, in the considered P-range, the slope of ε8-mRs[100]-2 is −0.059(2), whereas the slope of both ε8-mRs[100]-1 and ε8-mRs[010] is −0.052(3): The 8-mRs[100]-2 is, therefore, more affected by the P-induced deformation than 8-mRs[100]-1 and 8-mRs[010].

High-pressure evolution of some selected T–O–T angles. T2–O8–T2 in black squares, T1–O7–T3 red circles; T4–O1–T3 black squares, T4–O6–T2 red circles

High-pressure evolution of the ε8-mRs parameters; ε8-mRs[100]-2 in black squares, ε8-mRs[100]-1 red circles, ε8-mRs[010] blue triangles and ε8-mRs(001) cyan triangles
If the normalized O3–O3 and O7–O7 diameters of the 8-mRs[010] are plotted versus P, the trend are almost parallel up to P7 (Fig. 5). At P > P7, the diameter O3–O3 begins to decrease abruptly, whereas O7–O7 slightly increases. As the diameter O7–O7 is parallel to [100] and O3–O3 almost parallel to [001], this can explain the observed K a > K c at P > P7 (Table 10).

Evolution of the 8-mRs[010] diameters versus pressure. O7–O7 in black squares and O3–O3 in red circles
It is also interesting to note that at P8, the β angle reverses its negative trend with P and begins to increase. The change in β is somehow related to some T–O–T angles. We expect that the evolution of β might depend on the evolution of the inter-tetrahedral angles of the 8-mRs[010] and, in particular, on a combination of the T3–O1–T4 and T2–O6–T4 angles (Fig. 1). More in detail, if T3–O1–T4 and T2–O6–T4 were lying on the same plane (perpendicular to [010]), an increase in their values would correspond to a decrease in β. They actually do not lie on the same plane; however, if the value of the difference [(T2–O6–T4) − (T3–O1–T4)] is plotted versus pressure (Fig. 6), an almost overlapped trend to that of β is observed. This finding suggests that the combined effect of the T3–O1–T4 and T2–O6–T4 angles evolution can play a role in the evolution of the β angle and thus on the distortion of the (monoclinic) unit cell.

High-pressure evolution of the (normalized) difference between the inter-tetrahedral angles T3–O1–T4 and T4–O6–T2
Pressure-induced structural evolution: evolution of the extra-framework population
W sites
The first changes in the evolution of the extra-framework population are already observed at low-P regime. At P2, the analysis of the difference-Fourier maps of the electron density showed the presence of a maximum close (but distinct) to the position of W2. This peak, labeled as W2′ in this study, was not found in the analysis of the difference-Fourier maps at P1. Although we cannot exclude that the occupancies of the H2O sites are influenced by the constrains adopted for the DP values, it is possible that the sum of the occupancies of W2 and W2′ (which are mutually exclusive) at P2 is equal, within the e.s.d., to the occupancy of W2 site at P1. The distances W2′-O8 [i.e., 2.878(3) Å] and W2′-O2 [i.e., 2.893(3) Å] at P2 are shorter than the distances W2–O8 [i.e., 3.205(3) Å] and W2–O2 [i.e., 3.076(3) Å], and this suggests that the new W2′ site is involved in an energetically more favorable H-bonding network if compared to W2. At P9, the two maxima ascribable to W2 and W2′ were not distinctly observed anymore: Only one distinct electron-density peak was found. As the distance between the two subsites decreases significantly from P6 to P8 (from ~1 to ~0.75 Å, respectively), it is highly likely that they converge to one site at P9 (3.13 GPa), here re-labeled as W2 (Table 3, Fig. 7). It is worth to point out that the occupancy of W2 at P9 is slightly lower than the sum of the W2 and W2′ sites at P8 (Table 3).

Migration of W1 with P viewed down [010] (a, b, c), and general view of the P-induced changes into the structure (d, e). H2O molecules in small red spheres, K cations in yellow and Ca in dark gray
As pressure increases, the distance between W1 and Ca decreases (Table 5), toward non-realistic values expected for a Ca–W distance (Fig. 7). This finding suggests that Ca and W1 are actually two mutually exclusive sites, even in the low-P regime. An additional finding concerns the “migration” of W1 out of the 8-mRs[100]-1. Such a behavior implies a change in its H-bonding scheme. As reported in Table 5, at P1, the distances W1–O9 and W1–O3 are 2.940(3) and 3.081(3)Å, respectively. The same distances at P11 are 2.690(3) and 3.229(3) Å, respectively, which means that the distance W1–O9 decreases by ~9 %, whereas the W1–O3 increases by ~4.5 %.
The analysis of the difference-Fourier maps, based on the refinements at P10 and P11, revealed the presence of two new maxima, here labeled as W2″ and W1′, lying in the 8-mRs[100]-2 and 8-mRs[100]-1 rings, respectively (Table 3, Fig. 7), with partial site occupancy and mutually exclusive with the co-respective W1 and W2 (Table 3). These new sites have distances with some framework oxygen sites ascribable to H-bond interactions (e.g., W1′–O9 ~2.9 Å, W2″–O8 ~2.5 Å, W2″–O2 ~2.6 Å).
Additional effects of H2O migration among the W sites are observed at W4, W5 and W6. The fractional coordinates of the W3, W5 and W6 sites do not show any substantial change with the increase in pressure. The occupancy of the W4 sites decreases as pressure rises, whereas the occupancies of W5 and W6 increase. At P > P9, no evidence of the W4 site was found in the difference-Fourier maps, coupled with significantly higher densities at W5 and W6 than those observed at P1 (Fig. 8).

Clinographic view with the helicoidal configuration of the H2O molecule chain in the low (left) and high (right) compressional regime
A general view of the P-induced changes involving the extra-framework population is shown in Fig. 7.
Ca and K1–K2 sites
As reported in Table 5, the distance Ca–O7 increases with pressure, whereas the distances Ca–O4 and Ca–O3 decrease. This trend leads the Ca site toward the mirror plane located at y = ¼. This behavior might be correlated to the tilting of the TO4 tetrahedra and, in particular, to the closure of the O4–O7–O3 angle in the 8-mRs[100]-1.
The structural data show also a correlation between the distances K1/K2–O1 and K1/K2–O8 and the inter-tetrahedral angles T3–O1–T4 and T2–O8–T2, respectively. As explained in the previous section, the principal effect of the hydrostatic compression on the 8-mRs(001) is the tilting of the TO4 tetrahedra, which leads to a reduction of the T3–O1–T4 and T2–O8–T2 angles with pressure. The decrease in the aforementioned angles leads to a shortening of the K1/K2–O1 and K1/K2–O8 distances (Table 5).
Discussion and conclusions
This is the first experiment in which the high-P behavior of a natural phillipsite is described on the basis of single-crystal X-ray diffraction data (collected up to 9.4 GPa). The previous experiment on phillipsite was conducted on a polycrystalline sample up to 3.6 GPa (Gatta and Lee 2007). Gatta and Lee (2007) described the elastic behavior of phillipsite on the basis of powder data. However, the low quality of the powder data themselves, along with the modest P-range investigated (i.e., P max = 3.6 GPa), did not allow the authors to have a clear picture of the high-P behavior of this zeolite, and only one BM-EoS was used to model its compressibility. A potential change in the compressional behavior was already reported by Gatta and Lee (2007) at P > 2 GPa. In this study, we used the same natural sample previously used by Gatta and Lee (2007). In light of this, this work can be considered as an extension of the first study. Also in this case, we observe a change in the compressional behavior of phillipsite between 2.0 and 2.5 GPa, which is not apparently due to a potential penetration of the P-fluid molecules. The inversion of β-trend is likely the most evident effect of such a change. The diffraction patterns and the structure refinements confirmed that the space group P21 /m is preserved within the entire P-range investigated (i.e., 0–9.4 GPa). Therefore, the change in the elastic behavior does not reflect a phase transition, but rather a change in the deformation mechanisms at the atomic scale.
More specifically, phillipsite experiences a “softening” at P > 2–2.5 GPa, and the bulk modulus in the low-P regime is drastically higher than that of the high-P regime [i.e., 89(8) vs. 18.8(7) GPa, Table 10]. A similar behavior was previously observed in other open-framework materials (e.g., Gatta et al. 2006, 2008, 2009b, 2012). As the tetrahedra behave as rigid units (at least at a first approximation), the structure deformation is basically governed by tetrahedral tilting and by the rearrangement of the extra-framework population, as usually observed in zeolites (e.g., Gatta et al. 2008, 2014; Gatta and Lee 2014; Lotti et al. 2016).
The elastic anisotropic scheme of phillipsite is preserved within the P-range investigated, with K a, K c >> K b . The structure is, therefore, more compressible along [010]. From the low- to the high-P regime, the linearized bulk modulus K b decreases from 50(5) to 12(1) GPa. Such a behavior might be somehow influenced by a change in the extra-framework population and, more specifically, by the disappearance of the W4 site. W4 (along with W5 and W6) lies almost in the center of the 8-mRs[010] channel, in such a way that a helicoidal H2O chain occurs. The lack of W4 implies a reduced “pillar effect” of the H2O chain (Fig. 7, 8). As the H2O chain is perpendicular to (010), its weakening does not affect significantly the compressibility along [100] or [001]. However, if the ratio O7–O7/O3–O3 is plotted versus P, where O3–O3 and O7–O7 are two independent diameters of the 8-mRs[010] channel (Fig. 5), we observe an almost horizontal trend up to P7 and then a drastic increase at P > P7. Therefore, one of the reasons of the softening along [100] and [001] can be ascribed to the tilting of the 8-mRs[010]-tetrahedra, in response to the disappearance of W4. The tilting causes the deformation of the 8-mRs[010] channel and, in turn, the β-inversion.
An additional cause of the elastic anisotropy may be correlated to the migration of W1. The lack of W1 at high-P, in the 8-mRs[100]-1, leads to a significant shortening of the O8–O9 diameter, which is perpendicular to [001]. Therefore, the migration of W1 cannot affect K a or K b but only K c .
There is not a unique explanation about the H2O sites migration at high pressure and the occurrence of subsites, e.g., W1′, W2′, W2″ (Table 3). The lack of the proton positions does not allow a clear view of the H-bonding network. However, it appears that the framework deformation leads to energetically most favorable H-bonding connection with H2O molecules located in a slightly different positions than the parent ones (at room conditions), promoting split and, in general, H2O sites migration.
References
Agilent (2012) CrysAlis RED. Agilent Technologies Ltd, Yarnton
Angel RJ, Hazen RM, Downs RT (2000) High-temperature and high-pressure crystal chemistry. Rev Mineral Geochem 41:35–60
Angel RJ, Bujak M, Zhao J, Gatta GD, Jacobsen SD (2007) Effective hydrostatic limits of pressure media for high-pressure crystallographic studies. J Appl Crystallogr 40:26–32
Angel RJ, Alvaro M, Gonzalez-Platas J (2014) EosFit7c and a Fortran module (library) for equation of state calculations. Z Kristallogr 229:405–419
Armbruster T, Gunter ME (2001) Crystal structures of natural zeolites. Rev Mineral Geochem 45:1–57
Baerlocher C, McCusker LB, Olson DH (2007) Atlas of zeolite framework types, 6th edn. Elsevier, Amsterdam
Danisi RM, Armbruster T, Nagashima M (2015) Structural intergrowth of merlinoite/phillipsite and its temperature-dependent dehydration behaviour: a single-crystal X-ray study. Mineral Mag 79:191–203
Galli E, Loschi Ghittoni AG (1972) The crystal chemistry of phillipsites. Am Mineral 57:1125–1145
Gatta GD, Lee Y (2007) Anisotropic elastic behaviour and structural evolution of zeolite phillipsite at high pressure: a synchrotron powder diffraction study. Microporous Mesoporous Mater 105:239–250
Gatta GD, Lee Y (2014) Zeolites at high pressure: a review. Mineral Mag 78:267–291
Gatta GD, Comodi P, Zanazzi PF, Boffa Ballaran T (2005) Anomalous elastic behavior and high-pressure structural evolution of zeolite levyne. Am Mineral 90:645–652
Gatta GD, Nestola F, Boffa Ballaran T (2006) Elastic behavior, phase transition and pressure-induced structural evolution of analcime. Am Mineral 91:568–578
Gatta GD, Rotiroti N, Boffa Ballaran T, Pavese A (2008) Leucite at high-pressure: elastic behaviour, phase stability and petrological implications. Am Mineral 93:1588–1596
Gatta GD, Cappelletti P, Rotiroti N, Slebodnick C, Rinaldi R (2009a) New insights into the crystal structure and crystal chemistry of the zeolite phillipsite. Am Mineral 94:190–199
Gatta GD, Rotiroti N, Boffa Ballaran T, Sanchez-Valle C, Pavese A (2009b) Elastic behavior and phase stability of pollucite, a potential host for nuclear waste. Am Mineral 94:1137–1143
Gatta GD, Cappelletti P, Langella A (2010) Crystal-chemistry of phillipsites from the Neapolitan Yellow Tuff. Eur J Mineral 22:779–786
Gatta GD, Lotti P, Nestola F, Pasqual D (2012) On the high-pressure behavior of gobbinsite, the natural counterpart of the synthetic zeolite Na-P2. Microporous Mesoporous Mater 163:259–269
Gatta GD, Comboni D, Alvaro M, Lotti P, Càmara F, Domeneghetti MC (2014) Thermoelastic behavior and dehydration process of cancrinite. Phys Chem Miner 41:373–386
Gatta GD, Cappelletti P, de’ Gennaro B, Rotiroti N, Langella A (2015) New data on Cu-exchanged phillipsite: a multi-methodological study. Phys Chem Miner 42:723–733
Gottardi G, Galli E (1985) Natural zeolites. Minerals and rocks series. Springer, Berlin, p 409
Gualtieri AF, Caputo D, Colella C (1999a) Ion-exchange selectivity of phillipsite for Cs+ : a structural investigation using the Rietveld method. Microporous Mesoporous Mater 32:319–329
Gualtieri AF, Passaglia E, Galli E, Viani A (1999b) Rietveld structure refinement of Sr-exchanged phillipsites. Microporous Mesoporous Mater 31:33–43
Gualtieri AF, Passaglia E, Galli E (2000) Rietveld structure refinement of natural and Na-, K-, Ca-, and Ba-exchanged phillipsites. In: Colella C, Mumpton FA (eds) Natural zeolites for the third millennium. De Frede, Naples, pp 93–110
Langella A, Cappelletti P, de’ Gennaro R (2001) Zeolites in closed hydrologic systems. Rev Mineral Geochem 45:235–260
Lotti P, Gatta GD, Comboni D, Merlini M, Pastero L, Hanfland M (2016) AlPO4-5 zeolite at high pressure: crystal–fluid interaction and elastic behaviour. Microporous Mesoporous Mater 228:158–167
Mao HK, Xu J, Bell PM (1986) Calibration of the ruby pressure gauge to 800-kbar under quasi-hydrostatic conditions. J Geophys Res 91:4673–4676
Passaglia E, Sheppard RA (2001) The crystal chemistry of zeolites. Rev Mineral Geochem 45:69–116
Passaglia E, Galli E, Gualtieri AF (2000) Natural zeolites for the third millennium. De Frede, Naples, pp 259–267
Petříček V, Dušek M, Palatinus L (2014) Crystallographic computing system JANA2006: general features. Z Kristallogr 229:345–352
Rinaldi R, Pluth JL, Smith JV (1974) Zeolites of the phillipsite family. Refinement of the crystal structure of phillipsite and harmotome. Acta Crystallogr B 30:2426–2433
Rinaldi R, Smith JV, Jung G (1975) Chemistry and paragenesis of faujasite, phillipsite, and offretite from Sasbach, Kaiserstuhl, Germany. Neues Jahrb Miner Mon 10:433–443
Rothkirch A, Gatta GD, Meyer M, Merkel S, Merlini M, Liermann H-P (2013) Single-crystal diffraction at the Extreme Conditions beamline P02.2: procedure for collecting and analyzing high-pressure single-crystal data. J Synchrotron Radiat 20:711–720
Sani A, Cruciani G, Gualtieri AF (2002) Dehydration dynamics of Ba-phillipsite: an in situ synchrotron powder diffraction study. Phys Chem Miner 29:351–361
Steinfink H (1962) The crystal structure of the zeolite phillipsite. Acta Crystallogr 15:644–651
Stuckenschmidt E, Fuess H, Kvick A (1990) Investigation of the structure of harmotome by X-ray (293 K, 100 K) and neutron-diffraction (15 K). Eur J Mineral 2:861–874
Acknowledgments
The authors acknowledge the Italian Ministry of Education, MIUR-Project: “Futuro in Ricerca 2012—ImPACT—RBFR12CLQD.” DESY—PETRA III (Hamburg) is thanked for the allocation of beamtime. Y. Lee is thanked for the sample of phillipsite from Richmond, Victoria, Australia. Two anonymous reviewers are thanked for their constructive suggestions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Comboni, D., Gatta, G.D., Lotti, P. et al. On the P-induced behavior of the zeolite phillipsite: an in situ single-crystal synchrotron X-ray diffraction study. Phys Chem Minerals 44, 1–20 (2017). https://doi.org/10.1007/s00269-016-0832-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00269-016-0832-7