
Abstract
The evolution of the structural environment of \(\hbox {Cr}{^{3+}}\) along the solid solution \(\hbox {ZnAl}_{2-x}\hbox {Cr}_{x}\hbox {O}_4\) has been investigated using a multi-analytical approach. X-ray diffraction confirms that the system follows Vegard’s law. Diffuse reflectance spectra show a decrease of the crystal field parameter with the Cr content, usually related to the increase of the Cr–O bond length in a point charge model. This interpretation is discussed and compared to the data obtained by first-principle calculations based on density functional theory. X-ray absorption near edge structure spectra at the Cr K-edge show a pronounced evolution in the pre-edge with the Cr content, characterised by the appearance of a third feature. Calculations enable to assign the origin of this feature to Cr neighbours. The colour change from pink to brownish pink and eventually green along the solid solution has also been quantified by calculating the L*, a*, b* and x, y coefficients in the system defined by the International Commission on Illumination.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Spinels are common phases found in igneous, metamorphic and detrital rocks from the Earths upper mantle to the crust (Lenaz and Princivalle 2005; Evans and Frost 1975; Barnes and Roeder 2001). Although recognized as accessory minerals, spinels are widely considered to be important petrogenetic indicators (Irvine 1965, 1967; Lenaz and Princivalle 2005; Barnes and Roeder 2001; Balan et al. 2006; Perinelli et al. 2014). Extensive solid-solutions of chromium-bearing spinels can occur, playing an important role during fractional crystallization of the melt. For instance, the presence of Cr is known to strongly increase the stability of the spinel phase \(\hbox {MgAl}_{2}\hbox {O}_4\) (Klemme 2004; Brigida et al. 2007).
Moreover, due to their mechanical, optical and magnetical properties, spinels are of considerable technological importance as catalysts, refractories and pigments (Fernández-Osorio et al. 2012; Prim et al. 2013). Among these properties, colour has been widely investigated because addition of various chromophores (mainly transition elements) leads to a great variety of colouration, making spinel structure the basis of most ceramic pigments (Martos et al. 2007; Prim et al. 2013).
Recently, several studies have focused on the spinel solid solution gahnite-zincochromite \(\hbox {ZnAl}_{2-x}\hbox {Cr}_{x}\hbox {O}_4\) because of its change in colouration from pink to green with the chromium content (Halenius et al. 2010; Prim et al. 2013; Bosi et al. 2011). In order to understand the evolution of the colour in the solid solution, knowledge of the local structural environment of the chromophore cation (chromium) is required.
The spinel structure is based on an almost-perfect cubic close-packed array of oxygen atoms forming tetrahedral (T) and octahedral (M) cavities, with \(Fd\overline{\textit{3}}m\) space group. The general formula can be expressed as AB\(_2\)X\(_4\) where A and B are cations occupying respectively tetrahedral and octahedral sites, and X can be O2−, S2− or Se2− (Biagioni and Pasero 2014). The cation distribution in the \(\hbox {ZnAl}_{2-x}\hbox {Cr}_{x}\hbox {O}_4\) system is fully ordered, corresponding to normal spinel, with Zn2+ ions occupying the T sites and \(\hbox {Al}{^{3^+}}\) and \(\hbox {Cr}{^{3^+}}\) distributed on the M sites (Bosi et al. 2011; O’Neill and Dollase 1994; Kashii et al. 1999). Modifications of TO and MO bond lengths to accommodate cations with different sizes result in variations in the u oxygen fractional coordinate and the a unit-cell parameter (Lavina et al. 2002).
In Cr-bearing spinels, colour variation occurs even though Cr is essentially present in trivalent state in octahedral environments. The colour changes are related to the Al3+ by \(\hbox {Cr}{^{3+}}\) substitution. The ionic radius of \(\hbox {Cr}{^{3+}}\) (0.615 Å) being larger than the one of Al3+ (0.535 Å), the substitution of an element by the other implies structural modification. Besides X-ray diffraction, different techniques are often used to probe this structural modification: UV-visible absorption spectroscopy and chemically selective spectroscopies such as extended X-ray absorption fine structure (EXAFS) or X-ray absorption near edge structure (XANES). UV-visible spectra are usually interpreted within ligand field theory (Halenius et al. 2010). The colour change can be related to the diminution of the crystal field parameter \(\varDelta _0\) along the solid solution. In a point charge model, this modification of \(\varDelta _0\) is linked to the increase of the Cr–O bond length, R\(_{Cr-O}\), according to the relation (Burns 1993):
where q is the effective charge of the ligands and \(\langle r{^4 \rangle}\) is the mean value of the fourth power of the radial distance between d electrons and the nucleus. The \(q\langle r{^4 \rangle}\) quantity is considered constant. However, this interpretation differs from data obtained by EXAFS or XANES (Galoisy 1996). It has been demonstrated that differently from the interpretation of optical absorption spectra, the Cr–O distance determined by EXAFS in spinel containing dilute concentration of Cr MgAl\(_2\hbox {O}_4\):Cr or ruby \(\hbox {Al}_2\hbox {O}_3\):Cr is close to the one in the corresponding end-member, magniesochromite MgCr\(_2\hbox {O}_4\) (Juhin et al. 2007, 2008a) or eskolaite Cr\(_2\hbox {O}_3\) (Gaudry et al. 2003, 2005), respectively.
In this work, we aim to explore the modification in the UV-visible spectra and XANES spectra with the Cr concentration in the solid solution \(\hbox {ZnAl}_{2-x}\hbox {Cr}_{x}\hbox {O}_4\). This will bring information on the local structure of \(\hbox {Cr}{^{3+}}\) and on the origin of the colour modification with the chromium content.
Materials and methods
Sample preparation
The two end-members of the solid solution, i.e. ZnAl\(_2\hbox {O}_4\) and ZnCr\(_2\hbox {O}_4\), were first synthesised. Reagent-grade chemicals ZnO, \(\gamma\)-\(\hbox {Al}_2\hbox {O}_3\) and Cr\(_2\hbox {O}_3\) were weighted in the right stoichiometric proportions, mixed, ground in an agate mortar with ethanol and calcined in a platinum crucible at 1400 °C during 20 hours. Then, using the same procedure, nine compounds in the solid solutions \(\hbox {ZnAl}_{2-x}\hbox {Cr}_{x}\hbox {O}_4\) with x from 0.2 to 1.8 were synthesised from the end-members.
To support our interpretation, we also present XANES data measured on synthetic samples (Malézieux and Piriou 1988) belonging to \(\hbox {MgAl}_{2-x}\hbox {Cr}_{x}\hbox {O}_4\) solid solution, for which the Cr diluted compound has already been investigated (Juhin et al. 2008a). Five compositions were selected: x = 0.05, 0.5, 1.0, 1.5 and 1.95.
Analytical methods
X-ray powder diffraction (XRD) data were collected using a PANalytical X’Pert PRO diffractometer with Cu K\(\alpha\) radiations (\(\lambda _{{{\mathrm {Cu,K}}}\alpha _1}=1.54056\,\) Å, \(\lambda _{{{\mathrm {Cu,K}}}\alpha _2}=1.54439\,\) Å). Lattice parameters of the different \(\hbox {ZnAl}_{2-x}\hbox {Cr}_{x}\hbox {O}_4\) compounds were determined using the Rietveld method (Rietveld 1969) as implemented in the FullProf Suite program (Rodríguez-Carvajal 1993).
Diffuse reflectance spectra were measured using a Perkin Elmer Lambda 1050 spectrometer with an integrating sphere. The samples were finely grounded and the powders were deposited on a metallic sample holder. The spectra were recorded between 4000 and 36000 cm−1. The white reference was a perfectly flat tablet of BaSO\(_4\). Reflectance data were converted into absorption spectra using the Kubelka-Munk remission function \(F(\rho _\infty )\) (Kubelka and Munk 1931):
where K is the absorption coefficient, S the scattering coefficient, and \(\rho _\infty\) is the reflectance factor for a layer with an infinite depth, in practice an opaque layer. The spectra were recorded in absorbance A, so we firstly calculated the reflectance factor \(\rho _\infty\) = 10−A. Diffuse reflectance spectra do not provide a direct value of the molar extinction coefficient, but they can be used to compare one sample with one another.
We use the parameters L*, a*, b* to quantify the colour in the system defined in 1976 by the International Commission on Illumination (abbreviated CIE). The parameters were calculated from the experimental spectra, using the equation given by Wyszecki and Stiles (2008). The coefficients x and y were also calculated. They correspond to the coordinates of the compound in the chromaticity diagram.
XANES data were collected at the Cr K-edge (5989 eV) in transmission mode on the solid solution \(\hbox {ZnAl}_{2-x}\hbox {Cr}_{x}\hbox {O}_4\) at the ID21 beamline of the ESRF (Grenoble, France). Powders were fixed on a thin film XRF tape and covered with ultralene film. The monochromator position was calibrated using a Cr metallic foil. The incident beam was monochromatized using a Si(220) monochromator. The beam size on the sample was 200 μm. XANES spectra were recorded between 5.96 and 6.21 keV with a step of 0.2 eV and an accumulation time of 1 s/step. A total of 10 scans was recorded for samples less concentrated in Cr, and 5 scans were enough for the 4 more concentrated samples. For each sample, the scans were summed and the resulting spectrum was normalized using the software PyMCA (Solé and Papillon 2004). XANES data on the solid solution \(\hbox {MgAl}_{2-x}\hbox {Cr}_{x}\hbox {O}_4\) were collected at the Cr K-edge at the ID26 beamline of the ESRF in Total Fluorescence Yield. The sample were grounded and packed to form pellets. The incident beam was monochromatized using a Si(111) monochromator. The spectra were corrected from self-absorption using the software FLUO Haskel (1999).
XANES first-principles calculations
Structural relaxations and self-consistent field (SCF) calculations were done with the PWscf code (Giannozzi et al. 2009) and the XANES calculations with the XSpectra code (Gougoussis et al. 2009; Taillefumier et al. 2002), both included in the Quantum Espresso plane-waves based density functional theory (DFT) suite of codes. We used spin-polarised generalised gradient approximation (GGA) functionals with the Perdew-Burke-Ernzerhof (PBE) parameterisation (Perdew et al. 1996), and norm conserving pseudopotentials in the Kleinman-Bylander form. The parameters used for the pseudopotential generation are given by Juhin et al. (2007). The cut-off energies were determined from convergence tests and set to 90 Ry.
In order to understand the experimental XANES spectra obtained on the powdered samples, we have focused on two compounds and calculated the corresponding XANES spectra. The first one is a compound diluted in Cr, with the composition \(\hbox {ZnAl}_{1.875}\hbox {Cr}_{0.125}\hbox {O}_4\) which we use to interpret the origin of the spectral features visible in the spectrum of the most diluted sample, \(\hbox {ZnAl}_{1.8}\hbox {Cr}_{0.2}\hbox {O}_4\). The second compound is the end-member of the solid solution serie, ZnCr\(_{2}\hbox {O}_4\).
We started from structures of \(\hbox {ZnAl}_{2}\hbox {O}_4\) and ZnCr\(_{2}\hbox {O}_4\), which are obtained by a total energy minimization calculation. The lattice parameters were fixed to their experimental values, as determined by Rietveld refinement on the synthetic sample, while the atomic positions were allowed to vary to minimize the total energy and the interatomic forces. The cubic cells considered contained 56 atoms: 8 Zn, 32 O and respectively 16 Al or 16 Cr. For ZnCr\(_{2}\hbox {O}_4\), the collinear antiferromagnetic structure of Chan et al. (2007) was used. For the dilute compound, a Cr impurity was added in the structure \(\hbox {ZnAl}_{2}\hbox {O}_4\) by replacing one Al by one Cr and the structure was relaxed imposing S\(_z\) = 3/2. The atomic positions in the cell were allowed to vary in order to minimize the total energy and the inter-atomic forces. We used a single k-point sampling in the Brillouin zone. The Cr site, after relaxation, still exhibits a \(\textit{D}_{3d}\) symmetry.
From both \(\hbox {ZnAl}_{1.875}\hbox {Cr}_{0.125}\hbox {O}_4\) and ZnCr\(_{2}\hbox {O}_4\) relaxed structures, the Cr K-edge absorption cross sections were calculated in the electric dipole (E1) and electric quadrupole (E2) approximation. First, we calculated self-consistently the charge density of the system, with a 1s core hole on the substitutional Cr atom in \(\hbox {ZnAl}_{1.875}\hbox {Cr}_{0.125}\hbox {O}_4\) and a 1s core hole on one Cr among the 16 in ZnCr\(_{2}\hbox {O}_4\). Then, the all-electron wave functions were reconstructed within the projector augmented wave framework. The E1 and E2 absorption cross sections were computed as continued fractions using a Lanczos basis constructed recursively. We used one k-point for the self-consistent spin-polarized charge-density calculation including the core-hole, and a Monkhorst-Pack grid of \(2\times 2\times 2\) k-points in the Brillouin zone for the absorption cross-section calculation. For the convolution of the continued fraction, we used an energy dependent broadening parameter, which takes the main photoelectron damping modes into account. The broadening parameter used in this study is that of Bordage et al. (2012). The E2 cross-section is calculated as a combination of three well-chosen contributions according to Juhin et al. (2008a). Finally, in order to interpret the pre-edge features, partial density of states (DOS) are calculated on the systems having the core hole, using Löwdin projections, \(2\times 2\times 2\) Monkhorst-Pack k-points grid and a Gaussian broadening of 0.00368 eV.
Results and discussion
X-ray diffraction
The XRD patterns of the different samples indicate that all samples are single phase corresponding to spinel. The results of the Rietveld refinement are presented in Table 1 and in supplementary information. Figure 1 shows the structural refinement performed on the sample \(\hbox {ZnAl}_{1.4}\hbox {Cr}_{0.6}\hbox {O}_4\).

Rietveld refinement for sample \(\hbox {ZnAl}_{1.4}\hbox {Cr}_{0.6}\hbox {O}_4\). The red dots are the observed intensities, the black line is the calculated intensity. Blue vertical tick marks indicate Bragg positions in space group \(\textit{Fd}\overline{\textit{3}}m\) and the orange line plots the difference between observed and calculated intensities. R\(_{Bragg}\) = 2.17 % and \(\chi {^2}\) = 3.414 for this sample
We have determined the lattice parameter a as a function of the chromium content x in the \(\hbox {ZnAl}_{2-x}\hbox {Cr}_{x}\hbox {O}_4\) series (Fig. 2). There is a gradual increase in the lattice parameter when substituting Al by Cr. A linear regression points out that this solid solution obeys Vegard’s law with the following equation: \(a = 0.1215x + 8.086\) with a the cell parameter in Å, and x the molar fraction in chromium (Fig. 2). This is in good agreement with previous studies (Prim et al. 2013; Bosi et al. 2011). It was suggested that this variation is correlated with M-O bond length, though Zn-O bond lengths show also significant variations (Bosi et al. 2011; Ardit et al. 2012). This results from the difference in ionic radii sizes when Al\({^{3+}}\) substitutes for \(\hbox {Cr}{^{3+}}\). The \(\langle {{\mathrm {M-O}}} \rangle\) distance corresponds to an average value of Al–O and Cr–O distances (Table 1). This graph can be seen as a standard line, which can be used to determine the concentration of chromium in a gahnite phase doped with chromium (\(\hbox {ZnAl}_{2}\hbox {O}_4\):Cr) thanks to the determination of the lattice parameter.

In green, variation of the cubic lattice parameters a of \(\hbox {ZnAl}_{2-x}\hbox {Cr}_{x}\hbox {O}_4\) calculated with Rietveld refinement as a function of the chromium concentration x; in black the linear regression; for each composition the pictures of the powders is on the right, except for x = 0 (\(\hbox {ZnAl}_{2}\hbox {O}_4\)) which is the colourless gahnite
Optical spectroscopy
The colour of the samples varies with the chromium content (Fig. 2). The introduction of a few ppm of chromium in the colourless gahnite (ZnAl\(_2\hbox {O}_4\)) leads to a pink colour. This colour intensifies with Cr concentration x. From x = 1.0 a brownish hue appears which intensifies until x = 1.6. The powder at x = 1.8 appears brownish green and the zincochromite (ZnCr\(_2\hbox {O}_4\)) is green. This colour variation from pink to green with the Cr content was also observed on the solid solutions \(\hbox {MgAl}_{2-x}\hbox {Cr}_{x}\hbox {O}_4\) (Juhin et al. 2007), \(\hbox {Al}_{2-x}\hbox {Cr}_{x}\hbox {O}_3\) (Gaudry et al. 2003) and \(\hbox {MgAl}_{2-x}\hbox {Cr}_{x}\)Si\(_3\hbox {O}_{12}\) (Juhin et al. 2008b).
Figure 3 shows the remission function F(\(\rho _\infty\)) calculated from the diffuse reflectance spectra of the ten powdered samples \(\hbox {ZnAl}_{2-x}\hbox {Cr}_{x}\hbox {O}_4\). The overall intensity of the two most concentrated compounds (x = 1.8 and x = 2) is lower than the one of the other samples. This is due to the dependence of the intensity with the grain size distribution of the powder when reflectance data are acquired. The spectra show two main intense absorption bands, typical of trivalent chromium, \(\hbox {Cr}{^{3+}}\), in an octahedral symmetry. Their positions vary with the chromium content, from 18600 to 17500 cm−1 (\(\nu _1\)) and from 25200 to 24000 cm\({^{-1}}\) (\(\nu _2\)) approximately. The shift in energy is linked with the diminution of the crystal field parameter along the solid solution. The two main peaks can be assigned to the spin-allowed electronic d-d transitions in a regular octahedron between the \({^{4}}A_{2g}\) level and the \({^{4}}T_{2g}\) (\(\nu _1\)) and \({^{4}}T_{1g}\) (\(\nu _2\)) levels, respectively. The spectra also show two weak peaks at approximately 15000 and 29500 cm−1. They can be assigned to the spin-forbidden transitions from the energy levels \({^{4}}A_{2g}\) to \({^{2}}T_{1g}\) and \({^{2}}A_{1g}\). The two main absorption bands \(\nu _1\) and \(\nu _2\) present a shoulder at lower wave numbers, especially visible on \(\nu _1\). This is due to the distortion of the CrO\(_6\) octahedron. Indeed, because of the displacements of O atoms along the [111] direction, the symmetry around \(\hbox {Cr}{^{3+}}\) in spinel is lowered from O\(_h\) to D\(_{3d}\) creating a splitting of the energy levels \({^{4}}T_{1g}\) and \({^{4}}T_{2g}\) into two levels (Wood 1968; Ikeda et al. 1997). In agreement with the literature, the two intense broad bands of the spectra can then be assigned to the spin allowed \({^{4}}A_{2g}\) \(\rightarrow\) \({^{4}}E_{g}\) and \({^{4}}A_{2g}\) \(\rightarrow\) \({^{4}}A_{1g}\) transitions for \(\nu _1\), and to \({^{4}}A_{2g}\) \(\rightarrow\) \({^{4}}E_{g}\) and \({^{4}}A_{2g}\) \(\rightarrow\) \({^{4}}A_{2g}\) transitions for \(\nu _2\).

Diffuse reflectance spectra of the samples \(\hbox {ZnAl}_{2-x}\hbox {Cr}_{x}\hbox {O}_4\) with x between 0.2 and 2
To go further with these experimental data, we calculated the colour parameters L*, a*, b* and x, y (Table 2). We also placed x and y in a chromaticity diagram, which can be used to interpret the colour in terms of Cr concentration of more complexed materials composed of the solid solution \(\hbox {ZnAl}_{2-x}\hbox {Cr}_{x}\hbox {O}_4\) (Fig. 4). This representation was chosen because the relationship between the value of the parameter and the resulting colour is more straightforward than in the a* = f(b*) graph. The positions of the samples are consistent with the colour of the powders, varying from pink to green.

Chromaticity diagram (y as a function of x) with the 10 samples in the solid solution
XANES
Evolution of the XANES spectra with the Cr concentration
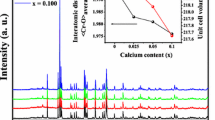
XANES spectra at the Cr K-edge of the solid solution \(\hbox {ZnAl}_{2-x}\hbox {Cr}_{x}\hbox {O}_4\) are shown in Fig. 5. All spectra present three main features, labelled a, b and c. While the position in energy of a and b is not much modified, c is shifted by 2.2 eV towards lower energies between the two extreme compositions \(\hbox {ZnAl}_{1.8}\hbox {Cr}_{0.2}\hbox {O}_4\) and ZnCr\(_{2}\hbox {O}_4\). The pre-edge region exhibits two or three peaks (\(\alpha\), \(\beta\), \(\gamma\)) according to the Cr concentration. Feature \(\alpha\) is at the same energy for all samples, whereas the position of the feature \(\beta\) is shifted to lower energies and feature \(\gamma\) appears as the chromium content increases (Fig. 6).

XANES spectra at the Cr K-edge for the different compositions \(\hbox {ZnAl}_{2-x}\hbox {Cr}_{x}\hbox {O}_4\), x between 0.2 and 2

Pre-edge region at the Cr K-edge for the different compositions \(\hbox {ZnAl}_{2-x}\hbox {Cr}_{x}\hbox {O}_4\), x between 0.2 and 2
Other solid solutions present interesting continuous colour changes such as the system \(\hbox {MgAl}_{2-x}\hbox {Cr}_x\hbox {O}_4\), very similar to the one studied here, which also varies from pink to green with the chromium concentration. For this solid solution, we observed the same evolution of the XANES spectra and pre-edge: a shift towards lower energies of the features c and \(\beta\), and the appearance of a third structure \(\gamma\) with the increasing chromium content (see Fig. 7). This \(\gamma\) feature was also observed in chromite FeCr\(_{2}\hbox {O}_4\) (Farges 2009). These spectra are then characteristic of \(\hbox {Cr}{^{3+}}\) in octahedral symmetry in spinels. Their evolution with the Cr content is particularly pronounced on the pre-edge region which will be interpreted in the next part.

XANES spectra at the Cr K-edge for different compositions in the solid solution \(\hbox {MgAl}_{2-x}\hbox {Cr}_{x}\hbox {O}_4\). The inset is a focus on the pre-edge region
Comparison with calculations
The calculated isotropic XANES spectra of the dilute compound \(\hbox {ZnAl}_{1.875}\hbox {Cr}_{0.125}\hbox {O}_4\) and the end-member compound ZnCr\(_{2}\hbox {O}_4\) are compared with the experimental spectra of the samples \(\hbox {ZnAl}_{1.8}\hbox {Cr}_{0.2}\hbox {O}_4\) and ZnCr\(_{2}\hbox {O}_4\) in Fig. 8. The agreement between the experimental and theoretical spectra is good since all the main features are well reproduced. The shift towards lower energies of the peak c on the calculated spectra of ZnCr\(_{2}\hbox {O}_4\) mimics the behavior of the experimental spectra. The inset in Fig. 8 is a focus on the pre-edge region. The peaks are quite well reproduced in the calculation for both the dilute and the concentrated samples. However, two peaks are distinguished on the feature \(\gamma\), while it is not as distinct on the experimental spectra. Furthermore, the pre-edge in the calculated spectrum is found at too high energies (i.e., too close to the main edge) because the calculation is limited in the modelling of electron-hole effects. This effect is well known and understood (Cabaret et al. 2010).

Comparison between the experimental (up) and the calculated (bottom) XANES spectra at the Cr K-edge of the dilute compound (pink line) and the end-member compound ZnCr\(_{2}\hbox {O}_4\) (green line). The calculated XANES spectra are aligned with the experimental ones with respect to the energy position of the main XANES peak. The inset is a focus on the pre-edge region
The upper panel in Fig. 9 shows the pre-edge area of the calculated spectrum of the dilute compound \(\hbox {ZnAl}_{1.875}\hbox {Cr}_{0.125}\hbox {O}_4\), and its decomposition into electric quadrupole transitions E2 and electric dipole transitions E1. As expected, it indicates that the pre-edge is only due to 1s \(\rightarrow\) 3d E2 transitions. Indeed, E1 transitions do not occur because the absorbing Cr site is centrosymmetric. E1 transitions only contribute to the background which is the tail of the absorption edge. In order to assign the E2 transitions, the density of states (DOS) projected on the absorbing Cr atom was plotted on the lower panel. It represents the empty states accessible by the photoelectron. In the monoelectronic picture in octahedron symmetry, the initial electronic configuration of \(\hbox {Cr}{^{3+}}\) 3d states is \((t{^{\uparrow }}_{2g}){^{3}}(e{_g}){^{0}}\). In the excited state, the photoelectron can probe the empty t\(_{2g}\) orbitals for spin down (minority spin), or the empty orbitals e\({_g}\) for spin up (majority spin) and spin down as detailed by Cabaret et al. (2010). These transitions are found on the calculated spectra: the first peak \(\alpha\) at 1.55 eV (E-E\(_F\) where E\(_F\) is the Fermi energy) is the result of the two transitions to the t\({^{\downarrow }}_{2g}\) and e\({^{\uparrow }}{_{g}}\) levels while the second feature \(\beta\) at 2.82 eV is due to the transition to the e\({^{\downarrow }}{_g}\) level. This interpretation is similar to the one given for the spinel \(\hbox {MgAl}_{2}\hbox {O}_4\):Cr (Juhin et al. 2008a). When the Cr concentration increases along the solid solution \(\hbox {ZnAl}_{2-x}\hbox {Cr}_{x}\hbox {O}_4\), the difference in energy position between peak \(\alpha\) and peak \(\beta\) decreases (Fig. 6) which is consistent with the diminution of the crystal field. Indeed, a rough estimate of crystal field splitting is given by the difference in energy between these two peaks. This will be discussed later in part 3.3.3.
The upper panel in Fig. 10 shows the pre-edge region of the calculated spectrum for the concentrated compound, ZnCr\(_{2}\hbox {O}_4\), and its decomposition into E1 and E2 transitions. Contrary to the dilute compound, E1 transitions are observed at 2.4 and 3.6 eV. These E1 transitions cannot be local, p–d mixing being forbidden because the absorbing atom site is centrosymmetric (D\(_{3d}\)). The observed E1 contributions in the pre-edge are then only due to non-local electric dipole transitions occuring when the empty p states of the absorbing atom are orbitally mixed with the empty 3d states of the nearest metal neighbours through the empty p states of the ligands (Cabaret et al. 2010). The lower panels of Fig. 10 show empty DOS projected on (i) the 4p absorbing Cr, (ii) the p neighbouring O and (iii) the 3d neighbouring Cr, all at the energies of feature \(\gamma\). The \(\gamma\) feature observed on the experimental pre-edge is then due to electric dipole transitions to the p states of the absorbing Cr, orbitally mixed with the 3d orbitals of the neighbouring Cr. Two peaks are contained in this feature because of the crystal field splitting of the neighbouring Cr 3d orbitals. Feature \(\gamma\) is absent from the experimental spectra of the dilute compound \(\hbox {ZnAl}_{1.8}\hbox {Cr}_{0.2}\hbox {O}_4\): it confirms the absence of Cr “clusters” in this sample.

Analysis of the pre-edge transition of the dilute compound. Upper panel calculated pre-edge spectra (brown) and its decomposition into electric quadrupole transitions E2 (red) and electric dipole transitions E1 (black). Lower panel projected density of states 3d of the Cr absorber

Analysis of the pre-edge transition of the concentrated compound. Upper panel calculated pre-edge spectra (brown) and its decomposition into electric quadrupole transitions E2 (red) and electric dipole transitions E1 (black). Lower panels projected density of states 3d (red) and p (black) projected on the Cr absorber, p (green) on O first neighbours and 3d (orange) on Cr next-nearest neighbours
Link with the UV-visible spectroscopy
Comparison of crystal field parameters \(\varDelta _0\) determined by optical absorption or XAS. In O\(_h\) symmetry, the \(\varDelta _0\) crystal field parameter is defined as the energy difference between an e\(_{g}\) orbital occupied by one electron and a t\(_{2g}\) orbital occupied by one electron (König and Kremer 1977). Values of \(\varDelta _0\) can be estimated from both optical and X-ray absorption spectroscopies, using respective approximations that are described in detail by Gaudry et al. (2005). From UV-visible data, the Tanabe-Sugano diagram in O\(_h\) symmetry equals the energy of the first absorption band \(\nu _1\) directly to \(\varDelta _0\). For the dilute and the concentrated compounds we find \(\varDelta _0\) (opt. abs.) equals to 2.31 and 2.15 eV respectively. Another estimate of \(\varDelta _0\) can be obtained from the pre-edge in XAS spectra. Indeed, as discussed in 3.3.2, the pre-edge region shows two peaks \(\alpha\) and \(\beta\), which can be attributed, respectively, to t\(_{2g}\) empty states for minority spin + e\({_g}\) empty states for majority spin, and to e\({_g}\) empty states for minority spin. We define \(\varDelta _0\) (XAS) as the energy separation between the \(\alpha\) and \(\beta\) peaks. \(\varDelta _0\) (XAS) can thus be understood as an estimate of the t\(_{2g}\)–e\(_{g}\) splitting in the presence of a 1s core-hole. \(\varDelta _0\) (XAS) of the dilute and the concentrated compound is determined from the experimental spectra (2.2 and 1.8 eV respectively) and from the calculated ones (1.25 and 1.09 eV respectively). All values are summarized in Table 3. The values of \(\varDelta _0\) (XAS) determined from the experiments and the calculations are significantly different due to the limitation of the calculation in the modelling of electron-hole interaction and interelectronic repulsions. Moreover the values of \(\varDelta _0\) (opt. abs.) and \(\varDelta _0\) (XAS) are also quite different. Ligand Field Multiplet calculations of the pre-edge have been performed in MgAl\(_2\hbox {O}_{4}{:}\hbox {Cr}{^{3+}}\) using different values of crystal field parameter and different reduction factors of the intra-atomic electronic repulsions (Juhin et al. 2008a; Juhin 2008). They indicate that the energy splitting between \(\alpha\) and \(\beta\) peaks in the pre-edge is not only due to the crystal field splitting but also to the multielectronic 3d-3d Coulomb repulsions. This effect is highly multielectronic and complicates the interpretation given by the DFT single particle picture. Hence, although their respective values are found relatively close (Table 3), \(\varDelta _0\) (XAS) is not directly related to \(\varDelta _0\) (opt. abs.). Nevertheless one can still notice that \(\varDelta _0\) (XAS) is decreased for a higher Cr content, a trend that is similar to the one measured for \(\varDelta _0\) (opt. abs.).
Determination of the relaxation parameter and link with \(\varDelta _0\). The Cr–O distance obtained by DFT calculations in the dilute compound \(\hbox {ZnAl}_{2}\hbox {O}_{4}{:}\hbox {Cr}{^{3+}}\) (1.98 Å) is close to the Cr–O distance obtained by Rietveld refinement in the concentrated compound ZnCr\(_{2}\hbox {O}_4\) (1.992 Å) (Table 4). The Al–O distance being 1.909 Å in \(\hbox {ZnAl}_{2}\hbox {O}_4\) (determined by Rietveld refinement), we calculated a relaxation parameter of \(\zeta\) = 0.85 as defined by Martins and Zunger (1984). From optical data, Halenius et al. (2010) found \(\zeta\) = 0.60. Both optical absorption and DFT calculations show a relaxation of the atomic positions when Al substitutes by Cr, but not in the same extent. The relaxation parameter is strongly related to the host structure (Ardit et al. 2014). The value we found from DFT calculation is consistent with other relaxation parameters found in the literature on systems belonging to the spinel group (Halenius et al. 2010; Juhin et al. 2007).
According to optical absorption data, the crystal field parameter decreases by 7.4 % between the dilute compound and the concentrated one. In the point charge model, the diminution of the crystal field parameter \(\varDelta _0\) is linked with the increase of the Cr–O bond length, R\(_{Cr-O}\), through the equation (1) detailed in the introduction. According to equation (1) the expected variation on the Cr–O distance is then 1.5 %, or 0.03 Å between the dilute compound and the concentrated one. However, according to DFT calculations, the variation of the Cr–O distance is slightly lower, which may indicate that the difference in crystal field splitting cannot be fully explained by the variation of the Cr–O distance. The environment beyond the first shell of O neighbours around the Cr absorber and the modification of the Cr–O chemical bond have to be taken into account to understand the modification on the crystal field parameter (García-Lastra et al. 2005, 2006). The electrostatic potential generated by other ions of the crystal contributes also to the separation between e\(_g\) and t\(_{2g}\) orbitals and therefore plays a key role for explaining the strong differences of \(\varDelta _0\) between ZnAl\(_2\hbox {O}_{4}{:}\hbox {Cr}{^{3+}}\) and ZnCr\(_2\hbox {O}_4\). It may also arise from changes in the orbital mixing between the Cr d states and the neighbouring O p states, as it is reflected in the value of the B Racah parameter measured from optical absorption (Table 3) using the formula \(B=((2E_{\nu _1}-E_{\nu _2})/3)(E_{\nu _2}-E_{\nu _1})/(9E_{\nu _1}-5E_{\nu _2})\) (\(E_{\nu _i}\) being the energy position of the band \(\nu _i\)) (Marfunin et al. 1979). The role of Cr neighbours has been emphasized in this study and the Cr–Cr pairs might have an impact on the colour along the solid solution \(\hbox {ZnAl}_{2-x}\hbox {Cr}_{x}\hbox {O}_4\).
Conclusion
The \(\hbox {Al}{^{3+}}\) by \(\hbox {Cr}{^{3+}}\) substitution and the Cr structural environment have been explored in the solid solution \(\hbox {ZnAl}_{2-x}\hbox {Cr}_{x}\hbox {O}_4\) using X-ray diffraction, UV-visible spectra, and X-ray absorption spectroscopy at Cr K-edge. The system follows Vegard’s law: a linear relationship is obtained between the cell parameter a and the chromium content x expressed by \(\textit{a = 0.1215x + 8.086}\) (\(\textit{a}\) in Å). UV-visible spectra indicate the presence of \(\hbox {Cr}{^{3+}}\) in octahedral sites and allow the calculation of chromaticity parameters. They show a continuous evolution of the colour with the Cr content which reproduces the visual aspect of the powders. The local environment of Cr has been characterized using XANES spectroscopy. Calculations based on the DFT reproduce the variation in the edge and the pre-edge as a function of the Cr content. They allow the assignment of a pre-edge feature to Cr–Cr pairs, visible in the spectra with high Cr content. These results indicate that the local Cr environment (coordination, Cr–O distance) is little modified by the Al/Cr substitution, while the second shell is increasingly composed of Cr neighbours with increasing Cr content. To conclude, this study provides detailed structural information on the solid solution \(\hbox {ZnAl}_{2-x}\hbox {Cr}_{x}\hbox {O}_4\) to understand the differences in colours, which can be useful to more complex heterogeneous materials such as pigments, enamels, and glasses.
References
Ardit M, Cruciani G, Dondi M (2012) Structural relaxation in tetrahedrally coordinated Co\({^{2+}}\) along the gahnite-Co-aluminate spinel solid solution. Am Mineral 97(8–9):1394–1401
Ardit M, Dondi M, Cruciani G (2014) On the structural relaxation around \(\text{ Cr }{^{3+}}\) along binary solid solutions. Eur J Mineral 26(3):359–370
Balan E, De Villiers JPR, Griet Eeckhout S, Glatzel P, Toplis MJ, Fritsch E, Allard T, Galoisy L, Calas G (2006) The oxidation state of vanadium in titanomagnetite from layered basic intrusions. Am Mineral 91(5–6):953–956
Barnes SJ, Roeder P (2001) The range of spinel compositions in terrestrial mafic and ultramafic rocks. J Petrol 42(12):2279–2302
Biagioni C, Pasero M (2014) The systematics of the spinel-type minerals: an overview. Am Mineral 99(7):1254–1264
Bordage A, Rossano S, Horn AH, Fuchs Y (2012) Site partitioning of \(\text{ Cr }{^{3+}}\) in the trichroic alexandrite BeAl\(_2\text{ O }_4\):\(\text{ Cr }{^{3+}}\) crystal: contribution from X-ray absorption spectroscopy. J Phys Condens Matter 24(22):225, 401
Bosi F, Andreozzi GB, Halenius U, Skogby H (2011) Zn-O tetrahedral bond length variations in normal spinel oxides. Am Mineral 96(4):594–598
Brigida C, Poli S, Valle M (2007) High-temperature phase relations and topological constraints in the quaternary system MgO-\(\text{ Al }_2\text{ O }{_3}\)-SiO\({_2}\)-Cr\(_2\text{ O }{_3}\): an experimental study. Am Mineral 92(5–6):735–747
Burns RG (1993) Mineralogical applications of crystal field theory, vol 5. Cambridge University Press, Cambridge
Cabaret D, Bordage A, Juhin A, Arfaoui M, Gaudry E (2010) First-principles calculations of X-ray absorption spectra at the K-edge of 3d transition metals: an electronic structure analysis of the pre-edge. PCCP 12(21):5619–5633
Chan K, Sau J, Zhang P, Cohen M (2007) Ab Initio calculations of phonon splitting in antiferromagnetic ZnCr\(_2\text{ O }_4\). Phys Rev B 75(5):054,304
Evans BW, Frost B (1975) Chrome-spinel in progressive metamorphisma preliminary analysis. Geochimica et Cosmochimica Acta 39(6–7):959–972
Farges F (2009) Chromium speciation in oxide-type compounds: application to minerals, gems, aqueous solutions and silicate glasses. Phys Chem Minerals 36(8):463–481
Fernández-Osorio A, Pineda-Villanueva E, Chávez-Fernández J (2012) Synthesis of nanosized (Zn1xCox)Al2O4 spinels: new pink ceramic pigments. Mater Res Bull 47(2):445–452
Galoisy L (1996) Local versus average structure around cations in minerals from spectroscopic and diffraction measurements. Phys Chem Minerals 23:217
García-Lastra J, Barriuso M, Aramburu J, Moreno M (2005) Origin of the different color of ruby and emerald. Phys Rev B 72(11):113,104
García-Lastra J, Aramburu J, Barriuso M, Moreno M (2006) Optical properties of \(\text{ Cr }{^{3+}}\)-doped oxides: different behavior of two centers in alexandrite. Phys Rev B 74(11):115,118
Gaudry E, Kiratisin A, Sainctavit P, Brouder C, Mauri F, Ramos A, Rogalev A, Goulon J (2003) Structural and electronic relaxations around substitutional \(\text{ Cr }{^{3+}}\) and Fe\({^{3+}}\) ions in corundum. Phys Rev B 67(9):094,108
Gaudry E, Sainctavit P, Juillot F, Bondioli F, Ohresser P, Letard I (2005) From the green color of eskolaite to the red color of ruby: an X-ray absorption spectroscopy study. Phys Chem Minerals 32(10):710–720
Giannozzi P, Baroni S, Bonini N, Calandra M, Car R, Cavazzoni C, Ceresoli D, Chiarotti GL, Cococcioni M, Dabo I, Dal Corso A, de Gironcoli S, Fabris S, Fratesi G, Gebauer R, Gerstmann U, Gougoussis C, Kokalj A, Lazzeri M, Martin-Samos L, Marzari N, Mauri F, Mazzarello R, Paolini S, Pasquarello A, Paulatto L, Sbraccia C, Scandolo S, Sclauzero G, Seitsonen AP, Smogunov A, Umari P, Wentzcovitch RM (2009) QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J Phys Condens Matter 21(39):395,502
Gougoussis C, Calandra M, Seitsonen A, Mauri F (2009) First-principles calculations of X-ray absorption in a scheme based on ultrasoft pseudopotentials: from \(\alpha\)-quartz to high-Tc compounds. Phys Rev B 80(7):075,102
Halenius U, Andreozzi GB, Skogby H (2010) Structural relaxation around \(\text{ Cr }{^{3+}}\) and the red-green color change in the spinel (sensu stricto)-magnesiochromite (MgAl\(_2\text{ O }_4\)-MgCr\(_2\text{ O }_4\)) and gahnite-zincochromite (ZnAl\(_2\text{ O }_4\)-ZnCr\(_2\text{ O }_4\)) solid-solution series. Am Mineral 95(4):456–462
Haskel D (1999) http://www.aps.anl.gov/~haskel/fluo.html
Ikeda K, Nakamura Y, Masumoto K, Shima H (1997) Optical spectra of synthetic spinels in the system MgAl\(_2\text{ O }_4\)-MgCr\(_2\text{ O }_4\). J Am Ceram Soc 80(10):2672–2676
Irvine TN (1965) Chromian spinel as a petrogenetic indicator: part 1. Theory. Can J Earth Sci 2(6):648–672
Irvine TN (1967) Chromian spinel as a petrogenetic indicator: part 2. Petrologic applications. Can J Earth Sci 4(1):71–103
Juhin A (2008) Propriétés électroniques et structurales du chrome en impureté dans les cristaux. Approche expérimentale et théorique. Sciences des matériaux. Paris, Paris 6. PhD thesis, thèse de doctorat
Juhin A, Calas G, Cabaret D, Galoisy L, Hazemann JL (2007) Structural relaxation around substitutional \(\text{ Cr }{^{3+}}\) in MgAl\(_2\text{ O }_4\). Phys Rev B 76(5):054,105
Juhin A, Brouder C, Arrio MA, Cabaret D, Sainctavit P, Balan E, Bordage A, Seitsonen A, Calas G, Eeckhout S, Glatzel P (2008a) X-ray linear dichroism in cubic compounds: the case of \(\text{ Cr }{^{3+}}\) in MgAl\(_2\text{ O }_4\). Phys Rev B 78(19):195,103
Juhin A, Calas G, Cabaret D, Galoisy L, Hazemann JL (2008b) Structural relaxation around substitutional \(\text{ Cr }{^{3+}}\) in pyrope garnet. Am Mineral 93(5–6):800–805
Kashii N, Maekawa H, Hinatsu Y (1999) Dynamics of the Cation Mixing of MgAl\(_2\text{ O }_4\) and ZnAl\(_2\text{ O }_4\) Spinel. J Am Ceram Soc 82(7):1844–1848
Klemme S (2004) The influence of Cr on the garnetspinel transition in the Earth’s mantle: experiments in the system MgOCr\(_2\text{ O }_3\)SiO\(_2\) and thermodynamic modelling. Lithos 77(1–4):639–646
König E, Kremer S (1977) Ligand field energy diagrams. Plenum Publishing Corporation, New York
Kubelka P, Munk F (1931) Ein Beitrag zur Optik der Farbanstriche. Z Tech Phys 12(11a):593–601
Lavina B, Salviulo G, Giusta AD (2002) Cation distribution and structure modelling of spinel solid solutions. Phys Chem Minerals 29(1):10–18
Lenaz D, Princivalle F (2005) The crystal chemistry of detrital chromian spinel from the southeastern alps and outer dinarides: the discrimination of supplies from areas of similar tectonic setting? Can Mineral 43(4):1305–1314
Malézieux JM, Piriou B (1988) Relation entre la composition chimique et le comportement vibrationnel de spinelles de synthèse et de chromites naturelles en microspectrométrie Raman. Bulletin de minéralogie 111(6):649–669
Marfunin AS, Egorova NG, Mishchenko AG (1979) Physics of minerals and inorganic materials: an introduction. Springer, Berlin
Martins J, Zunger A (1984) Bond lengths around isovalent impurities and in semiconductor solid solutions. Phys Rev B 30(10):6217–6220
Martos M, Martínez M, Cordoncillo E, Escribano P (2007) Towards more ecological ceramic pigments: study of the influence of glass composition on the colour stability of a pink chromium-doped ceramic pigment. J Eur Ceram Soc 27(16):4561–4567
O’Neill HSC, Dollase WA (1994) Crystal structures and cation distributions in simple spinels from powder XRD structural refinements: MgCr\(_2\text{ O }_4\), ZnCr\(_2\text{ O }_4\), Fe\(_3\text{ O }_4\) and the temperature dependence of the cation distribution in ZnAl\(_2\text{ O }_4\). Phys Chem Minerals 20(8):541–555
Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77(18):3865–3868
Perinelli C, Bosi F, Andreozzi GB, Conte AM, Armienti P (2014) Geothermometric study of Cr-spinels of peridotite mantle xenoliths from northern Victoria Land (Antarctica). Am Mineral 99(4):839–846
Prim S, García A, Galindo R, Cerro S, Llusar M, Folgueras M, Monrós G (2013) Pink ceramic pigments based on chromium doped M(\(\text{ Al }_{2-x}\text{ Cr }_x\))O\(_4\), M = Mg, Zn, normal spinel. Ceram Int 39(6):6981–6989
Rietveld HM (1969) A profile refinement method for nuclear and magnetic structures. J Appl Crystallography 2(2):65–71
Rodríguez-Carvajal J (1993) Recent advances in magnetic structure determination by neutron powder diffraction. Phys B Condens Matter 192(1–2):55–69
Solé VA, Papillon E (2004) PyMCA: X-Ray Spectra Visualization and Analysis in Python. In: NOBUGS 2004 conference, Paul Scherrer Institute, Villigen PSI, Switzerland
Taillefumier M, Cabaret D, Flank AM, Mauri F (2002) X-ray absorption near-edge structure calculations with the pseudopotentials: application to the K edge in diamond and \(\alpha\)-quartz. Phys Rev B 66(19):195,107
Wood DL (1968) Optical spectrum of \(\text{ Cr }{^{3+}}\) ions in spinels. J Chem Phys 48(11):5255
Wyszecki G, Stiles WS (2008) Color science: concepts and methods, quantitative data and formulae. Wiley, Chichester
Acknowledgments
This work was supported by the Réseau Francilien sur les oxydes fonctionnels (DIM Oxymore) and the Région Ile-de-France. We acknowledge the European Synchrotron Radiation Facility for provision of synchrotron radiation facilities at beamline ID21, through the proposal HG43. The calculations were partly performed using HPC resources from GENCI-IDRIS (100172-2015). L.V. acknowledges M. Chassé for his help with the chromaticity parameters and fruitful discussions. E.R. gratefully acknowledges the support of the Postdoctoral Fellowship of the Hungarian Academy of Sciences, the European Research Council (ERC Starting Grant No. 259709), and the French-Hungarian Balaton-TéT bilateral research program (Project No. TET_11_FR-XTHEOEXP) as well.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Verger, L., Dargaud, O., Rousse, G. et al. Spectroscopic properties of \(\hbox {Cr}^{3+}\) in the spinel solid solution \(\hbox {ZnAl}_{2-x}\hbox {Cr}_{x}\hbox {O}_4\) . Phys Chem Minerals 43, 33–42 (2016). https://doi.org/10.1007/s00269-015-0771-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00269-015-0771-8