
Abstract
The elastic and structure behavior of natural thaumasite compressed in methanol–ethanol–H2O up to 5 GPa was studied by synchrotron powder diffraction with a diamond anvil cell. In the pressure range between 0.0001 and 4.5 GPa, the compression is regular and slightly anisotropic, with a more rigid ab-plane coinciding with the orientation of hydrogen bonds and S–O, C–O bonds in anion groups. The corresponding bulk moduli derived from the third-order Birch–Murnaghan EoS fit are K a = 43(2), K c = 35(2), K T = 39(2) GPa. Rietveld refinements reveal some general features of the structure evolution of thaumasite, which are consistent with the observed elastic anisotropy. The compression within the ab-plane proceeds mainly at the expense of shortening of hydrogen bonds and much lesser decrease of C–O and S–O bonds. In the range of 0.0001–3 GPa the Ca–O polyhedra contract more rapidly along the c-axis as compared to the ab-plane. At about 5 GPa, thaumasite undergoes a reversible transformation to an amorphous phase. The observed behavior differs drastically with that studied previously using helium as the pressure medium, which suggests the effect of He penetration increasing the structure stiffness. Without helium support, the thaumasite structure is preserved only up to 4.5 GPa.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Thaumasite, Ca3Si(OH)6(CO3)(SO4)·12H2O, is a low-temperature mineral formed during hydrothermal alteration of mafic igneous and metamorphic rocks (Knill and Young 1960; Gross 1977). A special attention is drawn to thaumasite, because it is also the main product of sulfate attack (TSA) in concretes, which proceeds under the influence of sulfate-rich groundwater and leads to disintegration of the cement matrix (Crammond 1985; Hobbs and Taylor 2000; Zhang et al. 2009). Another remarkable feature is that thaumasite is the only mineral containing silicon in six coordination with hydroxyl (OH)− that is stable at low P–T conditions. Besides this mineral, two other synthetic silicates, both the high-pressure phases, are reported to contain VISi–OH: phase D, MgSi(OH)2O4 (Yang et al. 1997), and hydroxide perovskite MgSi(OH)6 (Wunder et al. 2012). The importance of thaumasite called forth a number of studies of its structure and non-ambient behavior (Edge and Taylor 1971; Effenberger et al. 1983; Jacobsen et al. 2003; Martucci and Cruciani 2006; Gatta et al. 2012; Ardit et al. 2014).
The thermal behavior of thaumasite has been investigated by low-temperature single-crystal X-ray and neutron diffraction (Jacobsen et al. 2003; Gatta et al. 2012), and high-temperature synchrotron time-resolved powder diffraction (Martucci and Cruciani 2006). No phase transitions were found between 22.5 and 417 K; a severe dehydration was observed at T > 393 K, leading to an irreversible collapse of the structure at T ≥ 417 K. The high-pressure (HP) powder diffraction study of thaumasite compressed in helium has revealed two isosymmetric phase transitions at 1.5–3 and 7.4–15 GPa, the structure being preserved up to 19.5 GPa (Ardit et al. 2014).
Thaumasite is enriched in volatile components; it contains about 42 wt% H2O, 13 wt% SO3, and 7 wt% CO2. Hexagonal structure of thaumasite (space group P63) is based on cylindrical columns of alternating Ca(OH)4(H2O)4 and Si(OH)6 polyhedra running parallel to [001], among which the sulfate and carbonate groups are hosted. The [Ca3Si(OH)6(H2O)12]4+ columns are connected with CO3 2− and SO4 2− groups through hydrogen bonds with H2O molecules, the majority of these bonds being sub-parallel to the ab-plane. A single-crystal neutron diffraction study of Gatta et al. (2012) provides a detailed picture of the arrangement of hydrogen bonds in the thaumasite structure and emphasizes their important role in the non-ambient behavior; in particular, the strengthening of H bonds [due to shorter d(O···O)] at low temperature helps to stabilize the thaumasite structure. Moreover, the temperature-dependent variation of the H bond strength appears to determine a zero or negative thermal expansion of carbonate groups, silicate octahedra, and sulfate tetrahedra (Jacobsen et al. 2003; Gatta et al. 2012). These data, as well as an irreversible amorphization of thaumasite upon dehydration (Martucci and Cruciani 2006), clearly suggest that the stability of thaumasite is mainly governed by the crystal-lattice H2O and hydrogen bonding system.
The peculiarities of the H bonds arrangement are also important for the high-pressure elastic properties of thaumasite. According to the data of Ardit et al. (2014), at pressure up to 8 GPa, the axial bulk modulus K a, corresponding to the main direction of H bonds lying within the ab-plane, is higher than K c corresponding to iono-covalent bonds along the [Ca3Si(OH)6(H2O)12]4+ columns. At that, the initial phase th-I is characterized by only slightly anisotropic compressibility (K a0/K c0 = 1.28), whereas for the first HP phase th-II, existing within the range of 3–8 GPa, the K a3.53 value is almost doubled compared to K a0, and K a0/K c0 becomes equal to 1.73 (Ardit et al. 2014).
The most striking result provided by Ardit et al. (2014) concerns the HP phase th-III existing between 14 and 19.5 GPa; it is characterized by the inversion of the axial bulk moduli (K a15.02/K c15.02 = 0.33) due to an enormous increase of the K c15.02 value up to 267 GPa at pressure between 9 and 14 GPa. The authors propose to explain such behavior by the change in the relative strengths of the iono-covalent bonds along the Ca,Si-columns parallel to the c-axis vs. the H bonds linking the columns within the ab-plane. However, without structural data, it is difficult to conceive a real structural mechanism which could provide such an anomalous inversion of the elastic properties. In addition, the data of Ardit et al. (2014) on the HP stability limit of thaumasite compressed in He (19.5 GPa) contradict the recent spectroscopic observations (Goryainov 2016) showing that in methanol–ethanol–H2O (MEW) mixture, thaumasite is stable only up to 5 GPa and exhibits reversible amorphization. Noteworthily, ettringite Ca6[Al(OH)6]2(SO4)3·26H2O, forming a solid solution with thaumasite, also transforms into a reversible amorphous phase at a relatively low pressure of about 3 GPa (Clark et al. 2008; Speziale et al. 2008). Moreover, a distinct difference between the compressibility in silicon oil and methanol:ethanol mixture is reported for ettringite (Clark et al. 2008). It appears that the composition of pressure transmitting medium can seriously influence the HP behavior of these minerals.
To check the role of different pressure media in the HP behavior of thaumasite, we studied the compressibility of thaumasite in MEW by X-ray powder diffraction. In this paper, we present the results of this study and compare them with the data of Ardit et al. (2014) obtained with helium. We provide the compressibility data for the pressure range up to 4.5 GPa, as well as discuss some general features of the HP structure behavior of thaumasite.
Experimental
A sample of thaumasite used in this study is from West Paterson (New Jersey, USA) and presents 1–5-mm-sized transparent prismatic crystal fragments. Electron-probe X-ray microanalysis in the energy-dispersive mode (MIRA 3 LMU scanning electron microscope with INCA Energy 450 XMax 80 microanalysis system) was used to determine the chemical composition of the sample. Only the elements nominally expected in thaumasite (Ca, Si, S) occurred within the detection limit, with the average content (from ten points): CaO 24.8 wt%, SiO2 9.4 wt%, and SO3 12.9 wt%. The silicon and sulfur contents are in good agreement with the ideal formula of thaumasite Ca3Si(OH)6(CO3)(SO4)·12H2O, whereas the calcium content is about 2% less, which is most probably related with a partial dehydration and cation migration under the focused electron beam (Gatta and Boffa Ballaran 2004). The Raman spectrum of our sample (the same as was studied by Goryainov 2016) is identical with that studied by Gatta et al. (2012).
The powdered sample, mixed with MEW in proportion 16:3:1, was placed into a gasket hole of a diamond anvil cell (DAC). The DAC is based on a modified Mao-Bell design (Fursenko et al. 1984) and employs two diamonds with 1 mm diameter culets. The pressure values were measured before and after the diffraction experiment from the singlet fluorescence line of SrB4O7:Sm2+ chips (Datchi et al. 1997; Rashchenko et al. 2013) using PRL spectrometer (BETSA). Three different series of the high-pressure experiments were performed using MEW as the pressure medium, the corresponding sets of data are listed in Table 1. Angle-dispersive X-ray diffraction was performed at the fourth beamline of the VEPP-3 storage ring of Novosibirsk Synchrotron Centre SSTRC (Ancharov et al. 2001; Piminov et al. 2016), with monochromatic beam (λ = 0.3685 Å) and MAR345 imaging plate detector (pixel dimension 100 μm). The sample to detector distance was 395 mm, and the focused X-ray beam was 100 × 100 μm2 in size. The program FIT2D (Hammersley et al. 1996) was used for integrating the two-dimensional images up to 2Θmax = 25o.
Rietveld refinements of the lattice parameters and crystal structure were performed using the package GSAS (Larson and Von Dreele 2000) and the EXPGUI graphical interface (Toby 2001) in the 2Θ range of 2°–20o. No symmetry change was observed up to 4.5 GPa, and the P63 structure model was used in all the refinements. To characterize the elastic behavior of thaumasite, we used cubed values of the lattice parameters to fit them with the same equation as used for the volume variation [third-order Birch–Murnaghan equation of state (Angel et al. 2014)]. All the data points from three sets of DAC experiments (listed in Table 1) were used in the fitting procedure.
The diffraction patterns collected at ambient pressure (hereafter 0 GPa), as well as at 1.53, 3.05, and 4.16 GPa were used for the structure refinements. The Bragg peak profiles were refined by a pseudo-Voight function. The instrumental background was approximated by a Chebyshev polynomial with 30 coefficients. Soft restraints were imposed on Si–O distances in Si(OH)6-octahedra (1.75–1.78 Å), C–O distances in CO3-groups (1.28–1.29 Å), and S–O distances in SO4-tetrahedra (1.44–1.47 Å), and gradually released after the initial stages of refinement. The isotropic displacement factors for all atoms were set according to the structural data of Jacobsen et al. (2003) and not refined. The crystal structure of thaumasite at 298 K reported by Jacobsen et al. (2003) was used as a starting model for the refinement at 0 GPa. For the refinements at high pressure, the refined structure at the previous pressure point was used as a starting model. The final refined parameters were the coordinates for all the atoms and the unit cell parameters.
Results and discussion
Elastic behavior
Upon the compression up to 4.5 GPa, no change in the set of diffraction peaks of thaumasite is observed (Fig. 1), which indicates the preservation of the original space group P63 throughout the whole pressure range. The pressure dependences of the lattice parameters and volume of thaumasite are presented in Fig. 2 in comparison with the data of Ardit et al. (2014) and Table 1. Within the range of 0–4.5 GPa the compressibility in MEW is monotonic and slightly anisotropic, the c direction being more compressible as compared to the ab-plane. The absence of distinct compression anomalies suggests no appreciable structure transformations. Fitting of the experimental data for 0–4.5 GPa pressure range to a third-order Birch–Murnaghan equation of state (Angel et al. 2014) gives: K a(0) = 43(2) GPa (K′ = 5.7), K c(0) = 35(2) GPa (K′ = 4.0), K T(0) = 39(2) (K′ = 5.2), V 0 = 1094.1(3).

X-ray powder diffraction patterns of thaumasite at different pressures

Normalized axial (a) and volume (b) compression curves of thaumasite for MEW (full symbols) and He medium (thin line + empty symbols). Thick lines show the third-order Birch–Murnaghan fit of the a/a 0, c/c 0, and V/V 0 data points
Within the initial pressure range of 0–1.4 GPa, the obtained K a, K c, and K T values are close to those found by Ardit et al. (2014) (41, 32, and 36 GPa, respectively), showing a similarity of the compression regimes in MEW and He at these P conditions. The compression anisotropy observed in both media agrees with thermo-elastic properties of thaumasite, with the structure being more expandable along [001] than on (001) due to the prevalence of iono-covalent bonds along the c-axis and H bonds within the ab-plane (Jacobsen et al. 2003; Gatta et al. 2012; Ardit et al. 2014).
Starting from about 1.5–2 GPa, the slopes of axial compression curves become clearly different for both media, the structure stiffness along a direction growing drastically upon the compression in He (Fig. 2). The bending of the a/P curve at pressures between 2 and 3 GPa, reported by Ardit et al. (2014), is a characteristic of the compression regime influenced by the penetration of additional molecules into the structure from an external medium (see, for example, Shen et al. 2011; Likhacheva et al. 2013). This suggests the penetration of He, making the thaumasite structure more stiff and decreasing the compressibility along the a direction. Note that within the pressure range of 9–14 GPa, the structure stiffness along c increases drastically upon the compression in He (Ardit et al. 2014). These compression anomalies observed within the range of 2–3 and 9–14 GPa, which are interpreted as isosymmetric phase transitions by Ardit et al. (2014), can apparently be related with two stages of He penetration into the thaumasite structure.
Starting from about 4 GPa, a gradual weakening of the diffraction peaks is observed upon the compression in MEW, indicating some structure disordering (Fig. 1). Within the range of 4.5–5 GPa, thaumasite becomes X-ray amorphous. At that, several weak maxima seen in the pattern of amorphized phase at 5.2 GPa are distinctly shifted from the most intense peaks observed in the pattern of initial phase at 4.5 GPa, for example, the maxima at 4.7° and 8.3°. This indicates that the amorphous phase is characterized by a different long-range structure as compared to the initial P63 phase. Such behavior agrees with the spectroscopic observations of Goryainov (2016), showing a series of drastic shifts and broadening of the main modes in the Raman spectrum of thaumasite at pressures between 4.4 and 5.2 GPa, leading to the appearance of a highly disordered phase. Note that the observed amorphization is related with the structure behavior of thaumasite, rather than it is caused by the deviation from hydrostatic conditions, since it was repeatedly observed within the same narrow pressure range in different media [MEW and KBr (Goryainov (2016)].
It should also be noted that the reversibility of the transition to amorphous phase is not full, since the proportions of the unit cell (a/c ratio) and volume measured upon the pressure release differ notably from the initial metrics (Fig. 2; Table 1). Though the peak intensities in the diffraction pattern collected on decompression regain the original values, the structure refinement did not converge to appropriate agreement factors due to instability of atomic coordinates, which suggests a largely disordered structure. A deviation from the initial metrics is also observed after the decompression from 4.5 GPa (prior to the transition pressure), which suggests some irreversible changes to the structure. Such sensitivity to the compression is probably a consequence of the predominance of hydrogen bonds in the structure. It is interesting that He medium apparently favors the preservation of the native thaumasite structure, since the unit cell metrics measured on the reverse are restored accurately (Ardit et al. 2014).
Structure evolution
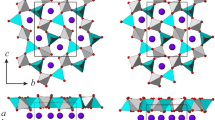
The X-ray structure refinements based on the data collected at 0, 1.53, 3.05, and 4.16 GPa confirm the structure model previously obtained (Jacobsen et al. 2003; Martucci and Cruciani 2006; Gatta et al. 2012) and show that the symmetry of thaumasite structure is preserved within the pressure range investigated. The refinement details are reported in Table 2, the atomic coordinates and bond distances are listed in Tables 3 (suppl.) and 4, respectively. An example of Rietveld plot (at 1.53 GPa) is presented in Fig. 3, and the fragment of the structure refined at 0 GPa is shown in Fig. 4.

Rietveld plot for thaumasite at 1.53 GPa. The observed and calculated profiles are indicated by (plus) and by a continuous line, respectively. The tick marks indicate the positions of allowed Bragg peaks in the respective symmetry. A difference curve is plotted at the bottom

The fragment of thaumasite structure refined at 0 GPa. The CO3 groups and SO4 tetrahedra are gray and dark-gray colored
In the initial structure at 0 GPa, the distances in the cation polyhedra and anion groups are in reasonable agreement with the previous single-crystal data (Jacobsen et al. 2003; Gatta et al. 2012). The mean C–O and S–O distances in anion groups are 1.290(10) and 1.472(12) Å, respectively, the S–O9 distances being relatively shorter than S–O6 (Table 4; Fig. 4). The distortion of SO4 tetrahedron is also evident from a larger angle ∠O6–S–O9 [110.7(6)o] as compared to ∠O6–S–O6 [108.2(6)o]. A slight aplanarity of CO3 group is seen from the deviation of ∠O5–C–O5 from 120o [114.9(8)o]. The mean Ca–(O3,O4) distances in calcium polyhedron are shorter compared to the mean Ca–(O1,O2) and Ca–(O7,O8) distances; the Si–O7 distances are relatively less than Si–O8 (Table 4; Fig. 4).
The evolution of interatomic distances with pressure is presented in Table 4 and partly in Fig. 5. The most noticeable feature is the preservation of the S–O and C–O distances in the anion groups at the initial level (within the accuracy limits) up to 1.5 GPa. The O···O distances corresponding to hydrogen bonds between SO4, CO3 groups and H2O molecules, on the contrary, experience an appreciable shortening within this pressure range. The contraction of Ca–O and Si–O polyhedra along the c-axis proceeds more or less monotonically throughout the whole pressure range, whereas the Ca–(O1,O2) distances corresponding to the ab-plane retain their initial value up to 3 GPa, and at P > 3 GPa they shorten more rapidly as compared to the Ca–(O3,O4) and Ca–(O7,O8) distances. On the whole, the anion polyhedra appear to be the most rigid units up to the highest pressure: the C–O bonds contract by 0.008%, and the S–O bonds contract by 0.004% at the expense of S–O9 bond, the S–O6 distance remaining at the initial level. Within the pressure range of 0–4.2 GPa, the Ca–O and Si–O distances shorten by 2 and 1.5%, respectively, whereas the O···O distances corresponding to hydrogen bonds experience the largest contraction by about 3%.

The pressure dependences of selected mean interatomic distances in thaumasite structure, based on the Rietveld refinements at 0, 1.53, 3.05, and 4.16 GPa (see Table 4). The plot of the mean \({\text{O}}_{{{\text{H}}_{ 2} {\text{O}}}}\)···\({\text{O}}_{{{\text{SO}}_{ 4} }}\) distance for hydrogen bonds between \({\text{SO}}_{4}^{2 + }\) anion and neighbor H2O molecules is shifted upright (as shown by arrow). The lines connecting the experimental points are visual guides
A relative incompressibility of CO3 and SO4 groups is in line with the structure properties of thaumasite revealed by single-crystal studies (Jacobsen et al. 2003; Gatta et al. 2012), as well as the structure behavior of other carbonate minerals (Martinez et al. 2004). In particular, during the contraction of thaumasite structure upon decreasing temperature, the SO4 tetrahedron even expands under the influence of the shortened H bonds (Gatta et al. 2012). On the other hand, linear high-frequency shifts of the C–O and S–O stretching modes in thaumasite, observed upon the pressure increase (Goryainov 2016), imply a regular contraction of the corresponding bonds. Therefore, the changes in the S–O, C–O, and O···O distances found within 0–1.5 GPa (Fig. 5; Table 4) can be regarded as showing a tendency towards the structure contraction at the expense of shortening of H bonds and much lesser decrease of C–O and S–O bonds. This tendency is most pronounced within the initial compression stage and is preserved throughout the whole pressure range.
The other main feature of the HP structure behavior of thaumasite is a more rapid contraction of Ca–O polyhedra along the c-axis [Ca–(O7,O8) distances] as compared to the ab-plane (Ca–(O1,O2) distances) in the P range of 0–3 GPa. It is probable that the Ca–(O1,O2) distances are preserved due to the shortened H bonds. On the whole, the structure models obtained at high pressure apparently present a rather realistic compression mechanism of thaumasite, consistent with the known elastic properties of anion and cation polyhedra composing its structure.
Conclusions
The compressibility of thaumasite in MEW within the P range 0–4.5 GPa is regular and slightly anisotropic (K a = 43(2), K c = 35(2), K T = 39(2) GPa); it agrees with thermo-elastic properties of thaumasite found previously. Within the initial P range of 0–1.5 GPa the compression regimes in MEW and He (Ardit et al. 2014) are similar, whereas at P > 1.5–2 GPa the divergence of the compression curves denotes the effect of He penetration, making the structure stiffer. Therefore, the MEW mixture appears to be more non-penetrating with respect to the thaumasite structure as compared to helium.
Rietveld refinements reveal some general features of the HP structure evolution of thaumasite, which are consistent with the observed elastic anisotropy. The contraction within the ab-plane proceeds mainly at the expense of shortening of H bonds and much lesser decrease of C–O and S–O bonds in anion groups. This tendency is most pronounced within the initial compression stage between 0 and 1.5 GPa. The Ca–O polyhedra contract more rapidly along the c-axis as compared to the ab-plane in the P range of 0–3 GPa.
In the absence of penetration effects, the initial structure of thaumasite exists only up to 4.5 GPa, similarly to the behavior of related mineral ettringite (Clark et al. 2008). The amorphization of thaumasite observed within the pressure range of 4.5–5.2 GPa involves a notable structural transformation and disordering, and presents a quite unusual example of transition between crystalline and amorphous phase, which is apparently a characteristic of this type of structures.
References
Ancharov AI, Manakov AYu, Mezentsev NA, Tolochko BP, Sheromov MA, Tsukanov VM (2001) New station at the 4th beamline of the VEPP-3 storage ring. Nucl Instrum Methods Phys Res A 470:80–83. doi:10.1016/S0168-9002(01)01029-4
Angel RJ, Gonzalez-Platas J, Alvaro M (2014) EosFit7c and a Fortran module (library) for equation of state calculations. Z Kristallogr 229(5):405–419. doi:10.1515/zkri-2013-1711
Ardit M, Cruciani G, Dondi M, Garbarino GL, Nestola F (2014) Phase transitions during compression of thaumasite, Ca3Si(OH)6(CO3)(SO4)·12H2O: a high-pressure synchrotron powder X-ray diffraction study. Miner Mag 8:1193–1208. doi:10.1180/minmag.2014.078.5.07
Clark SM, Colas B, Kunz M, Speziale S, Monteiro PJM (2008) Effect of pressure on the crystal structure of ettringite. Cement Concrete Res 38:19–26. doi:10.1016/j.cemconres.2007.08.029
Crammond N (1985) Thaumasite in failed cement mortars and renders from exposed brickwork. Cement Concrete Res 15:1039–1050
Datchi F, LeToullec R, Loubeyre P (1997) Improved calibration of the SrB4O7: Sm2+ optical pressure gauge: advantages at very high pressures and high temperatures. J Appl Phys 81:3333–3339. doi:10.1063/1.365025
Edge RA, Taylor FW (1971) Crystal structure of thaumasite, [Ca3Si(OH)6·12H2O](SO4)(CO3). Acta Cryst B27:594–601
Effenberger H, Kirfel A, Will G, Zobetz E (1983) A further refinement of the crystal structure of thaumasite, Ca3Si(OH)6CO3SO4·12H2O. Neu Jahrb Miner Monatsh 2:60–68
Fursenko BA, Litvin YA, Kropachev VD (1984) Apparatus with transparent anvils-windows for optical and X-ray studies at high pressures. Prib Tech Exp 5:174–178(in Russian)
Gatta GD, Boffa Ballaran T (2004) New insight into the crystal structure of orthorhombic edingtonite: evidence for a split Ba site. Miner Mag 68:167–175. doi:10.1180/0026461046810178
Gatta GD, McIntyre GJ, Swanson JG, Jacobsen SD (2012) Minerals in cement chemistry: a single-crystal neutron diffraction and Raman spectroscopic study of thaumasite, Ca3Si(OH)6(CO3)(SO4)·12H2O. Am Miner 97:1060–1069. doi:10.2138/am.2012.4058
Goryainov SV (2016) Raman study of thaumasite Ca3Si(OH)6(SO4)(CO3)·12H2O at high pressure. J Raman Spectr 47:984–992. doi:10.1002/jrs.4936
Gross S (1977) The mineralogy of the Hatrurim Formation, Israel. Geol Surv Israel Bull 70:1–80
Hammersley AP, Svensson SO, Hanfland M, Fitch AN, Hausermann D (1996) Two-dimensional detector software: from real detector to idealized image or two-theta scan. High Press Res 14:235–248
Hobbs DW, Taylor MG (2000) Nature of the thaumasite sulphate attack mechanism in field concrete. Cement Concrete Res 30:529–533
Jacobsen SD, Smyth JR, Swope RJ (2003) Thermal expansion of hydrated six-coordinate silicon in thaumasite, Ca3Si(OH)6(CO3)(SO4)·12H2O. Phys Chem Miner 30:321–329. doi:10.1007/s00269-003-0328-0
Knill DC, Young BR (1960) Thaumasite from Co., Down, Northern Ireland. Miner Mag 32:416–418
Larson AC, Von Dreele RB (2000) General structure analysis system (GSAS). Los Alamos National Laboratory Report, LAUR 86–748
Likhacheva AYu, Goryainov SV, Bul’bak TA (2013) An X-ray diffraction study of the pressure-induced hydration in cordierite at 4–5 GPa. Am Miner 98:181–186. doi:10.2138/am.2013.4199
Martinez I, Sanchez-Valle C, Daniel I, Reynard B (2004) High-pressure and high-temperature Raman spectroscopy of carbonate ions in aqueous solution. Chem Geol 207:47–58. doi:10.1016/j.chemgeo.2004.02.003
Martucci A, Cruciani G (2006) In situ time resolved synchrotron powder diffraction study of thaumasite. Phys Chem Miner 33:723–731. doi:10.1007/s00269-006-0124-8
Piminov PA, Baranov GN, Bogomyagkov AV, Berkaev DE, Borin VM, Dorokhov VL, Karnaev SE, Kiselev VA, Levichev EB, Meshkov OI, Mishnev SI, Nikitin SA, Nikolaev IB, Sinyatkin SV, Vobly PD, Zolotarev KV, Zhuravlev AN (2016) Synchrotron radiation research and application at VEPP-4. Phys Proc 84:19–26. doi:10.1016/j.phpro.2016.11.005
Rashchenko SV, Likhacheva AYu, Bekker TB (2013) Preparation of a macrocrystalline pressure calibrant SrB4O7: Sm2+ suitable for the HP-HT powder diffraction. High Press Res 33:720–724. doi:10.1080/08957959.2013.819098
Shen G, Mei Q, Prakapenka VB, Lazor P, Sinogeikin S, Meng Y, Park C (2011) Effect of helium on structure and compression behavior of SiO2 glass. PNAS 108(15):6004–6007. doi:10.1073/pnas.1102361108
Speziale S, Jiang F, Mao Z, Monteiro PJM, Wenk H-R, Duffy TS, Schilling FR (2008) Single crystal elastic constants of natural ettringite. Cement Concrete Res 38:885–889. doi:10.1016/j.cemconres.2008.02.004
Toby BH (2001) EXPGUI, a graphical user interface for GSAS. J Appl Crystallogr 34(2):210–213. doi:10.1107/S0021889801002242
Wunder B, Jahn S, Koch-Muller M, Speziale S (2012) The 3.65 A phase, MgSi(OH)6: structural insights from DFT-calculations and T-dependent IR spectroscopy. Am Miner 97:1043–1048. doi:10.2138/am.2012.4022
Yang H, Prewitt CT, Frost DJ (1997) Crystal structure of the dense hydrous magnesium silicate, phase D. Am Miner 82:651–654
Zhang FC, Ma BG, Yin G, Wu YY, Zhu YC (2009) Preparation and performance of sulfate resistance cement-based material. Key Eng Mater 400:195–201
Acknowledgements
The authors thank Prof. Yu.V. Seryotkin for the valuable discussion of the material, as well as we thank anonymous reviewers for their useful comments. This study is supported by the state assignment project (0330-2016-0004) and Russian Foundation of Basic Researches (Grant 15-55-45070). Diffraction experiments were carried out involving the equipment belonging to the Siberian Synchrotron and Terahertz Radiation Centre (SSTRC). The work was performed in part within the project 14.B25.31.0032 (the Ministry of Education and Science of Russian Federation).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Likhacheva, A.Y., Dementiev, S.N. & Goryainov, S.V. Compressibility and reversible amorphization of thaumasite Ca3Si(OH)6(CO3)(SO4)·12H2O pressurized in methanol–ethanol–H2O up to 5 GPa. Phys Chem Minerals 45, 219–226 (2018). https://doi.org/10.1007/s00269-017-0910-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00269-017-0910-5