
Abstract
Glioblastoma multiforme (GBM) is a highly malignant primary brain cancer with a dreadful overall survival and for which treatment options are limited. Recent breakthroughs in novel immune-related treatment strategies for cancer have spurred interests in usurping the power of the patient’s immune system to recognize and eliminate GBM. Here, we discuss the unique properties of GBM’s tumor microenvironment, the effects of GBM standard on care therapy on tumor-associated immune cells, and review several approaches aimed at therapeutically targeting the immune system for GBM treatment. We believe that a comprehensive understanding of the intricate micro-environmental landscape of GBM will abound into the development of novel immunotherapy strategies for GBM patients.
Similar content being viewed by others

Introduction
Primary brain cancer consists of tumors that originate from within the central nervous system (CNS) and comprises a myriad of different tumor types of benign to malignant status [1]. Unlike metastatic dissemination of cancers to the CNS, which are a far more common occurrence, primary brain cancer patients typically remain asymptomatic until overt clinical manifestation of tumor presence appears. These include headaches, seizures, nausea/emesis, syncope, neurocognitive dysfunction, personality changes, sensory loss, gait imbalance, urinary incontinence, hemiplegia, aphasia, hemispatial neglect, and visual field dysfunction. Of the ~50,000 newly diagnosed primary brain tumors each year in the United States, approximately 50% are histopathologically classified as gliomas of which the most aggressive type is glioblastoma multiforme (GBM). Glioblastomas are clinically classified as either primary GBMs (or de novo), i.e., without any prior symptomatic manifestation of the disease or secondary GBMs, which are the result of lower-grade gliomas that have degenerated in malignancy towards a higher grade GBM.
The efforts of The Cancer Genome Atlas (TCGA) have provided a detailed view of the genomic landscapes of lower grade gliomas and GBM’s [2,3,4,5,6]. TCGA’s extensive molecular characterization of gliomas has unveiled common genetic mutations and signaling abnormalities that are now recognized as drivers of uncontrollable growth, invasiveness, angiogenesis, and resistance to apoptosis [2,3,4,5,6].
GBMs are now classified into three distinct subtypes (Proneural, Classical, and Mesenchymal) based on gene expression profile and preponderance of driver gene mutations [2, 6,7,8]. GBMs of the neural subtype are now recognized as tumors with excessive adjacent neural tissue and this subtype is currently regarded as artifactual [8]. The clinical relevance to this classification, in terms of response to treatment and overall survival, has yet to be demonstrated. GBM tumors of the Classical subtype are characterized by aberrant expression of wild-type or mutated epidermal growth factor receptor (EGFR) in 100% of the cases, and are associated with homozygous deletion or mutation in the INK4a/ARF (CDKN2a) locus (in > 90% of cases) and loss of PTEN function (in ~ 37% of cases) [2, 6]. Genetically engineered mouse (GEM) models based on these events alone have proven sufficient to generate GBM tumors in mice [9,10,11]. The Proneural subclass of GBM is subdivided into two groups, those characterized by (1) overexpression of the receptor tyrosine kinase PDGFRα and loss of the p53 tumor suppressor gene and those with (2) recurrent mutations within the genes coding for isocitrate dehydrogenase (IDH1 and IDH2) [3, 4]. The latter GBMs are associated with a global hypermethylated genome (known as glioma CpG island methylator phenotype (G-CIMP)) and IDH mutant patients tend to have significantly prolonged survivals when compared to non-G-CIMP IDH wild-type Proneural GBMs [2]. IDH mutant GBMs are mostly secondary GBMs [3]. GEM models using genetic drivers corresponding to these events have recently been described [12, 13]. Overexpression of PDGF-A was shown to be sufficient to trigger gliomagenesis [13] but mutant IDH1 was not [12], reflecting our limited understanding of how IDH mutation can lead to glioma formation. Finally, the Mesenchymal subtype GBMs tend to be characterized by loss of Nf1 tumor suppressor gene function and several mouse models of Nf1 loss have demonstrated the driving nature of this lesion in GBM [14,15,16,17,18]. These models thus provide powerful platforms for advancements in genotype-specific treatments.
Despite our profound appreciation of the molecular drivers of GBM, targeted therapies against drivers of GBM have remained excessively inefficient (reviewed in [19, 20]). This is best exemplified by the use of EGFR kinase inhibitors in clinical settings. These clinical disappointments strongly support a precept by which oncogenic drivers are required for tumor initiation and maintenance of tumor growth, but either do not confer oncogenic addiction properties to GBMs [21] or there are significant pharmacokinetics barriers to CNS delivery [22] in addition to the blood–brain barrier (BBB). Thus far, there are no treatment modalities based on or specific to a given subtype or mutation status, and virtually all patients are given a standard of care treatment that consists of debulking surgery (when anatomically possible), followed by concomitant fractionated radiation (XRT) and temozolomide (TMZ) chemotherapy followed by adjuvant TMZ. With few exceptions, virtually all patients undergo surgical excision procedures. As such, post-surgical patients are administered steroids (dexamethasone (Dex)) for neurological symptomatic relief [23]. In addition, 20 to 40% of GBM patients are diagnosed after the sudden onset of seizures [24, 25], making the use of anticonvulsants (levetiracetam being a preferred agent due to its low toxicity profile) necessary and almost uniform for these patients. Despite this aggressive regimen, the median survival of GBM patients is ~15 months and <3% of patients survive longer than 5 years post-diagnosis [2]. This devastating prognosis is the product of neoplastic cells colluding with an intricate and highly heterogeneous tumor microenvironment, making neoplastic cell-centric therapeutic approaches unattainable.
It is clear that seeking alternative treatment approaches is of high priority for glioma. As new treatment paradigms are contemplated, significant considerations should be given to the effects that Dex, levetiracetam, and XRT/TMZ may have on the efficacy of the various treatments under development. Moreover, this cautionary statement also reinforces the need to study both tumor cells and the various components of the microenvironment in the context of standard of care.
Composition of the glioma tumor microenvironment
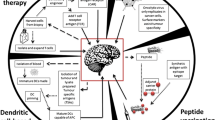
Surgically resected GBM tumor tissue consists of neoplastic cells and also contains non-transformed stromal cells. Estimates are that upwards of 30–40% of the cellular tumor bulk is composed of non-neoplastic stromal cells. There are many non-neoplastic cell types that contribute to the tumor stroma, including astrocytes, oligodendrocytes, endothelial cells (ECs), pericytes, and the multiple type of cells of the innate and adaptive immune system, all of which are present within a heavily modified extracellular matrix (ECM) (Fig. 1). During normalcy, these cells uniquely fulfill specific roles for proper brain functions. However, in cancer, the overarching principle is that transformed neoplastic cells coerce their surrounding cellular environment into enablers of cancer progression by acting on the various cellular components to alter their physiology towards pro-tumorigenic features. Glioma cells can produce and secrete a variety of cytokines and growth factors capable of promoting the infiltration of various cells including astrocytes, pericytes, and ECs. In addition, expanding tumors also trap different cell types and exert paracrine and physical influences on them. In this manner, bi-directional intercellular communications are established between neoplastic cells and the various components of the tumor microenvironment.

. Glioma tumors contain a highly diverse cellularity comprising of various resident and non-CNS-derived immune cells, each contributing in unique fashion to the tumor physiology. a Tumor-associated microglia and macrophages (TAMs) comprise upward of ~30% of the GBM tumor mass. Microglia (MG, CD11b+;CD45low) originate from the yolk sac and migrate to the CNS during development. Resting MGs take on a highly arborized anatomy, which is designed to constantly survey CNS tissue for damage or disease. Once activated, MGs adopt an amoeboid anatomy and assume a polarization phenotype consistent with a pro- or anti-inflammatory biochemisty. Bone marrow-derived macrophages (BMDMs, CD11b+;CD45high) originate peripherally (bone marrow) and migrate to and infiltrate GBM tumors responding to tumor-secreted chemokines and cytokines. Similarly, BMDMs have polarization capacities according to the identity of the intratumoral cytokines present. TAMs participate in substantial bi-directional crosstalk with neoplastic cells, which release cytokines and chemokines to recruit TAMs to the tumor microenvironment and to induce their polarization. TAMs in turn supply pro-tumorigenic growth factors and pro-survival cytokines. A highly exciting new approach to GBM treatment is to target polarization of TAMs using CSF-1R inhibitors, which function by reducing the levels of M2 (anti-inflammatory) polarized TAMs. b Myeloid-derived suppressor cells (MDSCs) originate from progenitor cells in the bone marrow and enter the circulation where they further differentiate into granulocytic or monocytic MDSCs (G-MDSC and M-MDSC, respectively) responding to tumor-derived factors. MDSCs migrate to lymphoid organs and to tumor sites. There, their function varies, but overall, MDSCs suppress T cell responses and NK cells cytotoxicity and induce Treg differentiation and expansion, all of which contribute to an immunosuppressive environment. HSC, hematopoietic stem cells; CMP, common myeloid progenitor and GMP, granulocyte-macrophage progenitor. c Tumor-infiltrating lymphocytes (TILs) are heavily influenced by the tumor microenvironment. Primed cytotoxic CD8+ T cells (TC) recognize neoplastic cells and mount anti-tumor responses. However, the presence of Treg cells and M2 polarized TAMs can suppress the effector function of TC cells leading to tumor outgrowth. d GBM vasculature and glioma stem cells (GSCs). GSCs are transformed glioma cells that express neuronal stem cell markers and have acquired some functional aspects of stem cell biology. GSCs are located within perivascular areas of the GBM microenvironment, which are built to promote self-renewal and maintain stemness. GSCs can promote an immunosuppressive environment by inhibiting effector T cell proliferation and activation, inducing T cell apoptosis, promoting Treg proliferation, and attracting and/or polarizing TAMs towards an M2 phenotype
Astrocytes
Astrocytes are the most abundant non-neuronal cells in the brain contributing upwards to ~50% of all cells in the cortex. They are generally viewed as essential for proper healthy brain function and physiology given their role in formation and maintenance of the blood–brain barrier, neurotransmission (synaptogenesis and support of synaptic transmission), nutritional and metabolic support, and regulation and maintenance of homeostasis [26, 27]. Astrocytes perform these multifaceted functions through a complex orchestration of localized proliferation and functional maturation.
The effects that normal non-neoplastic astrocytes may have on glioma physiology are likely to be significant, but have received relatively little attention. Astrocytes can trigger a physiological process called reactive gliosis in response to CNS injury such as stroke, cancer, neurodegenerative diseases and BBB damage to support neuronal viability and repair of brain tissue. This process involves the upregulation and phosphorylation of the intermediate filament protein GFAP, and secretion of ECM proteins, inflammatory cytokines as well as several growth factors (reviewed in [28]). In CNS injury models reactive astrocytes have been show to be harmful by inhibiting axonal regeneration after injury and by producing pro-inflammatory cytokines that exacerbate spinal cord injuries [29]. On the other hand, reactive astrocytes are critical for recovery after CNS trauma, ischemia, and in models of autoimmune encephalomyelitis [29]. A recent genomic analysis of reactive astrogliosis showed that 50% of reactive-mediated changes in gene expression profiles were dependent on the nature of the injury or disease [29], revealing an immense plasticity in the reactive astrocyte’s response towards injury and suggesting that in GBM, reactive astrogliosis is as unique as the patient’s tumor itself.
In response to GBM standard of care treatment, astrocytes have been shown to be able to protect neoplastic cells from TMZ treatment by forming heterotypic connexin43-positive gap junction with cancer cells to sequester cytoplasmic calcium and prevent apoptosis [30], a tactic that is similar to astrocytes’ known neuroprotection mechanism (reviewed in [31]). A question remains whether reactive gliosis unwittingly provides a cytokine-rich niche that stimulate neoplastic cell outgrowth and confers resistance to glioma therapy. Similarly, the influence that reactive astrocytes may have on the glioma immune fauna has yet to be determined.
Neurons and oligodendrocytes
Neurons are historically viewed as ultrasensitive entities that rapidly undergo cell death upon mild injury. In the context of malignant glioma, there is histopathological evidence of a much reduced density of healthy neurons within tumor mass. This is supported by tractography imaging techniques demonstrating massive loss of white matter neuronal tracks originating and projecting from tumors [32]. This suggests the presence of injured or distressed neurons closely associated with neoplastic cells. In fact, a recent report demonstrated that post-synaptic neurons can supply mitogenic signals to neoplastic cells through the upregulation of neuroligin-3, inducing a phosphoinositide 3-kinase signaling-mediated proliferative activity in glioma cells [33]. The effect of neuroligin-3 or the identity of any other growth factor or cytokine released by distressed neurons on the immune component of GBMs has not been studied.
Oligodendrocytes are support cells that are mainly responsible for the myelination of neuronal axons. They are produced from oligodendrocyte precursor cells (OPCs) in response to a variety of stimuli. Given the histopathological observation that neurons are absent within GBM tumors, less attention is paid to whether normal oligodendrocytes and OPCs are excluded or remain within tumor stroma. Moreover, markers of oligodendrocytes and OPCs are often shared with neoplastic GBM cells, rendering unambiguous identification difficult. As such, little is known regarding the influence (if any) of oligodendrocytes or OPCs on the immune component of GBM.
ECs, pericytes, and glioma stem cells
The brain vasculature is composed of ECs and pericytes, the latter being contractile cells that encircle ECs and are responsible to regulate vascular tone and blood flow. In addition, neurons and astrocytes make contacts with endothelia, pericytes, and other components of the microvasculature [34,35,36]. Studies in mice show that pericytes are necessary for vascular morphogenesis, endothelial quiescence in CNS and for the proper maintenance of the BBB [37,38,39,40,41,42]. Brain ECs are different from other ECs in that they physically interact with astrocytes making tight junctions, which are the physical basis of the BBB [43, 44]. The neurovascular unit therefore represents a microcosm of various cell types, growth factors, and molecules that orchestrate a continuous dynamism of transport of oxygen and nutrients, removal of cellular waste, and regulation of blood flow. Although highly physiologically active, ECs of the normal vasculature are mitogenically quiescent (only 0.01% of ECs dividing; [45]), a result in part by a balance between pro- and anti-angiogenic factors (e.g., vascular endothelial growth factor (VEGF) and thrombospondin, respectively). Thus, angiogenesis is tightly regulated by growth factors.
In contrast to normal blood vessels, the GBM vasculature is highly proliferative, which results in abnormal blood vessel structures [43, 45]. These tumor blood vessels are tortuous, often composed of blunted ends leading to significant hypoxic regions. In addition, GBM tumor blood vessels are unstable structures, which often causes hemorrhaging [43]. Less robust increase in blood vessel permeability are associated with increased edema, which is routinely observed in GBM [43]. Activated GBM ECs tend to express specific integrins that have been shown to enhance the capacity of immune cells to bind to the tumor vasculature and infiltrate tumor tissue [45].
GBMs’ neoplastic cellularity is highly heterogeneous and is composed of neoplastic cells with high to low differentiation potential. GBM cells that retain a high differentiation potential are commonly referred to as glioblastoma stem cells (GSCs) or tumor-initiating cells. These cells express markers of neuronal stem cells and are categorized as stem cells through a series of artificial ex vivo assays. They are however capable of tumor formation when orthotopically xenografted or allografted in serially diluted inocula when compared to “non-GSCs.” In vitro assays also demonstrate that these GSCs are slightly more resistant to radiation, DNA damage, and other therapeutic insults than non-GSCs are sensitive to. An amalgamation of these artificial attributes led to the concept that resistance of GBMs to many of the therapeutic options in current usage or under development is the result of the presence and/or activities of GSCs that are lurking within GBMs and are responsible for “repopulation” of GBMs during and post treatment.
The existence of a small population of neoplastic cells within GBMs that are endowed with unique capacities is undeniable. However, that this population is the source of resistance and repopulation at recurrence has yet to be demonstrated in an unequivocal manner in an in vivo system. Until then, the concept of GBM GSCs role in resistance remains circumstantial. However, it is nevertheless interesting that the GBM perivascular space has been shown to represent a niche where GSCs reside (reviewed in [46]). An interplay of cell surface receptors and membrane-bound and secreted ligands along with other factors secreted by ECs (e.g., nitric oxide) contribute to this area being optimal for GSC residence. One of the major pathways involved is Notch [47]. GSCs express Notch 1 and Notch 2 receptors and ECs express the Notch ligands JAG1 and DLL4 [47] and in vitro organotypic explant cultures confirmed the importance of Notch signaling in the interaction between GSCs and ECs [48]. Other growth factors and signaling pathways have been shown to create and maintain this GSC nurturing micro-environment (reviewed in [49, 50]).
Because of their unique characteristics, GSCs have been investigated for how they modulate the immune response to GBMs. Evidence for GSCs’ role in immunomodulatory reaction to GBM came from ex vivo studies demonstrating that GSCs but not their paired serum-grown counterparts cell lines inhibited T cell proliferation and activation, induced T regulatory cells (Tregs), and triggered T cell apoptosis. These effects were mediated by the activation status of signal transducer and activator of transcription 3 (STAT3) in GSCs [51, 52] and by hypoxia [53]. Similarly, GSCs were observed to influence innate immunity by inducing immunosuppressive characteristics of tumor-associated microglia/macrophages (TAMs), a capacity that depended on activated STAT3 [54]. Although shown to be immunosuppressive, GSCs are still recognized and destroyed by natural killer (NK) and CD8+ cytotoxic T (Tc) cells, perhaps due to their ability to process and present neo-antigens [55,56,57]. In patient-derived xenograft, periostin has been shown to be secreted by GSCs, which promote the recruitment of tumor-promoting M2-like polarized macrophage progenitors from the peripheral circulation [58]. Abrogation of periostin expression led to increased survival, mediated by a decrease in TAM density. Together, these studies demonstrate that GSCs, when compared to non-GSCs GBM cells, have a very different relationship with the immune system that is geared towards immunosuppression. Perhaps, GSCs’ true role in GBM biology is to abate immune recognition, thus enhancing tumor growth.
Extracellular matrix
The healthy brain ECM composition differs considerably from that of non-CNS tissues. The primary ECM components of the normal brain parenchyma primarily consist of a family of proteoglycans (known as lecticans (versican, aggrecan, neurocan, and brevican)) and two glycosaminoglycans to which they bind, hyaluronic acid and tenascins (TNCs) [59]. The ECM of glioma tumors differs quite considerably from that of normal brain due to the effect of ECM remodeling factors expressed and secreted by neoplastic cells and their complement of reprogrammed stromal cells. In fact, GBMs' highly invasive abilities reside in their heighten capacity to remodel the ECM and expression of cell–ECM adhesion molecules. How far ahead of the invasive front is the ECM remodeled is unknown and comprehensive analyses of ECM components (tumoral and peritumoral) and across GBM subtypes still remain limited. In other cancers, ECM-regulated mechanisms have been shown to contribute to T cell exclusion (reviewed in [60]). This represents a major challenge to immunotherapy-based treatments and similar principles are likely to be present in glioma. For example, high levels of TNC secreted by glioma have been shown to prevent T cell migration in vitro [61]. However, the significance of this observation remains to be determined in vivo.
Immune landscape of glioma
Several components of the immune system are present within malignant gliomas. These are microglia, peripheral macrophages, myeloid-derived suppressor cells (MDSCs), NK cells, leukocytes, CD4+ and CD8+ T cells, helper T (Th) cells, and Tregs [62]. These intratumoral immune cells are exposed to various cytokines and chemokines that are produced within GBMs and that are responsible to reprogram these infiltrating immune cells to acquire unique functional phenotypes and coax the immune system to perform inflammatory or anti-inflammatory functions with drastic consequences on glioma progression, invasion, and resistance to therapeutic intervention.
Tumor-associated microglia/macrophages
By far, the majority (up to ~30% of the tumor mass) of immune cells within gliomas are macrophages that arose from distinct ontogeneticity [63]. Brain tissue-resident microglia, bone marrow-derived macrophages (BMDMs), and extra-parenchymal macrophages are all observed within the tumor microenvironment (TME) [64, 65].
Microglia are yolk sac-derived myeloid cells [66, 67] that are found within the CNS and represent the major component of the innate immune system for the CNS. Microglia are not replenished after birth and maintenance in healthy adults is hypothesized to be achieved through localized proliferation and extended cellular longevity [67,68,69]. Extra-parenchymal macrophages represent another post-natally stable population of macrophages [70]; however, they are restricted to within the perivascular spaces, meninges, and are associated with the choroid plexus. Together with microglia, they function to protect the CNS tissue integrity and functionality through a variety of local immunological responses (recently reviewed in [71]). Microglia responds to local tissue injury through dramatic changes in physiology called polarization, a contextual process that results in pro-inflammatory or anti-inflammatory outputs [72].
In addition, changes in brain tissue homeostasis and pathological conditions recruits circulating monocytes to the brain parenchyma that give rise to BMDMs [73]. Unlike microglia, they are replenished via monocytes, especially in the context of glioma where the integrity of the BBB is compromised and rendered more permeable [74]. Peripheral macrophages are functionally flexible and can quickly adapt to various environmental cues by changing their phenotypes (a.k.a. polarization). Macrophages can alter their effector mechanisms along a spectrum comprised between a pro-inflammatory “M1” phenotype (portrayed by inflammatory, anti-tumor responses), and a cytoprotective, immunosuppressive “M2” phenotype (displayed by tissue repair and anti-inflammatory responses) [75]. Typical markers of M2-like polarized macrophages include high levels of interleukin-10 (IL-10), transforming growth factor-β (TGF-β), and low levels of IL-12. In addition, expression of Arg1, Mrc1, Chi3l3, Socs2, CD163, Fizz-1, and Ccl2 mRNAs are associated with M2 activation, whereas Nos2, IL12b, and Ciita are preferentially expressed in M1 macrophages [75]. Ex vivo M2 macrophages can be further induced to display discrete functional states (M2a, b, c) using specific cytokine stimulation [76], and to varying degrees of cytoprotective properties and immunosuppression. In recent years, such simplification of macrophage polarization distinction has been waning and rather a definition of discrete states of a spectrum is better accomplished using a set of functional features and gene expression profiles [77].
Similar to macrophages, microglia also exhibit remarkable plasticity, capable of polarization into M1 pro-inflamatory or M2 cytoprotective and immunosuppressive phenotypes depending on different stimulatory inputs.
Soon after non-cancerous brain injuries, microglia are activated and adopt mostly an M1 phenotype. They release compounds such as nitric oxide (NO), reactive oxygen species in addition to excitatory amino acids. They also produce and secrete the pro-inflammatory cytokines IL-1β, IL-12, TNF-α, and IL-6. On the other hand, M2-activated microglia produce and secrete high levels of CCL22, arginase-1 (Arg1), CCL17, mannose receptors, and scavenger receptors, and produce anti-inflammatory cytokines such as IL-10, IL-4, and TGF-β and low amount of IL-12 and NO (reviewed in [78, 79]).
Several studies in both humans and mouse model systems have demonstrated that gliomas are massively infiltrated with microglia and peripheral macrophages, collectively termed TAMs. The CD11b and Iba1 antigens are typically used as a common cell surface marker for microglia in both human and mouse tissues [80]. However, peripheral macrophages BMDMs and MDSCs also express CD11b and Iba1. Thus, distinguishing between the two cell types once they have infiltrated GBMs is hard to achieve experimentally and rely on a combination of markers. The most widely used combination to discriminate microglia from peripheral macrophages relies upon CD11b positivity together with levels of CD45 expression. CD11b+CD45high cells are considered BMDMs and CD11b+CD45low cells considered microglia [81,82,83,84,85]. This combination is adequate in murine models but does not accurately discriminate microglia from macrophages in human GBM samples. Until recently there was no microglia-specific marker identified that did not recognize macrophages in human. A recent report demonstrated however that the CD49d cell surface marker appears to discriminate between TAM microglia and peripheral macrophages in both human and murine GBMs [64].
Using immunohistochemical approaches, a few studies have shown higher abundance of TAMs in higher grade gliomas when compared to lower grade tumors, prompting the authors to correlate prognosis with TAM infiltration [86,87,88,89]. In fact, a recent study has identified transcriptomic signatures of immune and myeloid/macrophage gene expression that are associated with pathology, response to treatment, and overall survival [90]. However, given their ability to polarize, TAM numbers do not necessarily equate function and more recent flow cytometry studies with markers capable of deciphering polarization status of TAMs have better assessed functionality of GBM TAMs [77, 80, 91]. In human GBMs, TAMs were found to express significant levels of Toll-like receptors (TLRs), but engagement of TLR-4 did not result in TAM-mediated T cell proliferation. TAMs also do not produce any of the pro-inflammatory cytokines IL-1β, IL-6, and TNF-α. TAMs express major histocompatibility complex class II (MHC II); however, they do not express the co-stimulatory surface molecules CD86, CD80, and CD40, which are critical for T cell activation.
The outcomes of TAM depletion or targeting strategies in gliomas appear to be model and context dependent, which sometime leads to controversial outcomes. Nevertheless, the majority of studies support the concept that TAMs promote glioma growth by secreting growth and angiogenic factors as well as immune-suppressive cytokines (recently reviewed in [92]). Moreover, depletion strategies have also demonstrated a pro-tumorigenic role for TAMs [93,94,95] and recent studies have ascribed a significant role for colony-stimulating factor-1 receptor (CSF-1R) in glioma TAM biology. Depending on the mouse model, pharmacological inhibition of CSF-1R either depolarizes [96, 97] or depletes TAMs [98], both of which have the overall effect of reducing glioma growth and invasion.
It appears that while TAMs retain a few intact innate immune functions such as phagocytosis, cytotoxicity, and TLR expression, their capacity to be stimulated via TLRs, secrete cytokines, upregulate co-stimulatory molecules, and, in turn, activate anti-tumor effector T cells is not sufficient to initiate anti-tumor immune responses [80, 91]. A more recent and detailed characterization of CD11b+ TAMs from GBM patients reveal that microglia and MDSCs represented higher percentages of TAMs than peripheral macrophages [77]. More importantly, the report shed light on long-held beliefs that TAMs are mostly M2 (anti-inflammatory) polarized, demonstrating rather that TAMs assume phenotypes along a continuum of M1–M2 polarization with closer alignment to an undifferentiated or unpolarized M0 myeloid cell phenotype [77]. This observation is somewhat corroborated in different murine models of GBM where mouse TAMs do not fit into canonical model of M1 and M2 polarization [96, 99]; however, whether murine TAMs display M0 non-polarized features remains to be established. Overall, the observations made so far indicate that GBM TAMs display an attenuated pro-inflammatory response, thus acting as pro-tumorigenic in de facto. This concept opens the possibility of channeling TAMs towards a pro-inflammatory phenotype through pharmacological interventions that target M2 myeloid cells. In fact, ground-breaking work demonstrated the feasibility of such approach [96, 100], which has recently resulted in novel clinical treatment strategies for gliomas [101] (NCT02829723, phase I/II and NCT01790503, phase Ib/2).
MDSCs in gliomas
In both human and mice, MDSCs represent a heterogeneous population of myeloid progenitors and precursors found at various stages of differentiation towards granulocytes, macrophages, or dendritic cells (DCs). Experimentally, they are defined based on specific cell surface markers that are specific for each species (reviewed in [102]). In general, the immunosuppressive functions of MDSCs are multipronged. They have been shown to act on NK cells and to suppress their cytotoxic activities. MDSCs can also suppress the adaptive immune response of CD4+ and CD8+ T cells in an antigen-dependent or -non-dependent manner. MDSCs have also been shown to induce apoptosis in a subset of T cells. MDSCs are also capable of producing and secreting immunosuppressive cytokines and inducing Treg progression [102].
The functional role of MDSCs in gliomas has not often been addressed and their characterization remains mainly descriptive [103, 104]. There is a growing need to better define this type of cell in glioma since there’s increasing evidence that the mechanism of action of MDSCs is tumor type dependent. Efforts should be invested in determining abundance of the different subsets of MDSCs in different cancer types, including gliomas and even on a patient individual basis. Furthermore, systematic studies on functional impact MDSCs have on adaptive immune cells are required. Finally, transcriptomic characterizations of MDSCs need to be carried out separately from microglia and macrophages to ascertain their suppressive function and mechanisms of differentiation. These will reveal to be useful to determine if targeting MDSCs in glioma patients has significant clinical value.
Tumor-infiltrating leukocytes in gliomas
Th, Tc, and Treg cells
The microenvironment of gliomas is infiltrated with CD4+ Th cells, CD8+ Tc cells, and CD4+CD25+FoxP3+ Tregs [105,106,107,108]. A few studies have demonstrated a correlation between percentages of CD4+ and CD8+ tumor-infiltrating T cells and tumor grade and prognosis, importantly with regards to the extent of presence of CD8+(and not CD4+) associating with prolonged survival [105, 109]. Although tumor-infiltrating leukocytes (TILs) are present within the tumor microenvironment, they are lacking an anti-tumor T cell response due to the suppressive function of TGF-β and IL-10 cytokines that are secreted by glioma and other microenvironment cells. In addition, glioma cells tend to not express the co-stimulatory CD80/86 molecules, thus lacking in their ability to fully engage T cell receptor and T cell function. Glioma cells also have been shown to overexpress programmed death-ligand 1 (PD-L1), which strongly inhibits CD4+ and CD8+ T cell activation through engagement of the inhibitory checkpoint molecule programmed death-1 (PD-1) (reviewed in [110]).
Glioma cells also promote the infiltration and accumulation of immunosuppressive cells including Tregs and regulatory DCs within the microenvironment. Tregs are potent suppressors of the adaptive immune response through their capacity to inhibit the proliferation of any cytokine-secreting effector T cells. In normalcy, they are essential to confine and resolve the activation of the immune system. In glioma, accumulation of intratumoral Tregs correlates with poor prognosis [111] and both flow cytometry and immunohistochemical studies reported upward to 14% of CD4+ T cell population consisting of Tregs [112, 113]. These results are in sharp contrast to reports showing a lack of association between Tregs and prognosis [107, 112]. Other reports also revealed lower contribution (< 1%) of Tregs to the total T cell population [107, 111], perhaps reflecting variances in outcome when different methods and markers are used for cell type detection.
NK cells
NK cells are highly effective cytotoxic lymphocytes of the innate immune system. The activation of NK cells is tightly regulated by a sophisticated network of activating and inhibitory cell surface receptors. This network allows NK cells to distinguish normal from abnormal within seconds and target cell lysis is carried out through perforin-rich and granzyme-rich granules, when activating signals exceed inhibitory signals (reviewed in [114]). Normal cells express MHC I molecules, which interact with NK cell inhibitory receptor KIR (killer cell immunoglobulin-like receptor) and inhibits self-recognition and effective NK cell-mediated killing. In glioma, neoplastic cells express MHC I and are therefore resistant to recognition and destruction from NK cells [115]. In addition, GBM patients display reduced percentages of NK cells among peripheral blood mononuclear cell [116] and NK cells make up a minor portion of GBM-infiltrated CD45+ cell population [109, 117]. Those infiltrating NK cells were found to be non-functional, prompting the authors to suggest the potential role of TAMs, MDSCs, and Tregs in negatively regulating NK cells [117]. Recently, a feasibility study of chimeric antigen receptor (CAR) NK cells against ErbB2 for targeted therapy of GBM has been performed [118], bringing closer the clinical applications of NK cell-based treatments for gliomas.
The effects of standard of care treatment on the immune landscape of GBM
Following surgical removal of the tumor, GBM patients are exposed to an aggressive treatment regimen that consists of concomitant fractionated radiotherapy with TMZ (a DNA alkylating agent) chemotherapy, followed by adjuvant TMZ [119]. Craniotomy and tumor debulking neurosurgical procedures necessitate that patients be administered Dex to control post-surgical edema and anticonvulsants to prevent seizures. Therefore, the majority of patients undergo chemotherapy and radiotherapy treatments in the presence of those additional medications. There are few studies that address the effects of Dex on glioma biology and its standard of care treatment modalities and there are virtually no studies that include anticonvulsants (e.g., levetiracetam) in patient outcomes.
A significant number of chemotherapeutic agents have immunosuppressive effects when administered systemically and thus represent a major challenge for effective anti-cancer immunotherapy-based strategies. Most of our knowledge on the effect of radiation and TMZ treatment on the immune landscape of glioma microenvironment come from studies in the GL261 glioma mouse model. Curtin et al. [120] have demonstrated that radiation or TMZ-treated glioma cells undergoing apoptosis release the high mobility group box 1 protein, a Toll-like receptor 2 agonist that acts on DCs to cause their activation, which stimulate tumor antigen-specific T cell clonal expansion and anti-GBM immune response. In another study, a single radiation dose increased the ratio of CD8+ effector T cells to Tregs, an indirect measure of anti-tumor immune response. The treatment however had no statistically significant effect on survival [121].
Mathios et al. [122] have reported the immunosuppressive effects that systemic BCNU (Carmustine) alkylating agent chemotherapy treatment has on survival. Although BCNU treatment led to systemic and intratumoral lymphodepletion, survival was prolonged when compared to untreated controls, perhaps reflecting an unsuspected positive effect of BCNU on the innate immune system in this model or a minimal involvement of the adaptive immune system. BCNU is rarely used in clinical settings and therefore the significance of these observations to GBM patients remains debatable. BCNU is, however, used locally in the form of biocompatible wafers that are surgically positioned lining the surgical cavity during surgical removal of the tumor. Locally delivered BCNU in the GL261 model has been shown to have much less immunotoxicity (modest to no lymphodepletion) [122]. However, much like in patients [123], the effects on overall survival were very modest [122]. The effect of systemic TMZ treatment on the intratumoral immune fauna has yet to be assessed. Kim et al. [124] have studied the effect of low-dose TMZ treatment on CD4+ and CD8+ T cells from the spleen of GL261-bearing animals and showed that TMZ led to an increase in the secretion of INFγ from both cell types and also led to a decrease in the frequency of peripheral Tregs. Although these studies demonstrate that radiotherapy and chemotherapy do affect the immune system, a thorough and exhaustive examination of the effects of IR, TMZ, and combination of IR/TMZ—the standard of care for human GBM—on peripheral and intratumoral immune fauna has yet to be conducted.
A series of recent clinical and animal model studies have demonstrated that chemo-radiotherapy (TMZ/XRT) synergize with tumor vaccination immunotherapy to significantly increase survival. In some of these studies, TMZ-induced lymphopenia was observed but did not seem to negatively affect outcomes [125,126,127,128]. These results are in contrast to those reported by Litterman et al. [129], who showed that alkylating chemotherapy promotes deleterious effects on vaccination-induced immune responses [129]. It is possible that these discrepancies relate to the types of vaccines that target-specific tumor-associated neo-antigens that do not rely on Treg depletion, in contrast to TMZ [124].
The effects of TMZ on other immune cells of GBM have also been reported. For example, DCs and macrophages but not their precursor monocytes have been observed to be resistant to TMZ. Monocytes were shown to undergo apoptosis following TMZ treatment due to low levels of Ataxia telangiectasia mutated/ATM and RAD3-related (ATM/ATR) pathway activation and inability to repair double-strand breaks (DSBs), whereas DCs and macrophages were more capable of repairing TMZ-induced DSBs [130]. Current TMZ treatment for patients with GBM consists of sustained regimens. A few groups have studied the effects of low-dose metronomic TMZ regimen on Treg depletion in melanoma [131] and GBM [132] and demonstrated the beneficial advantages of Treg depletion.
GBM patients treated with TMZ/XRT often develop lymphopenia [133], which has been shown to negatively affect overall survival in elderly patients [134], presumably due to decreased immunity in these patients. In fact, a recent pilot study of GBM patients with lymphopenia showed that IL-2Rα blockade depleted Tregs and enhanced immunity so much so that tumor-specific antigen vaccination response was increased [135].
Steroids use and the immune system
Glucocorticoids (GCs) are an integral component in the treatment of gliomas [136]. GCs do not have a direct cytotoxic effect on glioma cells, but are used in order to diminish brain edema, prevent treatment-related hypersensitivity reactions, and suppress adverse effects such as nausea, emesis, and toxicity induced by chemoradiotherapy and to protect normal tissues [137]. Studies have demonstrated that GCs compromise survival in GBM [138, 139].
GCs have a well-established potent capacity to kill lymphoid cells of B and T cell origin, which has led to their inclusion in all chemotherapy protocols for lymphoid malignancies [140]. In addition to the effects on malignant lymphocytes, it is well established that exposure of T cell hybridomas and T cell clones to GCs, particularly Dex, causes programmed cell death [141]. Recently, the role of Dex on the expression of checkpoint inhibitors has started to be investigated. It has been found that Dex can enhance the expression of cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4) during T cell activation [142]. Dex can also enhance the expression of PD-1 both in mouse and human activated T cells in a dose-dependent manner [143]. This effect was mediated via the glucocorticoid receptor (GR) and was inhibited by the GR antagonist mifepristone (RU486). In parallel, Dex could suppress T cell functions by inhibiting production of cytokines such as IL-2, interferon-γ, and TNF-α and inducing apoptosis of primary human and mouse T cells. Interestingly, the ability of Dex to induce PD-1 expression was distinct among various T cell subsets, with the most prominent effect in memory T cells. These results are particularly interesting in the context of checkpoint immunotherapy, which has been shown to selectively expand memory T cells [144]. These findings suggest that Dex might compromise anti-tumor T cell immunity by inducing apoptosis or functional inactivation of tumor-specific T cells.
In addition to T cells, GCs significantly affect the properties and function of myeloid cells. Although the impact of steroids on the function of MDSC in the context of cancer has not been particularly investigated, studies regarding the effects of GCs in MDSC in autoimmunity have provided compelling evidence that the number and function of MDSCs is significantly altered. Specifically, GCs can induce a specific monocytic phenotype with anti-inflammatory properties in humans and a corresponding subset of monocytes in mice, which has been proved to resemble tumor-derived MDSCs [145]. High-dose Dex treatment in patients with immune thrombocytopenia (ITP) was found to increase MDSC numbers and to promote MDSC-suppressive function. The expression of IL-10 and TGF-β was also significantly upregulated in Dex-treated MDSCs, which inhibited the expansion of autologous CD4+ T cells and significantly attenuated CTL-mediated function [146]. Moreover, a 4-day regimen of high-dose dexamethasone in patients with ITP also expanded Treg and myeloid DCs [147].
Together, inactivation of memory T cells, expansion of Tregs, and generation of immunosuppressive MDSC by Dex will likely induce an immunosuppressive TME in patients with glioma. These findings indicate that Dex might suppress anti-tumor immune responses and facilitate tumor progression by multiple mechanisms. In addition, other than a cursory observation of no significant difference in the number of circulating monocytes between GBM patients given steroids or not prior to resection [77], there is a complete lack of understanding of the effects of Dex on intratumoral immune cells and function. Thus, the role of dexamethasone in the treatment of glioma in the era of immunotherapy should be fully re-evaluated.
Immunotherapies for cancer
Anti-cancer immunotherapy is a general term that encompasses various strategies that are intended to stimulate the patient’s immune system against her/his own cancer and to promote immune-mediated anti-tumor responses. Several approaches have been developed over the years and they include (but are not limited to) ADCC, cancer immunization, oncolytic viruses, CAR T cell therapy (CAR-T), cytokine treatment, DC therapy, and checkpoint blockade. Most of these approaches were originally developed for treatment of cancers other than brain cancer. Given the uniqueness of the brain in its accessibility and tissue/cellular composition, a certain degree of caution is warranted when considering application of these therapies and perhaps slight modifications in protocols will be necessary to achieve clinical success.
The basic scientific principle of ADCC is the targeting of cancer cells through the use of antibodies that recognize a specific antigen on the surface of cancer cells. The antibody’s Fab portion recognizes the target and cellular cytotoxicity is mediated by the Fc portion, which is primarily mediated by NK cells, although neutrophils and macrophages have also been shown to play a role. For primary CNS malignancies, antibodies against HER2/neu and tumor-specific gangliosides are being investigated for the treatment of gliomas [148, 149]. There is a requirement for knowledge of tumor expression of specific antigens for which optimized antibodies are clinically available, which restrict availability for some patients.
Immunization of cancer patients with antigens from their own tumor follows the biological principles of standard vaccination. Patients are immunized with specific peptides that are derived from his/her tumor and that are conjugated to a carrier protein. The patient develops an immune response generating humoral and cellular effects against the peptide epitope on the tumor cells. This technique is of great interest for the treatment of cancer often because of the uniqueness of tumor antigens, which are specific and thus minimizes side effects. For glioma, rindopepimut (Celldex) is a epidermal growth factor receptor vIII (EGFRvIII) peptide vaccine. Early clinical studies of rindopepimut demonstrated its safety and immunogenecity in a small cohort of patients. The development and optimization of Celldex has been extensively reviewed elsewhere [150]. Similar to ADCC, immunization using tumor cell-specific antigens requires prior knowledge of the genetic composition of the tumor and relies heavily on the immunogenic potential of the immunogen chosen, a characteristic that is still hard to predict.
The potential therapeutic application of viruses for the treatment of cancer is a mature concept. Viruses can hijack host cellular replication machinery upon infection to coax cells into amplifying their own genetic code, make progeny viruses, and inevitably inducing host cell death along with release of/and spread of viral progeny to initiate another cycle. The objective is to induce preferential viral infection of tumor cells over normal cells and to minimize immunosuppressive responses. There are various types of viruses that have been used throughout the year for the treatment of gliomas (recently reviewed in [151]), with the most popular ones being herpes simplex virus-1, adenovirus, poliovirus, parvovirus, reovirus, measles virus, Newcastle disease virus, and more recently Zika virus. Regardless of the approach, successful oncolytic virotherapy must reach a balance between supporting viral replication whilst promoting tumor immunity. With the recent emergence of immunotherapies in glioma, the research community is quickly gaining a clearer understanding of the composition and mechanisms of action of the glioma immune fauna. Studies aimed at establishing a better understanding of the immune responses to oncolytic viruses will be necessary in order to optimize the anti-glioma effects of oncolytic viruses.
The use of cytokines for the treatment of cancer typically involves systemic administration of a cytokine that is capable of stimulating the immune system. IL-2 for example is a critical cytokine necessary for antigen-specific T cell proliferation and has been shown to have efficacy in melanoma. IL-2 therapy showed only modest anti-tumor benefits in a small glioma clinical study [152].
DCs are myeloid-derived, highly potent antigen-presenting cells (APCs) that function to activate naïve cytotoxic CD8+ T cells to recognize and destroy cells with specific peptide antigens. Early studies in the brain demonstrated that unlike other tissues, DCs are not the predominant type of APCs rather microglia are [153,154,155,156]. Nevertheless, DC vaccine therapy relies on the isolation of a patient’s own DCs that are primed ex vivo with tumor-derived antigens, followed with re-implantation back into the host. This priming induces an antigen-specific Tc cell proliferation and when re-introduced into patients, boost T cell-mediated anti-tumor activities. There are several clinical efforts of dendritic cell therapy in gliomas (recently reviewed in [157]) with the furthest along (phase III trials; NCT00045968) in this respect being DCVax-L, which is an autologous tumor lysate-pulsed DC vaccine where early clinical trials report significant increases in median survival in GBM patients (31.4 months vs. 14.6 historical controls) [158].
This concept of modifying immune components ex vivo and re-introduce them back into patients is also the scientific premise in CAR-T-based therapies. In CAR-T, the patient’s own T cells are isolated, genetically engineered to express a chimeric antigen receptor that recognize a tumor antigen of interest, expanded ex vivo, and re-infused back into the patient. There are several chimeric antigen receptors aimed at various tumor-associated antigens. Promising results in leukemias and lymphomas [159, 160] have spearheaded investigations into the use of CAR-T for GBM. Currently, there are six GBM antigens that are the subject of targets for CAR-T cell therapy and are now undergoing clinical trials: EGFRvIII (NCT01454596, NCT02664363, NCT02209376), interleukin receptor 13Rα2 (IL-13Rα2) (NCT02208362), epidermal growth factor receptor 2 (HER2) (NCT02442297, NCT01109095), and ephrin type-A receptor 2 (NCT02575261). A case report of a GBM patient who demonstrated regression after autologous CAR-T therapy against IL-13Rα2 (intracavitary infusions of IL13BBζ-CAR) was recently published [161]. The clinical response, including dramatic improvements in quality of life, was observed for 7.5 months after initiation of therapy. The use of CAR-T cells for solid tumor treatment is associated with challenges including antigen validation, tumor heterogeneity, trafficking and infiltration, extensive retrieval protocols and genetic manipulation of T cells, and approaches to overcome the immunosuppressive microenvironment (reviewed in [162, 163]). Nevertheless, CAR-T-based therapies for the treatment of GBM is actively being pursued.
In healthy individuals, normal adaptive immunological homeostasis is exquisitely regulated by a balance of T cell co-stimulatory activating and inhibitory signals. These signals are transmitted to T cells by a series of cell surface receptors that are present on T cells called immune checkpoint receptors. The cognate ligands for these receptors are also cell surface molecules expressed in a variety of antigen-presenting cells such as regulatory Treg cells, Th cells, macrophages, and DCs (Fig. 2). In concert with an MHC-mediated activation of the T cell receptors (TCRs), these immune checkpoint receptors, when activated, elicit either negative or positive activities or influence on TCR activation and T cell physiology. These immune checkpoints therefore play a critical and fine tuning role in regulating T cell activation and effector function, but also control T cell homeostasis. Dysregulation of some of these checkpoints has been reported to be involved in autoimmune diseases, chronic infection, and cancer reinforcing their impact on effector T cell responses in the context of disease.

. T cells are activated upon engagement of their T cell receptor (TCR) by APCs (cancer cells, dendritic cells, TAMs etc.) to the TCR–CD3 complex in the presence of B7/CD28 co-stimulation. Many co-inhibitory pathways are upregulated upon T cell activation and are designed to attenuate TCR and co-stimulatory signals. Some of these ligand–receptor co-stimulatory and inhibitory complexes are expressed during initial activation of naïve T cells in lymph nodes, where dendritic cells are considered the main APCs, whereas others are expressed in peripheral tissues or tumor cells where they regulate the effector responses of T cells. Note that several ligands bind to multiple receptors with opposite effects on TCR signaling. Different ligand–receptor complexes are expressed on the surface of various APCs as well as in resting, naïve, and activated T cells. In addition to the distinct kinetics of expression, they have distinct affinities for their cognate binding partners. The extent of T cell activation is proportional to the strength of the TCR signaling, which is dictated by a multitude of factors that are highly spatiotemporal and context dependent. Binding ligands for VISTA have not been identified. APC antigen-presenting cell, B7RP1 B7-related protein 1, BTLA, B and T lymphocyte attenuator, GAL9 galectin 9, HVEM herpesvirus entry mediator, ICOS, inducible T cell co-stimulator, KIR killer cell immunoglobulin-like receptor, LAG3 lymphocyte activation gene 3, PD1 programmed cell death protein 1, PDL PD1 ligand, TIM-3 T cell–immunoglobulin–mucin domain 3
In cancer, there is an obligate evasion of immunosurveillance, the mechanistic details of which are starting to emerge. It is likely that small indolent tumors are kept from growing rapidly by a combination of cancer cell-centric effects (e.g., oncogene-induced senescence) and an anti-tumor immune system until a time at which cancer cells are either eradicated or develop mechanisms to progress beyond this negative pressure. Conceptually, there appears to exist a “biological switch” that converts an otherwise controlled tumor into an unrestrained one. Molecular events responsible for this progression are likely a combination of tumor cell-centric and humoral actions. We now know that tumor cells have evolved several strategies to overcome the negative influence of the host immune system by exploiting several aspects of the various interactions of tumor cells with the immune system.
The adaptive immune system’s efforts to eliminate neoplastic cells are mediated through a multi-step response cycle. The cycle starts with the production and release of tumor cell-associated antigens, presumably during neoplastic cell death. Various types of APCs internalize these antigens, process them while migrating to lymph nodes to present them (through MHC loading) to resident naïve T cells in order to activate TCRs and thus prime T cells against cancer-specific antigens (see [164] for a detailed review). The primed CD8+ T cells, now endowed with cytotoxic capacities, migrate towards and infiltrate tumor sites, specifically recognize cancer cells, and elicit tumor cell death, which in turn causes the release of more tumor-associated antigens, thereby continuing the cycle. This cycle between APCs and T cells and between tumor cells and T cells is intricately controlled by many ligand–receptor interactions (also known as checkpoint pathways) that are necessary to provide positive and negative signals to stimulate or inhibit T cell activation, and to regulate the duration and intensity of the immune response mounting against tumor cells.
So far, mechanistic details are mostly known on two crucial steps that are involved in the anti-tumor activation of the T cell response (Fig. 2). The first important interaction occurs during antigen presentation through the MHC to the TCR in the lymph node. APC’s CD80/86 ligands simultaneously interact with T cell’s CD28 co-stimulatory receptor to enhance TCR response and with T cell’s CTLA-4 co-inhibitory receptor to control the TCR activity. The balance between these co-stimulatory and co-inhibitory signals can be shifted dramatically by dampening the co-inhibitory CTLA-4 signal using approaches (e.g., blocking antibodies) that prevent the interaction between CD80/86 and CTLA-4. This results in a more robust MHC-TCR signal and a stronger priming of T cells. The second step in the process of anti-tumor T cell responses occurs at the tumor site. Primed T cells that have migrated to the tumor interact with a host of cells, including tumor cells, APCs, macrophages, NK cells, and astrocytes that are present in the TME. The strength of the MHC:TCR engagement between these TME cells and T cells determine the robustness of T cell cytotoxic effector function and it is dependent on the activation of co-stimulatory and co-inhibitory checkpoints. The co-inhibitory PDL1-PD1 complex has emerged as a crucial signal in effector T cell function and inhibition of this complex has been clinically proven to be a sound approach to boost the cytotoxic power of T cells.
Although CD28 serves as the prototype co-stimulatory whereas CTLA-4 and PD-1 serve as the prototype co-inhibitory receptors that regulate the outcome of T cell immune responses, it is now clear that numerous receptor ligand pairs on the surface of T cells and APC serve similar functions (Fig. 2) (recently reviewed in [165]). The evolutional requirement for the multiple checkpoint pathways is not entirely understood but it is possible that such pathways differentially dominate responses in the context of different microenvironments, in the presence of different antigens or at distinct phases of T cell activation at which they are differentially expressed. When the TCR is engaged, tyrosine phosphorylation of the TCR-associated CD3 chains recruits kinases and scaffold proteins leading to the formation of a supra-molecular complex that promotes activation of signaling cascades, generation of second messengers, and initiation of transcriptional events, which lead to T cell activation and differentiation programs [166]. These signaling pathways synergistically promote glycolysis and anabolic metabolism to support T cell clonal expansion and effector cell generation [167,168,169]. Co-stimulatory receptors, engaged simultaneously with the TCR, have a major impact on signaling events and a decisive role in the differentiation program of T cells.
Our understanding about the functional role of co-stimulation has evolved from the two-signal model proposed by Bretscher [170] and Lafferty et al. [171] to explain the activation of naïve T cells [170, 171]. Although T cell co-stimulatory pathways were envisioned as stimulators of T cell responses by that model, it is now clear that both stimulatory (co-stimulatory) and inhibitory (co-inhibitory) second signals exist and mediate their impact not only in naïve but also in effector, memory, and Treg cells [172,173,174]. These receptors are key regulators of T cell activation, tolerance, and exhaustion. As a consequence, these receptors are attractive therapeutic targets, which provide effective new treatment strategies in cancer, autoimmunity, infectious diseases, and allogeneic transplantation [175,176,177]. These pathways fall into two major families: the Ig superfamily, which includes the B7-CD28, TIM, CD226-TIGIT-CD96 families as well as LAG-3, and the TNF-TNF receptor superfamily [178,179,180]. Co-inhibitory receptors provide a balance on the activation and expansion of antigen-specific T cells upon encounter with antigen but also regulate T cell tolerance by restraining the initial activation of naïve self-reactive T cells and/or responses of harmful self-reactive T cells. Coinhibitory pathways also regulate the generation and function of thymic-derived Treg and Treg generated at peripheral sites [181, 182]. Ligands for various co-inhibitory receptors are expressed on APCs and also in non-hematopoietic cells [178,179,180]. The expression of co-inhibitory ligands on non-hematopoietic cells has a key role for the maintenance of tissue tolerance by suppressing the expansion and function of self-reactive T cells.
Tumor cells heavily exploit these immune checkpoint pathways as a means to evade immune detection. However, they provide a plethora of potential targets for the development of anti-cancer therapeutic agents aimed at boosting the anti-cancer immune responses [175]. Much of our knowledge on the function of these molecules in cancer has been derived from pre-clinical models of and clinical data from melanomas, lung, and renal cancers. The exact participation of checkpoint pathways in primary brain tumor pathogenesis is largely unknown and has just recently started to emerge. For many years, the CNS was viewed as an immune privileged organ incapable of surveillance by peripheral immunity because of the ostensible lack of a functional lymphatic system. This view has shifted considerably in the recent years. Seminal discoveries expanded our views on the role of the peripheral immune system and the brain. We now know that the brain is drained by classical lymphatic conduits that reside within the meninges [183, 184]. Lymphatics are typically designed to drain interstitial fluids out of tissues for degradation and removal into the circulatory system. During infection (and in cancer), lymphatic transport is essential for supplying antigens and APCs to draining lymph nodes, a very important step in the process of establishing a proper adaptive immune response. Additionally, there is evidence of a separate process in the brain called “glymphatics” whereby CSF (carrying extracellular proteins, antigens, and solutes) and interstitial fluid exchange extensively. This results in pushing interstitial fluid (ISF) into the perivenous space where it can collect and drain into the cervical lymph nodes (for a review see [185]). Together, these recent advances underscore the notion that the brain is indeed surveyed by the peripheral immune system.
Checkpoint inhibitors in GBM
Within the context of cancer treatment, activation of anti-tumor immunity by relieving the negative feedbacks exerted by CTLA-4 with a blocking antibody has proven to be successful. Several seminal studies have demonstrated that anti-CTLA-4 blocking antibodies can stimulate anti-tumor immune responses leading to regression of tumors and promoting long-lived immunity in mouse models of solid and hematologic cancers [186, 187]. These led to the clinical development of humanized antibodies aimed at efficient blocking of the interaction between CD80/86 and CTLA-4. CTLA-4 blockade has recently been approved in the United States for a spectrum of malignancies including melanoma and non-small-cell lung cancer. Despite promising clinical responses, our understanding of the mechanism(s) of anti-CTLA-4 anti-tumor immunity remains incomplete. The therapeutic effects of anti-CTLA-4 antibodies may not only be due to blocking CTLA-4 interaction with its ligands on T cells. In fact, recent work suggests that blocking CTLA-4 may also deplete intratumoral Tregs via an Fc receptor-mediated, antibody-dependent cellular cytotoxicity [187]. Further studies are necessary to fully understand the mechanism of action of anti-CTLA-4 therapy.
In a preclinical model of GBM, single agent anti-CTLA-4 blockade resulted in enhanced survival in the GL261 syngeneic mouse model. Reardon et al. [188] demonstrated that anti-CTLA-4 monotherapy leads to a 25% cure rate and increased median survival. Although positive, these responses to anti-CTLA-4 monotherapy were considered limited and treatment efficacy was drastically enhanced when administered in combination with anti-PD-1 blocking antibody, or radiation [188, 189]. Reardon and colleagues [189] demonstrated that dual blockade of PD-1 and CTLA-4 increased cure rates to 75%. CTLA-4 blocking therapy has also been used in combination with stimulation of 41-BB and radiation to achieve a 50% cure rate. Anti-CTLA-4 is currently tested in a phase III clinical trial for patients with recurrent GBM as a monotherapy or with anti-PD-1 blockade (Trial NCT02017717).
Another prototypical immune checkpoint is the cognate receptor ligand complex PD-1 receptor and its PD-L1 and PD-L2. PD-1 is a transmembrane receptor that exerts a major negative regulation in immune response by controlling T cell activation, T cell exhaustion, and T cell tolerance. PD-1 expression is tightly regulated, e.g., it appears at the surface of T cells shortly (< 24 h) after T cell activation and decreases with the elimination or clearance of antigen. Under conditions (such as chronic infection or cancer) of repetitive T cell stimulation by antigen, the levels of PD-1 expression remain high and T cells then experience multiple epigenetic modifications in addition to changes in transcription factor expression. These events result in a form of differentiation, channeling T cells into a state of exhaustion. It has been shown that exhausted T cells also can express multiple other inhibitory receptors, making them susceptible to blocking antibody inhibition of additional checkpoint pathways to rescue T cells from exhaustion. Supporting this phenomenon, Kim et al. [190] have co-targeted TIM-3 (T cell–immunoglobulin–mucin domain 3) simultaneously with PD-1 and demonstrated a much higher cure rates than with each modalities alone. However, rescue of exhaustion by inhibition of alternative coinhibitory receptor(s) in a sequential manner remains to be addressed.
PD-L1 and PD-L2 are both expressed on APCs in addition to other cell types, but PD-L1 appears to be more broadly expressed than PD-L2. Their expression is induced by pro-inflammatory cytokines. PD-L1 (or PD-L2) ligand binding to PD-1 results in tyrosine phosphorylation of the PD-1 cytoplasmic domain and recruitment of signaling complexes including the tyrosine phosphatase SHP-2. This leads to a reduced tyrosine phosphorylation of TCR signaling molecules and the attenuation of signaling pathways downstream of TCR, and an overall decrease in T cell activation and cytokine production. PD-1 signaling is therefore viewed as a negative modulator of T cell function to suppress effector immune responses. In normalcy, the PD-1 pathway restrains self-reactive T cells in target organs, maintaining tolerance in tissues and protecting them from immunopathology. For instance, mice lacking PD-1 or its ligands do not spontaneously develop autoimmune disease but rather accelerate or exacerbate autoimmunity, a phenotype that is much milder than that seen in the CTLA-4-knockout strain.
In cancer, tumor cells express PD-L1 (and PD-L2) and so do other cell types (e.g., fibroblasts, ECs, and other immune cells including TAMs). Experimental evidence demonstrates that tumor cells have hijacked this machinery. Tumor cells express elevated PD-L1 levels, causing effector function attenuation of TIL (Tumor Infiltrating Leukocytes) activity [191]. In addition, several groups have shown that activation of certain oncogenes and/or loss of tumor suppressor genes can result in higher expression levels of PD-L1 in tumor cells, thus further attenuating TILs. Not surprisingly, antibody blockade of PD-1/PD-L1 axis have demonstrated positive outcomes in advanced melanoma, lung cancer and renal cell carcinoma (reviewed in [175]). Anti-PD-1 monoclonal antibodies have been recently approved for cancer treatment while anti-PD-L1 is in late clinical stages. Despite the promising success of anti-PD-1 blockade in the clinic, responses are varied. Several studies are aimed at determining predictors of response to anti-PD-1 blockade. To date, both the presence of TILs and high expression of tumor PD-L1 within the tumor microenvironment are main prognostic factors of anti-PD-1 therapy.
In GBM, PD-L1 is expressed at high levels, as determined by western blotting, flow cytometry, mRNA, and immunohistochemistry [192, 193]. This suggests that anti-PD-1 therapy might be applicable. In fact, Parsa et al. [192] have demonstrated that PD-L1-expressing glioma cells are susceptible to T cell lysis, further reinforcing the concept that anti-PD-1 may have clinical benefit.
In the preclinical GL261 model, the success of anti-PD-1 monotherapy is dependent on dosage, with the best outcome reported being a cure rate of 50% [121, 188]. Not surprisingly, lesser posology appears to result in less robust effects [121]. Anti-PD-1 blocking antibody monotherapy treatments were shown to cause an increase in CD8 T cell to Treg ratio, which is indicative of a successful outcome [121, 188]. Inclusion of radiation or in combination with other checkpoint blockade led to enhanced efficacy of anti-PD-1 treatment [121, 188]. Although radiation has been shown to alter the immunogenicity profile of GL261 cells in vitro [121], the precise mechanism of synergism between radiation and anti-PD-1 therapy in GL261 and in GBM in general has yet to be explored. In melanoma it has been demonstrated that radiation promotes an oligo expansion of T cell receptors [194].
Currently, there are several ongoing trials utilizing anti-PD-1 or anti-PD-L1 for the treatment of GBM. Anti-PD-L1 treatments are in phase II trials with or without radiation and the VEGF-A neutralizing antibody bevacizumab (NCT02336165). A phase III trial of anti-PD-1 treatment in conjunction with TMZ chemotherapy with radiation in patients with newly diagnosed GBM is undergoing (NCT02617589) and a phase II trial of anti-PD-1 as neoadjuvant therapy is ongoing (NCT02550249).
Future directions
Virtually all of the various immunotherapies currently under development for GBM have utilized the GL261 syngeneic model for pre-clinical studies (recently reviewed in [195]). Although popular because of its ease of use, this model falls short of recapitulating critical aspects of human GBMs (reviewed in [196,197,198]). Notably, the genetics of the cell line (chemically induced KRas mutation) is not representative of human GBMs since Ras mutations are rarely observed in patients (< 1%, [2]). More importantly, the composition of the immune fauna in other tumors is not representative of that in GBMs [99]. Therefore, there is a need to conduct pre- and co-clinical studies in genetically accurate model systems and given the irrelevance of PDX immunodeficient models to study immunotherapeutics, GEM models offer unsurpassed relevance for immuno-oncology research. In addition, the recent creation of a comparative (canine to human) brain tumor consortium [199] expands the possibilities of pre- and co-clinical studies of immunotherapeutics to an additional species.
In using model systems for pre- and co-clinical studies, one has to incorporate standard of care treatments to their study design. Other than surgical resection, XRT/TMZ administration in brain tumor bearing mice is relatively easy to achieve. Surprisingly, there is no study reported so far that takes into account dexamethasone and levetiracetam in pre-clinical settings. Given the drastic effects that dexamethasone have on various immune functions, attempts at modeling immunotherapies for GBM in models that do not take into consideration steroid administration will likely lead to questionable outcomes that might not apply to GBM immunotherapy in humans.
Although reports of incidental responses to various types of immunotherapies for glioma are emerging, convincing clinical trial data has yet to emerge. The availability of biomarkers has greatly enhanced oncological practices and is now the basis of precision medicine for many cancers; however, potential predictive biomarkers of immunotherapies are unknown in glioma. Recent studies demonstrate a relationship among tumor mutational load, mismatch repair deficiencies, checkpoint receptor expression, neo-antigens processing and presentation, and response to checkpoint inhibitor therapy [200,201,202]. These observations predict that not every glioma patients are likely to benefit from monotherapy immune checkpoint inhibition [201]. Therefore, it is likely that additional predictor of response will be required to optimize treatment strategy.
Tumors are routinely genotyped against an ever-increasing panel of known gene mutations, which guide treatment decisions based on the prognostic significance of these genetic profiles, and, in many cases, provides the opportunity for targeted therapeutic agents that can be used or are under rapid clinical development. In the case of immunotherapies however, this concept remains nonexistent. Therefore, the development of cancer immunotherapies should include a mechanism for discovery, validation, and usage of biomarkers that will provide the means for candidate patient stratification for immunotherapy approaches. A starting point would be to immunophenotype all gliomas enrolled in clinical trials. This would provide information as to what type of immune infiltrate populations are present and their activation profiles. In addition, knowledge of the genetic drivers of each patient’s GBM will be necessary to address the influence of the tumor genotype on the identity of the immune fauna. This is a concept that can be molecularly dissected in the controlled scientific confine of GEMMs. The development of technological platforms that would accelerate tumor immunophenotyping without compromising specificity and sensitivity while reducing costs (when compared to standard flow cytometry) will be instrumental to successfully generate mechanistic links between immune infiltrates and treatment response. The ultimate goal is to stratify patients towards a precision immunocentric medicine tailored to their unique immune fauna.
The future of immunotherapies for glioma is cautiously bright. Analogous to prior new therapeutic approaches applied to gliomas in the past, glioma immunotherapies will be challenged by obstacles. Ultimately, success in the clinic will likely come from patient-tailored combination therapies, which can only be achieved through in-depth research on immune–tumor cell interactions and their responses to standard of care.
References
Louis DN, Ohgaki H, Wiestler OD, Cavenee, WK. HO classification of tumours of the central nervous system. Revised 4th ed, vol. 1. International Agency for Research on Cancer (IARC). Lyon; 2016.
Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–77.
Cancer Genome Atlas Research N, Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. 2015;372:2481–98.
Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H, et al. Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N Engl J Med. 2015;372:2499–508.
McLendon R, Friedman A, Bigner D, Van Meir EG, Brat DJ, Mastrogianakis M, et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008;455(7216);1061-8
Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110.
Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–73.
Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell. 2017;32:42–56 e46.
Acquaviva J, Jun HJ, Lessard J, Ruiz R, Zhu H, Donovan M, et al. Chronic activation of wild-type epidermal growth factor receptor and loss of Cdkn2a cause mouse glioblastoma formation. Cancer Res. 2011;71:7198–206.
Jun HJ, Acquaviva J, Chi D, Lessard J, Zhu H, Woolfenden S, et al. Acquired MET expression confers resistance to EGFR inhibition in a mouse model of glioblastoma multiforme. Oncogene. 2012;31:3039–50.
Zhu H, Acquaviva J, Ramachandran P, Boskovitz A, Woolfenden S, Pfannl R, et al. Oncogenic EGFR signaling cooperates with loss of tumor suppressor gene functions in gliomagenesis. Proc Natl Acad Sci USA. 2009;106:2712–6.
Bardella C, Al-Dalahmah O, Krell D, Brazauskas P, Al-Qahtani K, Tomkova M, et al. Expression of Idh1R132H in the murine subventricular zone stem cell niche recapitulates features of early gliomagenesis. Cancer Cell. 2016;30:578–94.
Ozawa T, Riester M, Cheng YK, Huse JT, Squatrito M, Helmy K, et al. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell. 2014;26:288–300.
Alcantara Llaguno SR, Wang Z, Sun D, Chen J, Xu J, Kim E, et al. Adult lineage-restricted CNS progenitors specify distinct glioblastoma subtypes. Cancer Cell. 2015;28:429–40.
Hambardzumyan D, Parada LF, Holland EC, Charest A. Genetic modeling of gliomas in mice: new tools to tackle old problems. Glia. 2011;59:1155–68.
Parada LF, Kwon CH, Zhu Y. Modeling neurofibromatosis type 1 tumors in the mouse for therapeutic intervention. Cold Spring Harb Symp Quant Biol. 2005;70:173–6.
Zhu Y, Guignard F, Zhao D, Liu L, Burns DK, Mason RP, et al. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell. 2005;8:119–30.
Zhu Y, Harada T, Liu L, Lush ME, Guignard F, Harada C, et al. Inactivation of NF1 in CNS causes increased glial progenitor proliferation and optic glioma formation. Development. 2005;132:5577–88.
Olson JJ, Nayak L, Ormond DR, Wen PY, Kalkanis SN, Ryken TC, et al. The role of targeted therapies in the management of progressive glioblastoma: a systematic review and evidence-based clinical practice guideline. J Neurooncol. 2014;118:557–99.
Prados MD, Byron SA, Tran NL, Phillips JJ, Molinaro AM, Ligon KL, et al. Toward precision medicine in glioblastoma: the promise and the challenges. Neuro Oncol. 2015;17:1051–63.
Hegi ME, Diserens AC, Bady P, Kamoshima Y, Kouwenhoven MC, Delorenzi M, et al. Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib—a phase II trial. Mol Cancer Ther. 2011;10:1102–12.
Vivanco I, Robins HI, Rohle D, Campos C, Grommes C, Nghiemphu PL, et al. Differential sensitivity of glioma- versus lung cancer-specific EGFR mutations to EGFR kinase inhibitors. Cancer Discov. 2012;2:458–71.
Omuro A, DeAngelis LM. Glioblastoma and other malignant gliomas: a clinical review. JAMA. 2013;310:1842–50.
Glantz MJ, Cole BF, Forsyth PA, Recht LD, Wen PY, Chamberlain MC, et al. Practice parameter: anticonvulsant prophylaxis in patients with newly diagnosed brain tumors. Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2000;54:1886–93.
Vecht CJ, Kerkhof M, Duran-Pena A. Seizure prognosis in brain tumors: new insights and evidence-based management. Oncologist. 2014;19:751–9.
Matyash V, Kettenmann H. Heterogeneity in astrocyte morphology and physiology. Brain Res Rev. 2010;63:2–10.
Molofsky AV, Deneen B. Astrocyte development: a guide for the perplexed. Glia. 2015;63:1320–9.
Placone AL, Quinones-Hinojosa A, Searson PC. The role of astrocytes in the progression of brain cancer: complicating the picture of the tumor microenvironment. Tumour Biol. 2016;37:61–69.
Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, et al. Genomic analysis of reactive astrogliosis. J Neurosci. 2012;32:6391–410.
Chen W, Wang D, Du X, He Y, Chen S, Shao Q, et al. Glioma cells escaped from cytotoxicity of temozolomide and vincristine by communicating with human astrocytes. Med Oncol. 2015;32:43.
Sofroniew MV. Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci. 2015;16:249–63.
Campanella M, Ius T, Skrap M, Fadiga L. Alterations in fiber pathways reveal brain tumor typology: a diffusion tractography study. PeerJ. 2014;2:e497.
Venkatesh HS, Johung TB, Caretti V, Noll A, Tang Y, Nagaraja S, et al. Neuronal activity promotes glioma growth through neuroligin-3 Secretion. Cell. 2015;161:803–16.
Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193–215.
Gerhardt H, Betsholtz C. Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res. 2003;314:15–23.
Sims DE. The pericyte—a review. Tissue Cell. 1986;18:153–74.
Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–61.
Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature. 2010;468:562–6.
Hellstrom M, Kalen M, Lindahl P, Abramsson A, Betsholtz C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126:3047–55.
Hellstrom M, Gerhardt H, Kalen M, Li X, Eriksson U, Wolburg H, et al. Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J Cell Biol. 2001;153:543–53.
Lindahl P, Johansson BR, Leveen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242–5.
Lindblom P, Gerhardt H, Liebner S, Abramsson A, Enge M, Hellstrom M, et al. Endothelial PDGF-B retention is required for proper investment of pericytes in the microvessel wall. Genes Dev. 2003;17:1835–40.
Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. Angiogenesis in brain tumours. Nat Rev Neurosci. 2007;8:610–22.
Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med. 2013;19:1584–96.
Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–57.
Jhaveri N, Chen TC, Hofman FM. Tumor vasculature and glioma stem cells: contributions to glioma progression. Cancer Lett. 2016;380:545–51.
Zhu TS, Costello MA, Talsma CE, Flack CG, Crowley JG, Hamm LL, et al. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res. 2011;71:6061–72.
Hovinga KE, Shimizu F, Wang R, Panagiotakos G, Van Der Heijden M, Moayedpardazi H, et al. Inhibition of notch signaling in glioblastoma targets cancer stem cells via an endothelial cell intermediate. Stem Cells. 2010;28:1019–29.
Charles N, Holland EC. The perivascular niche microenvironment in brain tumor progression. Cell Cycle. 2010;9:3012–21.
Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015;29:1203–17.
Di Tomaso T, Mazzoleni S, Wang E, Sovena G, Clavenna D, Franzin A, et al. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin Cancer Res. 2010;16:800–13.
Wei J, Barr J, Kong LY, Wang Y, Wu A, Sharma AK, et al. Glioblastoma cancer-initiating cells inhibit T-cell proliferation and effector responses by the signal transducers and activators of transcription 3 pathway. Mol Cancer Ther. 2010;9:67–78.
Wei J, Wu A, Kong LY, Wang Y, Fuller G, Fokt I, et al. Hypoxia potentiates glioma-mediated immunosuppression. PLoS ONE. 2011;6:e16195.
Wu A, Wei J, Kong LY, Wang Y, Priebe W, Qiao W, et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010;12:1113–25.
Alizadeh D, Zhang L, Brown CE, Farrukh O, Jensen MC, Badie B. Induction of anti-glioma natural killer cell response following multiple low-dose intracerebral CpG therapy. Clin Cancer Res. 2010;16:3399–408.
Brown CE, Starr R, Martinez C, Aguilar B, D’Apuzzo M, Todorov I, et al. Recognition and killing of brain tumor stem-like initiating cells by CD8+ cytolytic T cells. Cancer Res. 2009;69:8886–93.
Castriconi R, Daga A, Dondero A, Zona G, Poliani PL, Melotti A, et al. NK cells recognize and kill human glioblastoma cells with stem cell-like properties. J Immunol. 2009;182:3530–9.
Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol. 2015;17:170–82.
Ruoslahti E. Brain extracellular matrix. Glycobiology. 1996;6:489–92.
Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348:74–80.
Huang JY, Cheng YJ, Lin YP, Lin HC, Su CC, Juliano R, et al. Extracellular matrix of glioblastoma inhibits polarization and transmigration of T cells: the role of tenascin-C in immune suppression. J Immunol. 2010;185:1450–9.
Gieryng A, Pszczolkowska D, Walentynowicz KA, Rajan WD, Kaminska B. Immune microenvironment of gliomas. Lab Invest 2017;97:498–518.
Graeber MB, Scheithauer BW, Kreutzberg GW. Microglia in brain tumors. Glia. 2002;40:252–9.
Bowman RL, Klemm F, Akkari L, Pyonteck SM, Sevenich L, Quail DF, et al. Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Rep. 2016;17:2445–59.
Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci. 2016;19:20–27.
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–5.
Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518:547–51.
Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10:1538–43.
Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82:380–97.
Goldmann T, Wieghofer P, Jordao MJ, Prutek F, Hagemeyer N, Frenzel K, et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol. 2016;17:797–805.
Kabba JA, Xu Y, Christian H, Ruan W, Chenai K, Xiang Y et al. Microglia: housekeeper of the central nervous system. Cell Mol Neurobiol 2017;22.
Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91:461–553.
Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol 2011;11:762–74.
Weiss T, Weller M, Roth P. Immunological effects of chemotherapy and radiotherapy against brain tumors. Expert Rev Anticancer Ther. 2016;16:1087–94.
Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20.
Murray PJ, Wynn TA. Obstacles and opportunities for understanding macrophage polarization. J Leukoc Bio. 2011;89:557–63.
Gabrusiewicz K, Rodriguez B, Wei J, Hashimoto Y, Healy LM, Maiti SN et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight. 2016;1(2) 1–19
Kaminska B, Mota M, Pizzi M. Signal transduction and epigenetic mechanisms in the control of microglia activation during neuroinflammation. Biochim Biophys Acta 2016;1862:339–51.
Walker DG, Lue LF. Immune phenotypes of microglia in human neurodegenerative disease: challenges to detecting microglial polarization in human brains. Alzheimers Res Ther. 2015;7:56.
Hussain SF, Yang D, Suki D, Aldape K, Grimm E, Heimberger AB. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol. 2006;8:261–79.
Badie B, Schartner JM. Flow cytometric characterization of tumor-associated macrophages in experimental gliomas. Neurosurgery. 2000;46:957–61, discussion 961–52
Ford AL, Goodsall AL, Hickey WF, Sedgwick JD. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J Immunol. 1995;154:4309–21.
Gabrusiewicz K, Ellert-Miklaszewska A, Lipko M, Sielska M, Frankowska M, Kaminska B. Characteristics of the alternative phenotype of microglia/macrophages and its modulation in experimental gliomas. PLoS ONE. 2011;6:e23902.
Sedgwick JD, Schwender S, Imrich H, Dorries R, Butcher GW, ter Meulen V. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc Natl Acad Sci USA. 1991;88:7438–42.
Sedgwick JD, Schwender S, Gregersen R, Dorries R, ter Meulen V. Resident macrophages (ramified microglia) of the adult brown Norway rat central nervous system are constitutively major histocompatibility complex class II positive. J Exp Med. 1993;177:1145–52.
Geranmayeh F, Scheithauer BW, Spitzer C, Meyer FB, Svensson-Engwall AC, Graeber MB. Microglia in gemistocytic astrocytomas. Neurosurgery. 2007;60:159–66, discussion 166
Mieczkowski J, Kocyk M, Nauman P, Gabrusiewicz K, Sielska M, Przanowski P, et al. Down-regulation of IKKbeta expression in glioma-infiltrating microglia/macrophages is associated with defective inflammatory/immune gene responses in glioblastoma. Oncotarget. 2015;6:33077–90.
Morris CS, Esiri MM. Immunocytochemical study of macrophages and microglial cells and extracellular matrix components in human CNS disease. 1. Gliomas. J Neurol Sci. 1991;101:47–58.
Wierzba-Bobrowicz T, Kuchna I, Matyja E. Reaction of microglial cells in human astrocytomas (preliminary report). Folia Neuropathol. 1994;32:251–2.
Vauleon E, Tony A, Hamlat A, Etcheverry A, Chiforeanu DC, Menei P, et al. Immune genes are associated with human glioblastoma pathology and patient survival. BMC Med Genomics. 2012;5:41.
Hussain SF, Yang D, Suki D, Grimm E, Heimberger AB. Innate immune functions of microglia isolated from human glioma patients. J Transl Med. 2006;4:15.
da Fonseca AC, Badie B. Microglia and macrophages in malignant gliomas: recent discoveries and implications for promising therapies. Clin Dev Immunol. 2013;2013:264124.
Galarneau H, Villeneuve J, Gowing G, Julien JP, Vallieres L. Increased glioma growth in mice depleted of macrophages. Cancer Res. 2007;67:8874–81.
Nagai T, Tanaka M, Tsuneyoshi Y, Xu B, Michie SA, Hasui K, et al. Targeting tumor-associated macrophages in an experimental glioma model with a recombinant immunotoxin to folate receptor beta. Cancer Immunol Immunother. 2009;58:1577–86.
Zhai H, Heppner FL, Tsirka SE. Microglia/macrophages promote glioma progression. Glia. 2011;59:472–85.
Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19:1264–72.
Quail DF, Bowman RL, Akkari L, Quick ML, Schuhmacher AJ, Huse JT, et al. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science. 2016;352:aad3018.
Coniglio SJ, Eugenin E, Dobrenis K, Stanley ER, West BL, Symons MH, et al. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol Med. 2012;18:519–27.
Szulzewsky F, Pelz A, Feng X, Synowitz M, Markovic D, Langmann T, et al. Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS ONE. 2015;10:e0116644.
Yan D, Kowal J, Akkari L, Schuhmacher AJ, Huse JT, West BL, et al. Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene. 2017;36:6049–58.
Butowski N, Colman H, De Groot JF, Omuro AM, Nayak L, Wen PY, et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an ivy foundation early phase clinical trials consortium phase II study. Neuro Oncol. 2016;18:557–64.
Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest. 2015;125:3356–64.
Gielen PR, Schulte BM, Kers-Rebel ED, Verrijp K, Petersen-Baltussen HM, ter Laan M, et al. Increase in both CD14-positive and CD15-positive myeloid-derived suppressor cell subpopulations in the blood of patients with glioma but predominance of CD15-positive myeloid-derived suppressor cells in glioma tissue. J Neuropathol Exp Neurol. 2015;74:390–400.
Raychaudhuri B, Rayman P, Ireland J, Ko J, Rini B, Borden EC, et al. Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro Oncol. 2011;13:591–9.
Alexiou GA, Vartholomatos G, Karamoutsios A, Batistatou A, Kyritsis AP, Voulgaris S. Circulating progenitor cells: a comparison of patients with glioblastoma or meningioma. Acta Neurol Belg. 2013;113:7–11.
Fecci PE, Mitchell DA, Whitesides JF, Xie W, Friedman AH, Archer GE, et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006;66:3294–302.
Lohr J, Ratliff T, Huppertz A, Ge Y, Dictus C, Ahmadi R, et al. Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-beta. Clin Cancer Res. 2011;17:4296–308.
Wainwright DA, Dey M, Chang A, Lesniak MS. Targeting Tregs in malignant brain cancer: overcoming IDO. Front Immunol. 2013;4:116.
Heimberger AB, Abou-Ghazal M, Reina-Ortiz C, Yang DS, Sun W, Qiao W, et al. Incidence and prognostic impact of FoxP3+ regulatory T cells in human gliomas. Clin Cancer Res. 2008;14:5166–72.
Perng P, Lim M. Immunosuppressive mechanisms of malignant gliomas: parallels at non-CNS sites. Front Oncol. 2015;5:153.
Kmiecik J, Poli A, Brons NH, Waha A, Eide GE, Enger PO, et al. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J Neuroimmunol. 2013;264:71–83.
Jacobs JF, Idema AJ, Bol KF, Grotenhuis JA, de Vries IJ, Wesseling P, et al. Prognostic significance and mechanism of Treg infiltration in human brain tumors. J Neuroimmunol. 2010;225:195–9.
Wainwright DA, Sengupta S, Han Y, Lesniak MS. Thymus-derived rather than tumor-induced regulatory T cells predominate in brain tumors. Neuro Oncol. 2011;13:1308–23.
Leung W. Infusions of allogeneic natural killer cells as cancer therapy. Clin Cancer Res. 2014;20:3390–3400.
Wiendl H, Mitsdoerffer M, Hofmeister V, Wischhusen J, Bornemann A, Meyermann R, et al. A functional role of HLA-G expression in human gliomas: an alternative strategy of immune escape. J Immunol. 2002;168:4772–80.
Fadul CE, Fisher JL, Gui J, Hampton TH, Cote AL, Ernstoff MS. Immune modulation effects of concomitant temozolomide and radiation therapy on peripheral blood mononuclear cells in patients with glioblastoma multiforme. Neuro Oncol. 2011;13:393–400.
Poli A, Wang J, Domingues O, Planaguma J, Yan T, Rygh CB, et al. Targeting glioblastoma with NK cells and mAb against NG2/CSPG4 prolongs animal survival. Oncotarget. 2013;4:1527–46.
Zhang C, Burger MC, Jennewein L, Genssler S, Schonfeld K, Zeiner P et al. ErbB2/HER2-specific NK cells for targeted therapy of glioblastoma. J Natl Cancer Inst. 2016;108(5);1–12
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352:987–96.
Curtin JF, Liu N, Candolfi M, Xiong W, Assi H, Yagiz K, et al. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med. 2009;6:e10.
Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. 2013;86:343–9.
Mathios D, Kim JE, Mangraviti A, Phallen J, Park CK, Jackson CM, et al. Anti-PD-1 antitumor immunity is enhanced by local and abrogated by systemic chemotherapy in GBM. Sci Transl Med. 2016;8:370ra180.
Pallud J, Audureau E, Noel G, Corns R, Lechapt-Zalcman E, Duntze J, et al. Long-term results of carmustine wafer implantation for newly diagnosed glioblastomas: a controlled propensity-matched analysis of a French Multicenter Cohort. Neuro Oncol. 2015;17:1609–19.
Kim TG, Kim CH, Park JS, Park SD, Kim CK, Chung DS, et al. Immunological factors relating to the antitumor effect of temozolomide chemoimmunotherapy in a murine glioma model. Clin Vaccine Immunol. 2010;17:143–53.
Ardon H, Van Gool S, Lopes IS, Maes W, Sciot R, Wilms G, et al. Integration of autologous dendritic cell-based immunotherapy in the primary treatment for patients with newly diagnosed glioblastoma multiforme: a pilot study. J Neurooncol. 2010;99:261–72.
Fadul CE, Fisher JL, Hampton TH, Lallana EC, Li Z, Gui J, et al. Immune response in patients with newly diagnosed glioblastoma multiforme treated with intranodal autologous tumor lysate-dendritic cell vaccination after radiation chemotherapy. J Immunother. 2011;34:382–9.
Sampson JH, Aldape KD, Archer GE, Coan A, Desjardins A, Friedman AH, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro Oncol. 2011;13:324–33.
Wheeler CJ, Das A, Liu G, Yu JS, Black KL. Clinical responsiveness of glioblastoma multiforme to chemotherapy after vaccination. Clin Cancer Res. 2004;10:5316–26.
Litterman AJ, Dudek AZ, Largaespada DA. Alkylating chemotherapy may exert a uniquely deleterious effect upon neo-antigen-targeting anticancer vaccination. Oncoimmunology. 2013;2:e26294.
Bauer M, Goldstein M, Heylmann D, Kaina B. Human monocytes undergo excessive apoptosis following temozolomide activating the ATM/ATR pathway while dendritic cells and macrophages are resistant. PLoS ONE. 2012;7:e39956.
Ridolfi L, Petrini M, Granato AM, Gentilcore G, Simeone E, Ascierto PA, et al. Low-dose temozolomide before dendritic-cell vaccination reduces (specifically) CD4+CD25++Foxp3+regulatory T-cells in advanced melanoma patients. J Transl Med. 2013;11:135.
Banissi C, Ghiringhelli F, Chen L, Carpentier AF. Treg depletion with a low-dose metronomic temozolomide regimen in a rat glioma model. Cancer Immunol Immunother. 2009;58:1627–34.
Ellsworth S, Balmanoukian A, Kos F, Nirschl CJ, Nirschl TR, Grossman SA, et al. Sustained CD4+T cell-driven lymphopenia without a compensatory IL-7/IL-15 response among high-grade glioma patients treated with radiation and temozolomide. Oncoimmunology. 2014;3:e27357.
Mendez JS, Govindan A, Leong J, Gao F, Huang J, Campian JL. Association between treatment-related lymphopenia and overall survival in elderly patients with newly diagnosed glioblastoma. J Neurooncol. 2016;127:329–35.
Sampson JH, Schmittling RJ, Archer GE, Congdon KL, Nair SK, Reap EA, et al. A pilot study of IL-2Ralpha blockade during lymphopenia depletes regulatory T-cells and correlates with enhanced immunity in patients with glioblastoma. PLoS ONE. 2012;7:e31046.
Roth P, Happold C, Weller M. Corticosteroid use in neuro-oncology: an update. Neurooncol Pract. 2015;2:6–12.
Rutz HP. Effects of corticosteroid use on treatment of solid tumours. Lancet. 2002;360:1969–70.
Pitter KL, Tamagno I, Alikhanyan K, Hosni-Ahmed A, Pattwell SS, Donnola S, et al. Corticosteroids compromise survival in glioblastoma. Brain. 2016;139:1458–71.
Wong ET, Lok E, Gautam S, Swanson KD. Dexamethasone exerts profound immunologic interference on treatment efficacy for recurrent glioblastoma. Br J Cancer. 2015;113:232–41.
Schmidt S, Rainer J, Ploner C, Presul E, Riml S, Kofler R. Glucocorticoid-induced apoptosis and glucocorticoid resistance: molecular mechanisms and clinical relevance. Cell Death Differ. 2004;11:S45–55.
Wyllie AH. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature. 1980;284:555–6.
Xia M, Gasser J, Feige U. Dexamethasone enhances CTLA-4 expression during T cell activation. Cell Mol Life Sci. 1999;55:1649–56.
Xing K, Gu B, Zhang P, Wu X. Dexamethasone enhances programmed cell death 1 (PD-1) expression during T cell activation: an insight into the optimum application of glucocorticoids in anti-cancer therapy. BMC Immunol. 2015;16:39.
Ribas A, Shin DS, Zaretsky J, Frederiksen J, Cornish A, Avramis E, et al. PD-1 blockade expands intratumoral memory T cells. Cancer Immunol Res. 2016;4:194–203.
Varga G, Ehrchen J, Tsianakas A, Tenbrock K, Rattenholl A, Seeliger S, et al. Glucocorticoids induce an activated, anti-inflammatory monocyte subset in mice that resembles myeloid-derived suppressor cells. J Leukoc Biol. 2008;84:644–50.
Hou Y, Feng Q, Xu M, Li GS, Liu XN, Sheng Z, et al. High-dose dexamethasone corrects impaired myeloid-derived suppressor cell function via Ets1 in immune thrombocytopenia. Blood. 2016;127:1587–97.
Ling Y, Cao X, Yu Z, Ruan C. Circulating dendritic cells subsets and CD4+Foxp3+ regulatory T cells in adult patients with chronic ITP before and after treatment with high-dose dexamethasome. Eur J Haematol. 2007;79:310–6.
Fleurence J, Cochonneau D, Fougeray S, Oliver L, Geraldo F, Terme M, et al. Targeting and killing glioblastoma with monoclonal antibody to O-acetyl GD2 ganglioside. Oncotarget. 2016;7:41172–85.
Mineo JF, Bordron A, Quintin-Roue I, Loisel S, Ster KL, Buhe V, et al. Recombinant humanised anti-HER2/neu antibody (Herceptin) induces cellular death of glioblastomas. Br J Cancer. 2004;91:1195–9.
Paff M, Alexandru-Abrams D, Hsu FP, Bota DA. The evolution of the EGFRvIII (rindopepimut) immunotherapy for glioblastoma multiforme patients. Hum Vaccin Immunother. 2014;10:3322–31.
Maxwell R, Luksik AS, Garzon-Muvdi T, Lim M. The potential of cellular- and viral-based immunotherapies for malignant glioma-dendritic cell vaccines, adoptive cell transfer, and oncolytic viruses. Curr Neurol Neurosci Rep. 2017;17:50.
Danaila L, Ghyka G, Ursaciuc C. Interleukin-2 (IL-2) in the treatment of malignant brain tumors (glioblastomas). Rom J Neurol Psychiatry. 1993;31:195–206.
Hart DN, Fabre JW. Demonstration and characterization of Ia-positive dendritic cells in the interstitial connective tissues of rat heart and other tissues, but not brain. J Exp Med. 1981;154:347–61.
Hickey WF, Kimura H. Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science. 1988;239:290–2.
Lowe J, MacLennan KA, Powe DG, Pound JD, Palmer JB. Microglial cells in human brain have phenotypic characteristics related to possible function as dendritic antigen presenting cells. J Pathol. 1989;159:143–9.
Ulvestad E, Williams K, Bjerkvig R, Tiekotter K, Antel J, Matre R. Human microglial cells have phenotypic and functional characteristics in common with both macrophages and dendritic antigen-presenting cells. J Leukoc Biol. 1994;56:732–40.
Reardon DA, Mitchell DA. The development of dendritic cell vaccine-based immunotherapies for glioblastoma. Semin Immunopathol. 2017;39:225–39.
Prins RM, Soto H, Konkankit V, Odesa SK, Eskin A, Yong WH, et al. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin Cancer Res. 2011;17:1603–15.
Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–18.
Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33:540–9.
Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. 2016;375:2561–9.
Kakarla S, Gottschalk S. CAR T cells for solid tumors: armed and ready to go? Cancer J. 2014;20:151–5.
Priceman SJ, Forman SJ, Brown CE. Smart CARs engineered for cancer immunotherapy. Curr Opin Oncol. 2015;27:466–74.
Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10.
Patsoukis N, Weaver JD, Strauss L, Herbel C, Seth P, Boussiotis VA. immunometabolic regulations mediated by coinhibitory receptors and their impact on T cell immune responses. Front Immunol. 2017;8:330.
Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591–619.
Marko AJ, Miller RA, Kelman A, Frauwirth KA. Induction of glucose metabolism in stimulated T lymphocytes is regulated by mitogen-activated protein kinase signaling. PLoS ONE. 2010;5:e15425.
Rathmell JC, Fox CJ, Plas DR, Hammerman PS, Cinalli RM, Thompson CB. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol Cell Biol. 2003;23:7315–28.
Schultze SM, Hemmings BA, Niessen M, Tschopp O. PI3K/AKT, MAPK and AMPK signalling: protein kinases in glucose homeostasis. Expert Rev Mol Med. 2012;14:e1.
Bretscher PA. A two-step, two-signal model for the primary activation of precursor helper T cells. Proc Natl Acad Sci USA. 1999;96:185–90.
Lafferty KJ, Warren HS, Woolnough JA, Talmage DW. Immunological induction of T lymphocytes: role of antigen and the lymphocyte costimulator. Blood Cells. 1978;4:395–406.
Appleman LJ, Boussiotis VA. T cell anergy and costimulation. Immunol Rev. 2003;192:161–80.
Chambers CA, Allison JP. Co-stimulation in T cell responses. Curr Opin Immunol. 1997;9:396–404.
Collins AV, Brodie DW, Gilbert RJ, Iaboni A, Manso-Sancho R, Walse B, et al. The interaction properties of costimulatory molecules revisited. Immunity. 2002;17:201–10.
Baumeister SH, Freeman GJ, Dranoff G, Sharpe AH. Coinhibitory pathways in immunotherapy for cancer. Annu Rev Immunol. 2016;34:539–73.
Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–42.
Kamphorst AO, Ahmed R. Manipulating the PD-1 pathway to improve immunity. Curr Opin Immunol. 2013;25:381–8.
Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity. 2016;44:989–1004.
Schildberg FA, Klein SR, Freeman GJ, Sharpe AH. Coinhibitory pathways in the B7-CD28 ligand-receptor family. Immunity. 2016;44:955–72.
Ward-Kavanagh LK, Lin WW, Sedy JR, Ware CF. The TNF receptor superfamily in co-stimulating and co-inhibitory responses. Immunity. 2016;44:1005–19.
Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206:3015–29.
Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–5.
Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med. 2015;212:991–9.
Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523:337–41.
Jessen NA, Munk AS, Lundgaard I, Nedergaard M. The Glymphatic System: A Beginner’s Guide. Neurochem Res. 2015;40:2583–99.
Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–6.
Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, et al. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. 2013;1:32–42.
Reardon DA, Gokhale PC, Klein SR, Ligon KL, Rodig SJ, Ramkissoon SH, et al. Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model. Cancer Immunol Res. 2015;4:124–35
Belcaid Z, Phallen JA, Zeng J, See AP, Mathios D, Gottschalk C, et al. Focal radiation therapy combined with 4-1BB activation and CTLA-4 blockade yields long-term survival and a protective antigen-specific memory response in a murine glioma model. PLoS ONE. 2014;9:e101764.
Kim JE, Patel MA, Mangraviti A, Kim ES, Theodros D, Velarde E, et al. Combination Therapy with anti-PD-1, anti-TIM-3, and focal radiation results in regression of murine gliomas. Clin Cancer Res. 2017;23:124–36.
Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800.
Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13:84–88.
Wintterle S, Schreiner B, Mitsdoerffer M, Schneider D, Chen L, Meyermann R, et al. Expression of the B7-related molecule B7-H1 by glioma cells: a potential mechanism of immune paralysis. Cancer Res. 2003;63:7462–7.
Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015;520:373–7.
Yeo AT, Charest A. Immune checkpoint blockade biology in mouse models of glioblastoma. J Cell Biochem. 2017;118:2516–27.
Maes W, Van Gool SW. Experimental immunotherapy for malignant glioma: lessons from two decades of research in the GL261 model. Cancer Immunol Immunother. 2011;60:153–60.
Oh T, Fakurnejad S, Sayegh ET, Clark AJ, Ivan ME, Sun MZ, et al. Immunocompetent murine models for the study of glioblastoma immunotherapy. J Transl Med. 2014;12:107.
Szatmari T, Lumniczky K, Desaknai S, Trajcevski S, Hidvegi EJ, Hamada H, et al. Detailed characterization of the mouse glioma 261 tumor model for experimental glioblastoma therapy. Cancer Sci. 2006;97:546–53.
LeBlanc AK, Mazcko C, Brown DE, Koehler JW, Miller AD, Miller CR, et al. Creation of an NCI comparative brain tumor consortium: informing the translation of new knowledge from canine to human brain tumor patients. Neuro Oncol. 2016;18:1209–18.
Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M, et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol. 2016;34:2206–11.
Hodges TR, Ott M, Xiu J, Gatalica Z, Swensen J, Zhou S, et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro Oncol. 2017;19:1047–57.
Johanns TM, Miller CA, Dorward IG, Tsien C, Chang E, Perry A, et al. Immunogenomics of hypermutated glioblastoma: a patient with germline POLE deficiency treated with checkpoint blockade immunotherapy. Cancer Discov. 2016;6:1230–6.
Acknowledgements
This work was supported by NIH/NCI grants CA183605, CA183605S1, and CA212605 and the DoD grant PC140571 (to V.A.B.) and NIH/NCI grants CA185137, CA179563, and CA069246 (to A.C.)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Vassiliki A. Boussiotis has patents on the PD-1 pathway licensed by Bristol-Myers Squibb, Roche, Merck, EMD-Serono, Boehringer Ingelheim, AstraZeneca, Novartis, and Dako. The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Boussiotis, V.A., Charest, A. Immunotherapies for malignant glioma. Oncogene 37, 1121–1141 (2018). https://doi.org/10.1038/s41388-017-0024-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-017-0024-z
- Springer Nature Limited
This article is cited by
-
Pan-cancer multi-omics analysis of PTBP1 reveals it as an inflammatory, progressive and prognostic marker in glioma
Scientific Reports (2024)
-
Advanced immunotherapies for glioblastoma: tumor neoantigen vaccines in combination with immunomodulators
Acta Neuropathologica Communications (2023)
-
PSRC1 Regulated by DNA Methylation Is a Novel Target for LGG Immunotherapy
Journal of Molecular Neuroscience (2023)
-
Single-cell RNA sequencing reveals evolution of immune landscape during glioblastoma progression
Nature Immunology (2022)
-
The complex role of tumor-infiltrating macrophages
Nature Immunology (2022)