
Abstract
Pediatric glioblastoma is relatively rare compared with its adult counterpart but is associated with a similarly grim prognosis. Available data indicate that pediatric glioblastomas are molecularly distinct from adult tumors, and relatively little is known about the pediatric glioblastoma tumor microenvironment (TME). Cancer immunotherapy has emerged as a new pillar of cancer treatment and is revolutionizing the care of patients with many advanced solid tumors, including melanoma, non-small cell lung cancer, head and neck cancer, and renal cell carcinoma. Unfortunately, attempts to treat adult glioblastoma with current immunotherapies have had limited success to date. Nevertheless, the immune milieu in pediatric glioblastoma is distinct from that found in adult tumors, and evidence suggests that pediatric tumors are less immunosuppressive. As a result, immunotherapies should be specifically evaluated in the pediatric context. The purpose of this review is to explore known and emerging mechanisms of immune evasion in pediatric glioblastoma and highlight potential opportunities for implementing immunotherapy in the treatment of these devastating pediatric brain tumors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Central nervous system (CNS) tumors are the leading cause of cancer-related death in children, having surpassed leukemia in recent years [1]. Pediatric high-grade gliomas (pHGG), in particular, are aggressive primary brain tumors. Historically, the World Health Organization (WHO) used histological features to classify pediatric HGGs as anaplastic astrocytomas (AA; WHO Grade III) or glioblastomas (GBM; WHO Grade IV). More recently, molecularly defined subsets of pediatric HGGs, including the H3 K27M mutant diffuse midline glioma (WHO Grade IV), were also incorporated into the WHO classification system [2].
While GBM is a common intrinsic brain tumor in adults, pediatric glioblastoma (p-GBM) accounts for only 15% of pediatric brain tumors [3] and exhibits a number of unique features (Table S1). P-GBM is usually reported in the second decade of life (with the highest incidence occurring in patients that are 15–19 years of age), though some cases have been reported in neonates and children under the age of 5 years, in whom better outcomes have been observed [4]. To date, ionizing radiation is the only environmental factor known to have a significant association with p-GBM [5]. Li-Fraumeni syndrome, constitutional mismatch repair deficiency syndrome (CMMRD), neurofibromatosis type I, and Turcot syndrome have also been linked to p-GBM [6].
The current treatment for p-GBM includes maximal safe resection followed by radiation therapy (in children > 3 years of age), with variable chemotherapy regimens currently being explored [3]. However, despite emerging knowledge that these tumors have a distinct molecular profile compared to their adult counterparts [7], reports have shown that most patients with p-GBM have similarly poor survival outcomes [8]. New, effective treatment strategies are needed. Cancer immunotherapy has emerged as a new focus in cancer treatment and is revolutionizing the care of patients with many advanced solid tumors. However, the rational design of immunotherapy regimens for the treatment of p-GBM will require a deeper understanding of intratumoral heterogeneity [9] and multi-faceted mechanisms of immune evasion [10]. The aim of this review is to explore the known and potential mechanisms of immune evasion and discuss therapeutic opportunities that may be unique to p-GBM (Table 1).
Immune evasion in pediatric versus adult GBM
Effective treatments for p-GBM are currently limited by an incomplete understanding of immune evasion by tumor cells, as well as the interaction between the tumor microenvironment and the host immune system. Though the immune system can specifically identify and eliminate tumor cells based on their expression of tumor-specific antigens, a process known as immunosurveillance [11], immunologic recognition of GBM antigens appears to be poor from the start. These tumor cells escape the immune response and continue to proliferate [12] resulting in ‘tumor immunoediting’, whereby tumor cells become less immunogenic, further facilitating their continued evasion from the immune system. An immunosuppressive tumor microenvironment subsequently develops and contributes to tumor progression.
Mechanisms of innate immune evasion
Innate immunity refers to patterned defense mechanisms that are rapidly activated upon the appearance of an antigen. In the CNS, innate immunity is mediated by microglia, macrophages, natural killer (NK) cells, and granulocytes [10]. GBM evades these components of the innate immune system through a variety of unique mechanisms.
Microglia and macrophage-mediated immune evasion
Microglia migrate to the brain during embryogenesis and serve as the brain’s resident phagocytes. Adult GBM is highly infiltrated by microglia, which along with monocyte-derived macrophages differentiate into tumor associated macrophages (TAMs) when exposed to the hypoxic microenvironment and represent up to 30% of the tumor mass [13,14,15]. TAMs are generally thought to promote tumor progression through the release of cytokines that impact cell proliferation, survival, motility, and immunosuppression. Accordingly, high TAM density has been correlated with a poor prognosis in adults [13]. On the other hand, although a small subset of p-GBMs demonstrate increased expression of microglia/macrophage genes and high levels of TAMs [16, 17], the enrichment of macrophage-related genes has not yet been correlated with outcome in patients with p-GBM.
TAMs adopt different phenotypes based on the microenvironment [18]. Historically, two dominant phenotypes were described: M1 and M2 macrophages. Activated M1 macrophages produce high levels of pro-inflammatory cytokines (IL-1β, TNF-α, and IL-12), up-regulate expression of cell surface molecules involved in antigen presentation (MHC II, CD80 and CD86), and stimulate a Th1 response, which promotes a tumoricidal effect. Conversely, M2 macrophages produce immunosuppressive cytokines (IL-10, IL-6, IL-23, and TGF-β), promote tissue remodeling, and downregulate MHC II, CD80 and CD86, thereby facilitating tumor progression [19]. Later stages of disease progression are associated with a transition from an M1 to an M2 phenotype, whereas therapeutic conversion back to an M1 phenotype is an area of active exploration [19]. More recent studies, however, have suggested that the polarization of glioma TAMs into M1 and M2 phenotypes is an oversimplification. Rather, TAMs appear to express a continuum of phenotypes, with the ability to co-express M1 and M2 markers [20, 21]. Further studies are needed to better characterize the population of TAMs present in p-GBM.
NK cell-mediated immune evasion
NK cells are cytotoxic effectors of the innate immune system, playing an important role in the elimination of malignant cells. NK cell activation can be induced through interactions between surface receptors, such as the natural killer group 2d (NKG2D), and their ligands [22]. Extracellular lactate dehydrogenase 5 (LDH5) produced by GBM cells induces the expression of NKG2D ligands including MHC class I related gene B (MICB) and the UL-16 binding protein (ULBP-1) on host monocytes [23]. Ligand binding results in increased NK cell anti-tumor activity through the release of perforin and granzyme, which are required for effective tumor cell lysis [22]. However, a recent study showed that pediatric HGG did not exhibit increased NKG2D ligand expression relative to normal adjacent brain tissue, nor significant myeloid or NK cell infiltration compared to adult tumors–possibly due to poor trafficking of immune cells to the tumor site, and thus a lack of immune surveillance [22]. Interestingly, an examination of pediatric glioma samples indicated that transcriptional expression of NKG2D ligands only partially predicted protein abundance, suggesting intrinsic molecular mechanisms that reduce NKG2D ligand expression by the tumor, and therefore prevent activation of NK cells [24].
Toll-like receptor-mediated immune evasion
Toll-like receptors (TLRs) are membrane spanning receptors expressed on cells of the innate immune system, which recognize pathogen-associated molecular patterns. There is increasing evidence that TLRs might play a role in tumor progression and the maintenance of cancer stem cells (CSCs) [25]. For example, increased expression of TLR2 by TAMs has been shown to promote glioma invasion and growth by upregulating the expression of matrix metalloproteinase-9 (MMP9), which plays a role in extracellular matrix degradation and facilitates the migration of tumor cells through the extracellular space [26]. In addition, TLR2 activation triggers the downstream MAPK Erk1/2 signaling pathway and ultimately causes the downregulation of MHC class II molecules by TAMs. Impaired MHC class II expression limits the activation and maintenance of helper T cells, thereby diminishing T-cell–dependent anti-tumor immunity [26].
TLR4 expression, on the other hand, plays a role in tumor cell differentiation and promotes a non-CSC phenotype. GBM CSCs do not express TLR4, whereas differentiated cells do; one explanation is that without TLR4, the innate immune system remains suppressed, thereby providing CSCs a survival advantage in the hostile tumor-immune microenvironment [25]. Upregulation of selected TLRs in tumors might therefore represent a potential therapy to sensitize GBM tumor cells to immune responses. However, the expression of TLRs in p-GBM has not been well characterized.
Mechanisms of adaptive immune evasion
MHC-mediated immune evasion
The adaptive immune system involves antigen-specific processes that are associated with a lag period but result in immunologic memory. The adaptive immune system is mediated by T cells and B cells. CNS antigen presentation is thought to occur at so-called CNS ‘immune gateways’ in deep cervical lymph nodes [27] and at the blood–brain barrier (BBB), where MHC molecules are expressed by antigen presenting cells (APCs) [28, 29]. Human MHC molecules are encoded by human leukocyte antigen (HLA) genes [30]. MHC I molecules are expressed by all nucleated cells and display both “self” and “non-self” antigens to cytotoxic T cells, while MHC II molecules are only found on APCs, present pathogenic antigens, and activate helper T cells.
In the context of a tumor, activation of the immune system requires that tumor antigens be recognized as foreign. Therefore, two mechanisms for immune evasion involve: 1) the loss of expression of MHC I molecules by tumor cells, and 2) the downregulation of the MHC I antigen presentation pathway [31, 32]. Either mechanism allows the tumor cells to “hide” from cytotoxic T lymphocytes (CTLs).
Interestingly, p-GBM cells highly express the classical MHC class I peptides, suggesting that pediatric glioma cells might require a third mechanism for evading the immune system [33]. Recent studies have shown that p-GBM expresses HLA-G and HLA-E–non-classical MCH molecules that promote inhibitory pathways [34, 35]. HLA-G has been implicated in immune escape by adult glioma cells and protects targeted cells from NK cell cytolysis as well as antigen-specific lysis through interactions with CTL surface molecules [36]. Similarly, HLA-E binds to CD94/NKG2A, an inhibitory receptor expressed on both NK cells and CTLs [10, 37], and upregulation of HLA-E may enhance tumor cell resistance to NK cell-mediated toxicity.
Treg-mediated immune evasion
An alternative mechanism of immune invasion involves regulatory T cells (Tregs), which are a subset of T cells that suppress the immune system, thereby maintaining homeostasis and promoting tolerance of self-antigens. Tregs come from four potential sources in the tumor milieu: 1) trafficking from the thymus, bone marrow, lymph nodes, or blood, 2) differentiation through modification of APCs, 3) expansion from dysfunctional dendritic cell stimulation, and 4) conversion of T cells by TGF-β [38]. Tregs shift the tumor cytokine milieu toward immunosuppression [32], and upregulated levels of Tregs have been reported in adult GBM [39]. Of note, indoleamine 2,3 dioxygenase 1 (IDO1) and tryptophan dioxygenase (TDO) contribute to Treg recruitment and have been reported in both pediatric HGGs and adult GBM [40, 41]. Therapeutic inhibition of IDO1 and/or TDO therefore represents a promising strategy for the treatment of p-GBM.
T cell dysfunction-mediated immune evasion
T cell dysfunction has been shown to play a role in tumor progression and may occur through T cell exhaustion, sequestration, anergy, and/or deletion. T cell exhaustion occurs in the setting of prolonged antigen stimulation and significantly limits the subsequent immune response [42]. Exhausted T cells demonstrate a loss of effector function, sustained upregulation of inhibitory molecules, and an altered metabolic profile. The Programmed Death Ligand-1 (PD-L1)/Programmed Cell Death-1 (PD-1) pathway serves as a central regulator of T-cell exhaustion. PD-1 engagement with its ligand PD-L1 downregulates cytokine production and causes decreased proliferation and inactivation of T cells. Glioma cells express PD-L1 on their cell surface and upregulate PD-L1 expression on monocytes through an IL-10-dependent mechanism [42], contributing to T cell inhibition. Inhibition of the PD-L1/PD-1 pathway is therefore being explored as a therapeutic strategy for adult patients. However, the recent CheckMate 143 phase III trial did not demonstrate a survival benefit for adult patients with recurrent GBM [43]. Furthermore, PD-L1 expression has been found to be relatively low in p-GBM, suggesting that inhibition of this pathway may be even less likely to be successful in pediatric patients [44, 45]. Further studies are needed to understand the alternate pathways that mediate T cell infiltration in p-GBM.
T cell depletion is accompanied by tumor-imposed loss of sphingosine-1-phosphate receptor 1 (S1PR1or S1P1) [46], one of five G protein–coupled receptors that binds the lipid second messenger, sphingosine-1-phosphate (S1P) [46]. S1P1 is located on the cell surface of lymphocytes and is required for trafficking out of the lymph node or thymus. The concentration of S1P is higher in blood and lymph and low in most other tissues, therefore establishing a gradient that directs T-cell egress from lymphoid organs into the circulation [46, 47]. S1P-S1P1 binding promotes the rapid internalization and degradation of the receptor and mediates lymphocyte trafficking from lymphoid organs to the circulation [47]. GBM cells downregulate the expression of these receptors, thereby resulting in sequestration of these naïve T lymphocytes in the bone marrow [46].
Alternatively, T cell anergy may occur, reflecting a state of unresponsiveness of the immune system. There are two types of anergy: clonal anergy from defective co-stimulation resulting in RAS/MAPK dysfunction, and adaptive anergy from continuously low levels of antigen exposure and deficient Zap70 kinase activity [48]. T cell anergy is mediated in part by cytotoxic T-lymphocyte associated protein-4 (CTLA-4), a protein found on CTLs, Tregs and T helper cells. When T cells recognize an antigen without strong co-stimulation, the T cell receptors may lose their ability to deliver activating signals. Alternatively, the T cells may engage inhibitory receptors (e.g. CTLA-4) that block activation either by competitively preventing CD28 binding to B7 co-stimulators or by generating inhibitory signals that attenuate activation via the T cell receptor and CD28. While adult brain tumors have shown increased CTLA-4 expression [49], CTLA-4 expression has not been well characterized in pediatric patients.
Deletional tolerance may also prevent an anti-tumor response at the central or peripheral level. Deletion refers to the elimination of T cells and B cells through apoptosis. Deletional tolerance has been observed in patients with GBM, and T-cell apoptosis is induced via a FasL-mediated deletion of invading lymphocytes [46]. An increased T-cell propensity for apoptosis results from a cooperative interaction between CD70 and gangliosides [32]. CD70, in turn, mediates T-cell apoptosis through interactions with CD27, a member of the TNF receptor family. However, the role of deletional tolerance in p-GBM immune evasion remains unclear.
Contribution of the pediatric GBM immune microenvironment
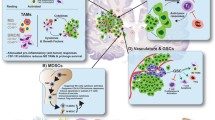
The above mechanisms of immune evasion rely on the interface between the tumor microenvironment and the host immune system (Fig. 1). Among children, glioma cells create an immunosuppressive environment through several parallel pathways involving the release of soluble factors and hypoxia induction. For example, TGF-β suppresses T cell growth and IL-2 production, activates Tregs [50], and downregulates NKGD2 [38]. Microglia secrete TGF-β in response to brain tumors, resulting in local and systemic brain tumor-mediated immunosuppression [51]. Macrophage migration inhibitory factor (MIF), another cytokine, is primarily upregulated in necrotic areas and adjacent to blood vessels, particularly in the presence of a TP53 mutation. MIF acts as an immune checkpoint regulator, enhancing autophagy in GBM and contributing to the escape of dendritic cell surveillance [52].

Immune altering mechanisms in pediatric glioblastoma. Created with BioRender.com CD80, cluster of differentiation 80; CD86, cluster of differentiation 86; CD94, cluster of differentiation 94; CSC, cancer stem cell; CTL, cytotoxic T lymphocyte; DC, dendritic cells; HIF-1, hypoxic inducible factor 1; HLA, human leucocyte antigen; IDO1, indolamine 2,3 dioxygenase 1; IL-6, interleukin 6; IL-10, interleukin 10; LDH5, lactate dehydrogenase 5; MICB, MHC class I related gene B; MIF, macrophage migration inhibitory factor; miR, microRNA; MMP9, matrix metalloproteinase-9; PD-1, programmed cell death protein 1; PDL-1, programmed death ligand 1; p-GBM; pediatric glioblastoma; PlGF, placental growth factor; S1P1, sphingosine 1 phosphate; S1P1R, sphingosine 1 phosphate receptor; STAT-3, signal transducer activator of transcription-3; TCR, T cell receptor; TDO, tryptophan dioxygenase; TLR, toll-like receptor; Tregs, regulatory T cells
The tumor milieu skews the inflammatory response away from a cell-mediated Th1 response towards a humoral Th2 response [10]. Elevated levels of IL-10, a Th2 cytokine, have been associated with enhanced tumor growth, inhibition of IFN-γ and tumor necrosis factor-alpha (TNF-α) production by the immune system, downregulation of MHC class II expression by monocytes, and induction of T cell anergy [53]. A recent study involving p-GBM, however, showed that although IL-10 was not expressed by these tumors, other immunosuppressive cytokines were detected in the tumor microenvironment [24].
The tumor microenvironment is also often characterized by hypoxia due to morphologically irregular blood vessels, irregular blood flow, and rapid oxygen consumption by proliferating cells [54]. Hypoxia promotes angiogenesis, and molecular markers of hypoxia such as hypoxic inducible factor 1 (HIF-1) and VEGF expression levels are upregulated in adult GBM, though not typically in p-GBM [55]. Hypoxia also activates the signal transducer and activator of transcription-3 (STAT3), which triggers HIF-1 synthesis [32, 50] and induces Treg activation. Elevated STAT3 levels promote Treg proliferation, as well as the expression of FOXP3, TGF-β and IL-10, all of which inhibit CTL differentiation and dendritic cell maturation [10].
Overall, these findings suggest that the p-GBM microenvironment might be less immunosuppressive than that of adult GBM. However, a better understanding of the p-GBM microenvironment and its interaction with the immune system is necessary in order to develop effective immunotherapies for this patient population.
Immunotherapeutic strategies for pediatric GBM
Cancer immunotherapy has demonstrated efficacy in a number of advanced solid tumors, including melanoma, non-small cell lung cancer, head and neck cancer, and renal cell carcinoma. Unfortunately, immunotherapy has so far had limited success among adult patients with GBM. However, due to the unique characteristics of p-GBM, there is a growing interest in exploring immunotherapeutic strategies for the pediatric population, including immune checkpoint inhibitors, cancer vaccines, oncolytic viral therapy, and adoptive cellular immunotherapy (Table 2).
Immune checkpoint inhibitors
Immune checkpoint blockade functions by interfering with inhibitory pathways that attenuate the activity of CTLs and can be used either in isolation to bolster the native immune response or in tandem with other therapies that would otherwise be inhibited by the immune system [56]. PD-1 inhibitors have emerged as one option. PD-L1 tumor positivity, a high tumor mutation burden (TMB), and high T cell density are associated with a tumoral response to PD-1 blockade [57]. In 2017, the FDA approved pembrolizumab (KEYTRUDA®), a humanized monoclonal antibody PD-1 inhibitor, for adult and pediatric patients with unresectable or metastatic TMB-high solid tumors, as well as microsatellite instability-high or mismatch repair (MMR) deficient solid tumors [58, 59]. A pediatric phase I trial studying pembrolizumab for pediatric patients with recurrent, progressive, or refractory gliomas or medulloblastomas is in the recruitment phase (NCT02359565). Unfortunately, a recent phase III clinical trial (CheckMate 143) found that nivolumab, an alternate PD-1 inhibitor, did not result in improved survival relative to bevacizumab in adult patients with recurrent GBM [43]. Another phase III trial (CheckMate 548: NCT02667587) investigating temozolomide plus radiation therapy with nivolumab vs. placebo is currently ongoing for adults with newly diagnosed MGMT-methylated GBM, but pediatric patients have not yet been studied.
Macrophage checkpoint blockade represents another option. Humanized monoclonal antibodies have been developed that target the macrophage immune checkpoint CD47, which has been shown to be particularly enriched in p-GBM. CD47 is overexpressed on the cell surface of tumor cells and inhibits the phagocytosis of tumor cells by macrophages upon binding of its ligand [60]. While the signaling cascade downstream of CD47 is not well understood, inhibitors targeting CD47 have been developed and are currently under investigation in adults with hematologic malignancies [60]. Pre-clinical investigations have demonstrated that by inhibiting CD47, macrophages can be “unlocked” to phagocytize tumor cells in several pediatric brain tumor models [58].
Immune checkpoint inhibition is less effective in tumors with a low TMB. Although GBM is typically associated with a low mutational load [61], p-GBM that arises in the context of CMMRD is characterized by a particularly high TMB [62]. Due to the high frequency of genomic mutations that are present in patients with CMMRD, there may be an increased likelihood of generating immunogenic tumor neoantigens by the host immune system, and CMMRD + tumors may therefore be more likely to benefit from PD-1 blockade [63, 64]. In particular, this therapy could be beneficial for a subset of pediatric patients showing evidence of microsatellite instability [65].
Cancer vaccines and oncolytic viral therapy
Cancer cell vaccines are designed to elicit an endogenous immune response using synthetic peptides, fragments of DNA/RNA, or tumor cell lysate as immunogens to boost and/or elicit long-term immune responses [66]. Although peptide vaccines have shown promise in pre-clinical and clinical studies, they are usually restricted to a specific HLA haplotype, therefore limiting the number of patients who can be treated. Alternatively, autologous dendritic cell (DC) and DNA/RNA vaccines involve loading patient derived DCs with tumor antigens from cell lysates or exposing them to tumor cDNA libraries. Though this approach has been explored in the pediatric population [67], an ongoing challenge involves eliciting an immune response against relatively weak “self” tumor antigens [58]. Clinical trials involving p-GBM patients include a phase I trial evaluating a tumor lysate pulsed dendritic cell vaccine (NCT01808820). A trial that evaluated a heat shock protein vaccine (HSPPC-96) was terminated due to a lack of efficacy (NCT02722512).
Oncolytic viral therapies also aim to activate the immune system, converting an immunosuppressed environment to a pro-inflammatory environment. Mechanistically, oncolytic viral therapy effects an anti-tumor response via two main mechanisms: 1) direct lysis of cancer cells due to viral replication, and 2) activation of an immune response against the tumor because of the release of neoantigens into the microenvironment following immunogenic cell death. The latter mechanism also has the potential to establish long-term immunological memory that may prevent tumor recurrence. Oncolytic viral therapy HSV G207 is currently being evaluated for the treatment of children with recurrent GBM [68].
Adoptive cellular immunotherapy
Adoptive cellular therapies involve manipulating effector immune cells ex vivo before reintroducing them to the patient. One approach consists of harvesting tumor-infiltrating lymphocytes (TILs) from the tumor bed, or alternatively immune cells isolated from peripheral blood or lymph nodes. The cells are stimulated ex vivo with cytokines and/or tumor antigen, and then infused back into the patient. Despite reports of immune activation, this therapy was clinically ineffective at mounting an anti-tumor immune response in patients with progressive primary or recurrent malignant gliomas [69].
Another approach to adoptive cellular immunotherapy involves the use of chimeric antigen receptors (CARs), which are artificial fusion proteins that incorporate an extracellular ligand recognition domain, a transmembrane domain, and an intracellular signaling domain to induce T cell activation upon antigen binding [55]. CAR T-cell therapy consists of isolating the patient’s T cells. The collected T cells are engineered to express the CAR receptor, which “reprograms” the T cells to target tumor cells. The modified CAR T-cells are expanded in the lab and subsequently infused back into the patient. CAR T-cell therapy has been successful in the treatment of malignancies such as acute lymphoblastic leukemia and chronic myeloid leukemia [70]. Recently, CAR T-cells have shown promise in mediating anti-tumor activity in adult patients with GBM by targeting HER2 or IL13Ra2 [58]. CAR T-cell therapy targets cell surface neoantigens, which may be limited in the case of p-GBM. However, recent studies demonstrated that driver mutations can be targeted with bispecific antibodies and CAR T-cells [71]. This approach could potentially be applied to common oncogenic mutations that are otherwise difficult to target in patients with p-GBM.
Current limitations and future directions
The existing challenges to finding effective treatments for p-GBM are multifactorial, and include: 1) inadequate animal models, 2) tumor heterogeneity and plasticity, and 3) an incomplete understanding of the tumor microenvironment. A general lack of experimental models that recapitulate intratumoral heterogeneity remains a major limitation of p-GBM research. However, a recently developed novel alpha thalassemia/mental retardation syndrome X-linked (ATRX)-deficient GBM mouse model has been promising [72]. These mice begin to develop brain tumors in the first few weeks of life, and demonstrate similar mutations as children and adolescent patients with the disease. Furthermore, the ATRX – deficient mouse model exhibits a functional immune system, thereby facilitating the development and implementation of immunotherapy strategies for the treatment of p-GBM [72].
Tumor heterogeneity and plasticity represents another limitation, particularly with regard to CAR T-cell therapy, as the loss of tumor-associated antigens (TAAs) allows some tumor cells to escape the CAR T-cells. As a result, CAR T-cells that target single antigens often result in antigen-negative relapses in pre-clinical and clinical studies [73]. Such relapses have prompted the development and design of CARs that can target multiple TAAs, such as bi-specific, trivalent, tandem, split, and synNotch CARs [73]. The first three strategies have been explored for adult patients, while bi-specific antigens are currently being explored in pediatric patients with refractory and recurrent GBM (NCT02208362).
An incomplete understanding of the p-GBM microenvironment remains a key limitation to effective treatment. Adult GBM models do not accurately recapitulate the pediatric tumor microenvironment, and as described above, recent work has suggested that pHGG might be less immunosuppressive than adult tumors. The presence of distinct immunophenotypes suggests a need for different immunotherapeutic approaches in adult vs. pediatric patients [74].
Ultimately, an improved understanding of the tumor biology and epigenetic landscape of p-GBM has provided an opportunity to better assess patient prognosis, but the translation of this knowledge into the development of new therapies has proven difficult. Despite its limitations, immunotherapy may represent a promising new line of treatment for these patients and remains an innovative focus for future studies.
Conclusion
While the immune infiltrate of adult GBM has been extensively studied, less is known about the tumor microenvironment and immune response in p-GBM. There is growing evidence that p-GBM undergoes less infiltration by immune cells and may also employ fewer immunosuppressive strategies compared with adult GBM. Until recently, the treatment of the p-GBM population has primarily been based on data obtained from adult trials. However, a growing understanding of the molecular and cellular features of p-GBM has demonstrated that optimal treatment requires the identification of biomarkers and therapies specific to pediatric patients. An expanded and updated understanding of the immunosuppressive processes and the tumor microenvironment of p-GBM will facilitate the development of pediatric-specific therapeutic strategies.
Availability of data and material
Not applicable.
References
Curtin SC, Miniño AM, Anderson RN (1999) Declines in Cancer Death Rates Among Children and Adolescents in the United States
Louis DN et al (2021) The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol 23(8):1231–1251
Das KK, Kumar R (2018) Chapter 15 Pediatric Glioblastoma. p. 1–14
Liu M et al (2018) National cancer database analysis of outcomes in pediatric glioblastoma. Cancer Med
Immanuel V et al (2017) Variegated colors of pediatric glioblastoma multiforme: what to expect? Rare Tumors 9(2):81–84
Solomon DA, Perry A (2018) 22 - Familial Tumor Syndromes. Elsevier Inc. p. 505–545
Amin SB et al (2020) Comparative molecular life history of spontaneous canine and human gliomas. Cancer Cell 37(2):243-257.e7
Nikitović M et al (2016) Pediatric glioblastoma: a single institution experience. Childs Nerv Syst 32(1):97–103
Sewell J, Pan E (2016) Current challenges in the treatment of glioblastoma. Clin Oncol 1:1–2
Rolle CE, Sengupta S, Lesniak MS (2012) Mechanisms of immune evasion by gliomas. Adv Exp Med Biol
Smyth, J.B.S.a.M.J. (2007) Immune surveillance of tumors. Clin Investig. 117: 1137–46
Burkholder B et al (2014) Tumor-induced perturbations of cytokines and immune cell networks. Elsevier. p. 182–201
Coniglio SJ, Segall JE (2013) Review : molecular mechanism of microglia stimulated glioblastoma invasion. Matrix Biol 32(7–8):372–380
Bowman RL et al (2016) Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Rep 17(9):2445–2459
Chen Z et al (2017) Cellular and molecular identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res 77(9):2266–2278
Ross J, Chen Z, Szulzewsky F, Schniederjan M, Becher O, Hambardzumyan D (2018) PDTM-43. The role of tumor associated macrophages in pediatric high-grade glioma | Neuro-oncology. Neuro-Oncology. 20(Issue:suppl_6):vi213–vi213:
Engler JR et al (2012) Increased microglia/macrophage gene expression in a subset of adult and pediatric astrocytomas. PLoS ONE 7(8):e43339–e43339
Maher EA, Bachoo RM (2014) Glioblastoma. Rosenberg's Molecular and Genetic Basis of Neurological and Psychiatric Disease: fifth Edition : p. 909–917
Mignogna C et al (2016) Pathology–research and practice original article a reappraisal of macrophage polarization in glioblastoma: histopathological and immunohistochemical findings and review of the literature. Pathol-Res Pract 212(6):491–499
Simonds EF et al (2021) Deep immune profiling reveals targetable mechanisms of immune evasion in immune checkpoint inhibitor-refractory glioblastoma. J Immunother Cancer 9(6):002181
Klemm F et al (2020) Interrogation of the microenvironmental landscape in brain tumors reveals disease-specific alterations of immune cells. Cell 181(7):1643-1660.e17
Haberthur K et al (2016) NKG2D ligand expression in pediatric brain tumors. Cancer Biol Ther 17:1253–1265
Zussman BM, Deibert CP, Engh JA (2015) A previously unidentified mechanism of immune evasion in glioblastoma. Neurosurgery 76:N20–N21
Farrell K et al (2018) Pediatric glioblastoma cells inhibit neurogenesis and promote astrogenesis, phenotypic transformation and migration of human neural progenitor cells within cocultures. Exp Cell Res 362(1):159–171
Mulkearns-hubert EE et al (2017) Glioblastoma cancer stem cells evade innate immune suppression of self-renewal through article glioblastoma cancer stem cells evade innate immune suppression of self-renewal through reduced TLR4 expression. Stem Cell 20(4):450-461.e4
Qian J et al (2018) TLR2 promotes glioma immune evasion by downregulating MHC class II molecules in microglia. Cancer Immunol Res 6(10):1220–1233
Cserr HF, Knopf PM (1992) Cervical lymphatics, the blood-brain barrier and the immunoreactivity of the brain: a new view. Immunol Today. p. 507–512
Brown NF et al (2018) Harnessing the immune system in glioblastoma
Sampson JH et al (2020) Brain immunology and immunotherapy in brain tumours
Janeway CAPTMW, Shlomchik M (2002) Immunobiology
Dhatchinamoorthy K, Colbert JD, Rock KL (2021) Cancer immune evasion through loss of MHC class I antigen presentation. Front Immunol 12:636568
Razavi S-M et al (2016) Immune evasion strategies of glioblastoma. Front Surg 3:1–9
Yeung JT et al (2013) Increased expression of tumor-associated antigens in pediatric and adult ependymomas: implication for vaccine therapy. J Neurooncol 111(2):103–111
Houke H et al (2019) IMMU-14. Implications for t-cell immunotherapy: cell surface antigen and HLA class I expression in pediatric brain tumors are heterogenous. Neuro-Oncology. 21(Supplement_2): p. ii96-ii96.
He W et al (2014) Proteomic comparison of 3D and 2D glioma models reveals increased HLA-E expression in 3D models is associated with resistance to NK cell-mediated cytotoxicity. J Proteome Res 13(5):2272–2281
Wiendl H et al (2002) A functional role of HLA-G expression in human gliomas: an alternative strategy of immune escape. J Immunol 168(9):4772–4780
Wu Z et al (2020) HLA-E expression in diffuse glioma: relationship with clinicopathological features and patient survival. BMC Neurol 20(1):59–59
Mangani D, Weller M, Roth P (2017) The network of immunosuppressive pathways in glioblastoma. Biochem Pharmacol 130:1–9
Ooi YC et al (2014) The role of regulatory T-cells in glioma immunology. Clin Neurol Neurosurg 119:125–132
Lauing KL et al (2016) TB-28immunosuppressive ido1 and tdo are expressed in pediatric central nervous system tumors. Neuro-Oncology 18:iii173.4–iii173
Scutti JAB (2018) Importance of immune monitoring approaches and the use of immune checkpoints for the treatment of diffuse intrinsic pontine glioma: from bench to clinic and vice versa (Review). Int J Oncol 52(4):1041–1056
Mirzaei R, Sarkar S, Yong VW (2017) T Cell exhaustion in glioblastoma : intricacies of immune checkpoints. Trends Immunol 38(2):104–115
Reardon DA et al (2020) Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the checkMate 143 phase 3 randomized clinical trial. JAMA Oncol 6(7):1003–1010
Pinto N et al (2017) Pediatric Blood & Cancer Patterns of PD-1, PD-L1, and PD-L2 expression in pediatric solid tumors
Kabir TF et al (2018) Immune checkpoint inhibitors in pediatric solid tumors: status in 2018. Ochsner Clin 18:370–376
Chongsathidkiet P et al (2018) Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nature Medicine 24(September)
Garris CS et al (2014) Sphingosine-1-phosphate receptor 1 signalling in T cells: trafficking and beyond. Immunology 142:347–353
Woroniecka KI et al (2018) T-cell dysfunction in glioblastoma: applying a new framework 24(16): 3792–3803
Eric K, Ring J.M.M.G.Y.G, Gregory KF (2018) checkpoint proteins in pediatric brain and extracranial solid tumors: opportunities for immunoherapy. 23(2): 342–350
Wei J et al (2011) Hypoxia potentiates glioma-mediated immunosuppression. PLoS ONE. 6
Jackson CM et al (2016) Systemic tolerance mediated by melanoma brain tumors is reversible by radiotherapy and vaccination. Clin Cancer Res 22(5):1161–1172
Mangano K et al (2018) Pathogenic role for macrophage migration inhibitory factor in glioblastoma and its targeting with specific inhibitors as novel tailored therapeutic approach 9(25): 17951–17970
Van Meir EG (1995) Cytokines and tumors of the central nervous system. Glia 15(3):264–288
Fuchs Q et al (2020) Hypoxia inducible factors' signaling in pediatric high-grade gliomas: role, modelization and innovative targeted approaches. Cancers (Basel). 12(4)
Wang SS, Bandopadhayay P, Jenkins MR (2019) Towards immunotherapy for pediatric brain tumors. Elsevier Ltd. p. 748–761
Lyon JG et al (2017) Engineering challenges for brain tumor immunotherapy ☆. Adv Drug Deliv Rev 114:19–32
Le DT et al (2017) Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357(6349):409–413
Sayour EJ, Mitchell DA (2017) Immunotherapy for pediatric brain tumors. MDPI 35(21):2450–2456
Shin K et al (2020) Multidisciplinary care for a patient with syndromic craniosynostosis: a case report with 20 years of special care. Spec Care Dentist 40(1):127–133
Zhang W et al (2020) Advances in anti-tumor treatments targeting the CD47/SIRPα Axis. Front Immunol 11:18
Hoffman M et al (2019) Intratumoral genetic and functional heterogeneity in pediatric glioblastoma. Cancer Research
Abedalthagafi M (2018) Constitutional mismatch repair-deficiency: Current problems and emerging therapeutic strategies. Impact Journals LLC. p. 35458–35469
Samstein RM et al (2019) Tumor mutational load predicts survival after immunotherapy across multiple cancer types, in nature genetics. Nature Publishing Group. p. 202–206
Bouffet E et al (2018) Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair De fi ciency 34(19)
Wagner LM, Adams VR (2017) Targeting the PD-1 pathway in pediatric solid tumors and brain tumors. Onco Targets Ther 10:2097–2106
Ratnam NM, Gilbert MR, and Giles AJ, Immunotherapy in CNS cancers : the role of immune cell trafficking. 2018(May): p. 1–10
Lasky JL et al (2013) Autologous tumor lysate-pulsed dendritic cell immunotherapy for pediatric patients with newly diagnosed or recurrent high-grade gliomas. Anticancer Res 33(5):2047–2056
Friedman GK et al (2021) Oncolytic HSV-1 G207 immunovirotherapy for pediatric high-grade gliomas. N Engl J Med 384(17):1613–1622
Plautz GE et al (1998) Systemic T cell adoptive immunotherapy of malignant gliomas. J Neurosurg 89(1):42–51
June CH et al (2018) CAR T cell immunotherapy for human cancer. Science 359(6382):1361–1365
Douglass J et al (2021) Bispecific antibodies targeting mutant. Sci Immunol 6(57)
Koschmann C et al (2016) ATRX loss promotes tumor growth and impairs nonhomologous end joining DNA repair in glioma. Sci Transl Med 8(328):328ra28
Land CA et al (2020) Chimeric antigen receptor T-cell therapy in glioblastoma: charging the T cells to fight. J Transl Med 18(1):428
Griesinger AM et al (2013) Characterization of distinct immunophenotypes across pediatric brain tumor types. J Immunol 191(9):4880–4888
Funding
No funds, grants, or other support were received.
Author information
Authors and Affiliations
Contributions
RN: conceiving, writing, and revising; CJ: editing, and revising; GW: conceiving, editing, and revising; DH: editing and revising. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Njonkou, R., Jackson, C.M., Woodworth, G.F. et al. Pediatric glioblastoma: mechanisms of immune evasion and potential therapeutic opportunities. Cancer Immunol Immunother 71, 1813–1822 (2022). https://doi.org/10.1007/s00262-021-03131-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-021-03131-y