
Abstract
Tumour microenvironment (TME), which consists of widely diverse immune and stromal cells and the factors that they secrete, cultivates a chronic inflammatory, immunosuppressive, and pro-angiogenic intratumoural atmosphere, which has been reported to correlate with patient outcomes and treatment efficacy. In this study, we characterized TME pattern through the “Estimation of STromal and Immune cells in MAlignant Tumours using Expression data” (ESTIMATE) algorithm and build a TME-related signature (TMERS), which is serving as an independent prognostic factor in MIBC. Moreover, we found that the TMERS was highly positive correlated with immune infiltration, the expression of immune checkpoints and high malignancy molecular subtypes such as basal, infiltrated and basal/SCC-like. The value of the TMERS in assessing the immunotherapy response was evaluated using the tumour immune dysfunction and exclusion (TIDE) algorithm and confirmed in several cohorts treated with immune checkpoint inhibitors (ICIs). Furthermore, the TMERS had a negative correlation with the tumour mutation burden (TMB), which is a potential predictive biomarker of immunotherapy response. Remarkably, combining TMERS and TMB was more effective for survival and ICI response prediction. In conclusion, we established a novel TMERS which depicts the TME pattern and acts as a robust independent prognostic factor and predictive biomarker for the response to ICIs when combined with the TMB.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bladder cancer (BLCA) is a urinary tract malignancy with a high incidence and is the 10th most common cancer, with nearly 549,000 new cases and 200,000 deaths in 2018 worldwide [1]. Notably, BLCA was one of the first cancers identified to be immunogenic when Alvaro Morales reported successful intravesicular administration of BCG in 9 patients to prevent recurrence [2]. BCG was the second immunotherapeutic agent approved by the United States Food and Drug Administration (FDA) after interferon-alpha (IFN-α) for its efficacy in the treatment of non-muscle-invasive bladder cancer (NMIBC), especially contaminated with carcinoma in situ (CIS). However, most patients do not respond to BCG as a frontline immunotherapeutic agent for NMIBC, and ~ 50% of responders relapse. Moreover, non-responders will finally recur or even invade into the muscle membrane and progressed to MIBC, which make the therapy of BLCA more complex. MIBC, which do not have a favourable prognosis with a five-year survival rate < 50%, are more prone to metastasize and need systemic therapy combined with radical surgery and chemotherapy [3, 4]. Even with the rapid development of clinical imaging, chemotherapy and surgery, the treatment outcome of MIBC is not that satisfactory based on the current staging and grade system. However, dozens of studies focusing on BCG have illustrated the oncogenic immune biology of BLCA to some extent and pushed us to find new strategy for MIBC.
Recently, many teams focused on investigating molecular classification of BLCA and established many molecular subtypes based on transcriptomic or mutational landscape, which indicated BLCA is a far more complex disease than previously recognized [5]. So far, five classification systems have been widely accept and described. Lund group first defined five major subtypes: UroA, genomically unstable (GU), infiltrated, UroB and basal/SCC-like based on expression of cell-cycle genes, receptor tyrosine kinases as well as FGFR3, RB1 and TP53 mutation frequency [6]. Then UNC group have divided MIBC into basal-like and luminal subtypes by characterizing differential stages of urothelial differentiation [7]. Furthermore, MDA group found another p53-like, which was consistently resistant to neoadjuvant MDAC chemotherapy and distinct from basal-like and luminal subtypes [8]. Moreover, MDA group made a comprehensive analysis of mRNA data in TCGA-BLCA cohorts and established another classification system called TCGA mRNA clusters including basal squamous, luminal, luminal infiltrated, luminal papillary, and neuronal. TCGA group produced a four-class system with groups referred to I, II, III and IV [9]. Every subtype established by different groups displayed a unique characteristic and overlap between each other. But they do not consider the tumour microenvironment (TME) pattern when establishing the classification system.
The last few decades have witnessed a revolution in cancer treatment, with a transition away from chemotherapy and radiation targeting tumours broadly towards antibody-based immunotherapies that modulate immune responses against tumours more precisely. The human immune system can efficiently distinguish and eradicate abnormal tumour cells when it is intact and undisturbed. However, tumour cells can hijack a number of potent negative regulators to attenuate immune surveillance and survive [10, 11]. Immune checkpoint inhibitors (ICIs) represent the first generation of antibody-based immunotherapy. ICIs play non-redundant roles by blocking the receptor and/or ligand interactions of molecules, such as cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and the programmed death-1 (PD-1)/programmed death-ligand-1 (PD-L1) pathway, which are involved in priming T cell activation or function and rescuing immune surveillance [12]. Some clinical trials based on therapeutic antibodies blocking the PD-1/PD-L1 pathway, such as CheckMate, KEYNOTE and IMvigor, indicated robust and durable responses in patients with various cancers [13, 14], including advanced and metastatic MIBC [15]. However, only a minority of patients with advanced cancer benefit from ICIs, with a response rate only modestly above the historical 10% response rate to traditional chemotherapies.
Tremendous efforts have been dedicated to identify predictive biomarkers for immunotherapy. PD-L1 immunohistochemistry (IHC)-based tissue testing might be the most widely accepted method. Although many studies reported higher response rates in patients with stronger PD-L1 expression, quantification of the cut-off level, which is very complex, has limited the utility of this indicator. With the popularity of high-throughput sequencing, we can easily assess the genetic features of individuals. Tumour genetic mutation, a usual genetic disorder, could trigger dysfunction of some proteins or peptides, inducing the formation of neo-antigens, which can be recognized by immune cells and initiate immune-mediated tumour cells clearance. Thus, recent studies have shown the potential of the tumour mutation burden (TMB) as a predictive biomarker for immunotherapy due to its reflection of the overall neo-antigens load [16, 17]. More multi-centre clinical trials based on the TMB should be performed to determine its clinical utility.
Despite the research focus on genetic and epigenetic alterations in cancer cells that drive malignancy, TME has been increasingly accepted to play an integral and indispensable role in tumour anatomy and physiology. Recognizing the nature of TME in the evolution of cancer has changed our understanding of cancer development from a cancer cell-centric perspective to a view considering a more complex tumour ecosystem that supports tumour growth and metastasis [18]. Instead of working alone, cancer cells construct their specific TME by interacting closely with the extracellular matrix (ECM) and stromal cells [19]. A wide variety of immune and non-immune cells within the TME infrastructure, together with the factors that they secrete, cooperate to create a chronic inflammatory, immunosuppressive, and pro-tumoural environment [20]. Cancer cells can adapt and grow in such environments to disguise themselves as normal cells to escape detection and eradication by host immune surveillance. Many studies have indicated that the TME composition has a strong correlation with immune response and chemotherapy benefit. Changes in the abundance of immune cells infiltrating TME, such as CD8 + T cells, dendritic cells (DCs), tumour-associated macrophages (TAMs), and regulatory T cells (Tregs), can influence the clinical outcomes of various malignancies, including melanoma, lung cancer, breast cancer and MIBC. Accordingly, the immune cells infiltrating TME constitute a vital element of tumour tissue. A growing body of evidence clarifies the clinicopathological significance of TME infiltrates in predicting prognoses and therapeutic effects. Nonetheless, no studies have reported a comprehensive transcriptome analysis of the TME in MIBC and investigated its role in predicting the response to immunotherapy.
In the present study, we integrated the genomic information of several cohorts to systematically analysed TME pattern and established a TME-related signature (TMERS), which was significantly associated with cellular, genomic, molecular, and clinicopathological characteristics of MIBC. As a result, we fortunately found that the TMERS was a robust independent prognostic factor and predictive biomarker for the response to ICIs in MIBC when combined with the TMB.
Materials and methods
Data collection and processing
Publicly available transcriptomic cohorts for BLCA with full clinical annotations were systematically searched. Samples without complete prognosis information were removed from further evaluation. In total, three microarray cohorts, GSE13507, GSE32548, GSE32894 and one RNA-sequencing (RNA-seq) cohort, TCGA-BLCA, were enrolled in our study. All raw data and clinical information from microarray cohorts were downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). Then, the raw data were processed via RMA background correction, log2 transformation and normalization by the package “Affy” in R [21]. The TCGA-BLCA pancancer normalized data and clinical information were downloaded from UCSC Xena (https://tcga.xenahubs.net) or Supplementary table from Robertson et al. [22]. Detailed information for patient clinicopathological characteristics in each cohort can be found in our previous study [23] and Supplementary Table 1–4. The ENSEMBL ID was annotated with the highest expression as the gene symbol expression in RNA-seq data. And the probes were annotated with the highest expression as the gene symbol expression in microarray data. The TCGA-BLCA somatic mutation data, which were identified using MuTect2, were obtained from UCSC Xena (https://tcga.xenahubs.net). The called somatic variants determined by TCGA were utilized as the raw mutation count. Mutation status was analysed and visualized by the packages “maftools” [24] and “complexheatmap” [25] in R. Then, the TMB per megabase of each sample was calculated as the total number of mutations counted in the exome territory. An estimate of the exome size was 38 Mb according to a previous study [26]. Data were analysed with R (version 3.5.2) and Bioconductor packages.
DEGs associated with the TME pattern
The TCGA-BLCA cohort was utilized as a training cohort to identify TME patterns in MIBC. Immune scores, stromal scores and ESTIMATE scores were calculated for each sample by applying the ESTIMATE algorithm [27]. To clarify the genes correlated with TME patterns, we divided the patients into two groups based on immune scores and stromal scores at the median cut-off. DEGs between the immune scores and stromal scores were determined using the package “limma” in R [28], which estimates gene expression changes by implementing an empirical Bayesian approach. The significance criteria for DEGs were set as an adjusted p value < 0.05 and |logFC| > 1.5. Finally, all upregulated and downregulated DEGs were merged into a single collection of TME DEGs.
Establishment and validation of the prognostic TMERS
Kaplan–Meier (KM) survival analyses were used to screen out prognostic related candidate genes among the TME DEGs. Then, LASSO Cox regression analysis based on the package “glmnet” in R was applied to build an optimal TME prognostic signature for MIBC by using the candidate genes mentioned above [29]. The Cox regression model with LASSO was used for dimension reduction to reduce noise or redundant genes. The optimal values of the penalty parameter λ were determined through 10 cross-validations. The TMERSscore of our model for each sample was defined by the expression of each TME-related gene and its associated Cox coefficient. The TMERSscore = \(\sum_{i = 1}^{n}\) (coefi × Expri), where Expri is the expression of the gene in the signature for patient i and coefi is the LASSO Cox coefficient of gene i. According to the TMERS, we grouped all patients into low or high TMERS groups at the median cut-off in each cohort. To reflect the prediction accuracy and ability of the TMERS model, the area under the curve (AUC) for 1-year, 3-year and 5-year overall survival (OS) and disease-free survival (DFS) was measured through time-dependent receiver operating characteristic (ROC) curve analysis using the package “survivalROC” in R [30]. KM survival analyses were used to assess OS and DFS between the high or low TMERS groups and the indicated stratified clinicopathological characteristics.
Gene set variation analysis (GSVA) and functional annotation
GSVA, in a non-parametric and unsupervised method, is commonly employed for estimating the variation in pathway and biological process activity in the samples of an expression cohort. The gene set files of “h.all.v6.2.symbols” containing 50 critical gene sets were downloaded from the MSigDB of Broad Institute. To investigate the difference on biological process based on TMERS, the common activated/suppressed pathways between the high and low TMERS groups were identified with a cut-off of p < 0.05 and t value > 2 using the package “limma” and “GSVA” in R [31]. We also performed GSEA analyses on the above hallmark gene sets via javaGSEA to show the common GSEA plot.
Construction and validation of a predictive nomogram
The TMERS, TMB and other clinicopathological characteristics were subjected to univariate and multivariate cox regression analyses to identify independent prognostic factors, and the results were visualized with the package “forestplot” in R. Then, the identified independent prognostic factors were utilized to establish a nomogram with the packages “rms”, “nomogramEx” and “regplot” in R[32]. Next, calibration curves and decision curve analysis (DCA) were used to determine whether our established nomogram was suitable for clinical use.
Evaluation of infiltrating immune cells in the TME
We used the single-sample gene set enrichment analysis (ssGSEA) algorithm to evaluate the relative abundance of infiltration immune cells in MIBC TME. The marker gene set for TME infiltration immune cell type was obtained from Bindea et al. [33]. Both innate immune cells (DCs, immature DCs [iDCs], activated DCs [aDCs], plasmacytoid DCs [pDCs], eosinophils, mast cells, macrophages, natural killer cells [NKs], NK CD56dim cells, NK CD56bright cells, and neutrophils) and adaptive immune cells (B cells, T cells, T helper cells, T helper 1 [Th1], Th2, T gamma delta [Tγδ], CD8 + T, T central memory [Tcm], T effector memory [Tem], T follicular helper [Tfh] cells, T helper 17 (Th17) cells, regulatory T (Treg) cells and cytotoxic cells) were investigated. The enrichment scores calculated by ssGSEA were used to represent the relative abundance of each TME infiltrating cell in MIBC.
Evaluation of the immune checkpoint inhibitors (ICIs) response
The tumour immune dysfunction and exclusion (TIDE) algorithm was used to evaluate the predictive efficiency of TMERS and/or TMB for the ICIs response in MIBC [34]. Moreover, four independent cohorts containing clinical information related with immunotherapy were considered to validate our model: the IMvigor210 (mUC) trial of patients with metastatic urothelial cancer (mUC) receiving PD-L1 blockade with atezolizumab (transcriptomic and clinical data were retrieved using the package “IMvigor” in R) [35]; The detailed clinical information of IMvigor210 (mUC) cohort could be found in Supplementary Table 5; the GSE78220 cohort of patients with metastatic melanoma receiving PD-1 blockade with pembrolizumab or nivolumab [36]; the GSE35640 cohort of patients with metastatic melanoma receiving MAGE-A3 antigen-based immunotherapy [37]; and the GSE117358 cohort of mice receiving antibodies against CTLA-4 and PD-L1 [38].
Statistical analyses
The statistical significance for mean value of variables between two groups was estimated by unpaired Student’s t tests. In addition, for variables in more than two groups, one-way ANOVA test was used. The χ2 test was applied to analyse correlations between the TMERS and clinicopathological parameters. KM survival curves were generated to calculate survival advantage, and the significance of differences between survival curves was determined using the log-rank test by the package “survminer” in R. Correlation coefficients were computed using Spearman’s and distance correlation analyses. Two-sided Fisher’s exact tests were used to analyse contingency tables. Univariate and multivariate cox proportional hazard models were used to estimate the hazard ratios of variables and determine independent prognostic factors. A nomogram and calibration curves were generated, and DCA was performed according to Iasonos’ suggestion [32]. Time-dependent ROC curves were performed to assess the predictive accuracy of the prognostic models. All statistical analyses were performed with R software 3.5.3. Statistical significance was set at p < 0.05.
Results
Estimation of tumour microenvironment (TME) patterns and identification of TME-related prognostic genes
A flow diagram and design of our study can be viewed in Supplementary Fig. 1. Immune scores, stromal scores and ESTIMATE scores, which were obtained via the ESTIMATE algorithm, were representative of components within the TME and were used to characterize the TME patterns in the TCGA-BLCA cohort. The detailed information of the scores in each sample was listed in Supplementary Table 6. Then, results showed that all the scores displayed a tendency for a positive correlation with tumour grade. Moreover, the stromal scores and ESTIMATE scores were significantly correlated with pathological TNM stage (Supplementary Fig. 2a–e). The KM curves demonstrated that patients with low stromal scores lived longer than patients with high stromal scores, but no significant differences in immune scores and ESTIMATE scores were observed (Supplementary Fig. 2f–h and Supplementary Table 6). To depict the TME patterns through transcriptomic data, DEGs of immune scores and stromal scores were screened and merged to serve as TME DEGs (Supplementary Fig. 2i–j and Supplementary Tables 7, 8). After KM survival analyses, 141 of 778 TME DEGs, which were related with prognosis, were finally identified and defined as TME-related prognostic candidate genes (Supplementary Table 9).
Establishment and validation of the TMERS
To develop a gene signature that could better represent the TME pattern, the survival-related TME DEGs were submitted to LASSO cox regression analysis for dimension reduction. Then, we established a TME-related signature (TMERS) consisting of 7 genes to predict OS in the TCGA-BLCA training cohort, and the formula was calculated as follows: expression of ALDH1L2 * 0.0404 + expression of CD109 * 0.0859 + expression of FER1L4 * (− 0.0008) + expression of FLRT2 * 0.0167 + expression of KANK4 * 0.0259 + expression of NCAM1 * 0.0690 + expression of TLL1 * 0.0065 (Supplementary Fig. 3 and Supplementary Table 10). The KM curves indicated that high TMERS group was significantly associated with poorer OS, while low TMERS group was associated with better OS (Log-rank test, p < 0.0001, Fig. 1a, b). To assess the predictive value of the TMERS, its performance was measured in some other independent cohorts, including 1 external cohort (GSE13507) for OS validation as well as 1 internal cohort (TCGA-BLCA) and 3 external cohorts (GSE13507, GSE32548 and GSE32894) for DFS validation. Consistent with the outcomes for OS prediction in TCGA-BLCA cohort, we found that the TMERS worked well in all validation cohorts and that high TMERS groups were associated with a poorer prognosis (GSE13507, Log-rank test, p = 0.018, Fig. 1c, d and TCGA-BLCA cohort, Log-rank test, p = 0.013, Supplementary Fig. 4a, b; GSE13507, Log-rank test, p = 0.00028, Supplementary Fig. 4d, e; GSE32548, Log-rank test, p = 0.00028, Supplementary Fig. 4 g, h; GSE32894, Log-rank test, p = 0.0035, Supplementary Fig. 4j, k). Moreover, time-dependent ROC curves showed a high accuracy of TMERS in prediction prognosis in MIBC (Supplementary Fig. 4c, f, i, l and Supplementary Fig. 5a, b).

TMERS is significantly associated with established molecular subtypes. a, b KM survival curves and risk score of OS based on TMERS in TCGA-BLCA training cohort. KM curves with Log-rank p value < 0.0001 showed a significant survival difference between low and high TMERS groups. The low TMERS group showed significantly better OS than high TMERS group. c, d KM survival curves and risk score of OS based on TMERS in GSE13507 cohort. KM curves with Log-rank p value = 0.018 showed a significant survival difference between low and high TMERS groups. The low TMERS group showed significantly better OS than high TMERS group. f The bar plots demonstrated the relationship between TMERS and molecular subtypes, as well as alteration in TP53, RB1, FGFR3, KDM6A and PPARγ. The correlation between TMERS and those factors was measured with the χ2 test. The asterisks represented the statistical p value (*P < 0.05; **P < 0.01; ***P < 0.001). f Alluvial diagram showing the changes of TMERS, indicated molecular subtypes and vital status in TCGA-BLCA cohort. g–i) Differences in TMERS between different molecular subtypes. The upper and lower ends of the boxes represented interquartile range of values. The lines in the boxes represented median value. One-way ANOVA tests were used to compare the statistical difference between MDA (g), TCGAcluster (h) and Lund (i) molecular classification systems. (p < 2.2e−16)
Correlations between TMERS and molecular classification as well as clinicopathological characteristics
As so many classification systems have been established, we next aimed to investigate their association with TMERS. TMERS was dichotomized at median cut-off into high or low TMERS groups. Surprisingly, we found that low TMERS group was concentrated on the subtypes of luminal, luminal papillary, TCGA I, II, uroA, and GU, which were all related with low malignancy and better survival. However, basal, basal squamous, TCGA IIII, IV, infiltrated and basal/SCC-like, characterized by high malignancy and worse prognosis, significantly accumulated in high TMERS group (Fig. 1e, f). Moreover, we also found that TMERSscore increased in the high malignancy subtypes and decreased in low malignancy subtypes, which was consistence with above findings (Student t tests or one-way ANOVA tests, p < 2.2e–16, Fig. 1g–i and Supplementary Fig. 6a–d). Then the correlations of TMERS with clinicopathological characteristics were further evaluated. In terms of clinical features, TMERSscore was markedly increased in patients who were elderly, as well as non-papillary, lymphovascular invasion, and more advanced stages and grades, which again revealed TMERS is indicator of MIBC malignancy (Supplementary Fig. 7a–i and Supplementary Table 11). The cluster heat map showed that the expression of the TME prognostic genes ALDH1L2, CD109, FLRT2, KANK4, NCAM1 and TLL1 was upregulated in high TMERS group, while the expression of FER1L4 was downregulated in high TMERS group. Immune scores, stromal scores and ESTIMATE scores were all strongly upregulated in high TMERS group compared to low TMERS group, indicating that TMERS can better represent the TME patterns in MIBC (Fig. 3a). As the TMERSscore was highly correlated with the above clinical features, we sought to clarify whether the TMERS had prognostic value independent of these clinicopathological characteristics. Stratified survival analyses showed that TMERSscore could efficiently predict the survival rate of patients in most of the subgroups (Supplementary Fig. 8a–r).
The TMERS can be utilized as an independent prognostic factor in MIBC
As the TMERS was significantly correlated with high malignancy and played a vital role in MIBC tumorigenesis, we sought to clarify whether our selective TMERS was a clinically independent prognostic factor for MIBC patients through univariate and multivariate cox regression analyses. The TMERS and TMB, together with other clinical features, including age, sex, histological subtype, grade, lymphovascular invasion, the number of positive lymph nodes by HE staining, pathological T stage, pathological N stage, pathological M stage and pathological tumour stage, were enrolled as covariates to perform the analysis. The results demonstrated that the TMERS, TMB, pathological N stage and age were the only four independent factors that could be utilized to predict the prognosis of MIBC patients (Fig. 2a, b and Supplementary Fig. 9). Combining the four prognostic factors, we constructed a nomogram that serves as a clinically relevant quantitative method by which clinicians can predict mortality in MIBC patients (Fig. 2c). Every patient is assigned a total point value by adding the point for each prognostic parameter. Higher total points correspond to a worse clinical outcome of patients. Furthermore, calibration curves indicated that the nomogram had similar performance to that of an ideal model (Fig. 2d, e). DCA also revealed that the nomogram had high potential clinical utility (Fig. 2f, g). Moreover, we also validated TMERS was still an independent factor when compiling with TMB in IMvigor210 (mUC) cohort (Supplementary Fig. 10a–c).

TMERS is an independent prognosis factor in the nomogram. a, b Forest plot summary of the univariate and multivariable cox analyses of the TMERS and clinicopathological characteristics. Four independent prognosis factors including age, pathologic N stage, TMB and TMERS were indicated. The blue diamond squares on the transverse lines represent the HR, and the black transverse lines represent the 95% CI. The p value and 95% CI for each clinical feature are displayed in detail. c Nomograms for predicting the probability of patient mortality at 3- or 5-year OS based on TMERS. d–e Calibration curves of the nomogram for predicting the probability of OS at 3-, and 5-years. f, g Decision curve analyses (DCA) curve of the nomogram based on TMERS for 3-years and 5-years OS prediction
The landscape of TME immune infiltration in MIBC
Due to the potential role of the TMERS in representing the TME, we next prepared to depict the TME immune landscape, which is one of the most important components within the TME in MIBC. Thus, the abundance of TME immune infiltration cells were calculated via ssGSEA according to the specific reference gene sets (Supplementary Table 12). We established an immune cell network characterizing a comprehensive landscape of TME immune cell interactions, cell clusters, and their prognostic effect on the OS of MIBC patients (Fig. 3b; Supplementary Tables 13, 14). Four clusters were identified within the immune cells. The results showed that Treg cells were strongly positively correlated with almost all other immune cells but negatively correlated with NK CD56bright cells. KM survival analyses demonstrated that the infiltration of certain innate immune cells (DCs, eosinophils, and NK CD56bright cells) and adaptive immune cells (T cells, CD8 + T cells, TFH cells, Th17 cells, and cytotoxic cells) was a favourable prognostic factor, while the infiltration of other innate immune cells (mast cells, macrophages, and NK CD56dim cells) had an unfavourable impact on the prognosis of MIBC patients (Supplementary Fig. 11). As the variations in immune infiltration allowed us to more clearly observe the intrinsic traits of individual differences, the relationship between the TMERS and immune infiltration was measured. We found that high TMERS patients had high levels of immune infiltration (Fig. 3a and Supplementary Table 11). Moreover, the TMERS, stromal scores, immune scores, and ESTIMATE scores were related to high infiltration of almost all immune cells except for NK CD56bright cells and TH17 cells (Fig. 3c, d).Then we performed GSVA to explore the biological behaviours based on TMERS. We found high TMERS group was remarkably enriched in angiogenesis, complement, epithelial-mesenchymal transition (EMT), inflammatory response, KRAS signalling up, which implied highly chronic inflammation. While, low TMERS group only presented enrichment in DNA repair pathway, which hinted the immune activation (Supplementary Fig. 12 and Supplementary Tables 15–18). Therefore, comprehensive consideration of function annotations supported the hypothesis that the TMERS could represent TME patterns and is associated with immune regulation in MIBC.

The landscape of TME immune cell infiltration and its correlation with TMERS. a Cluster heat map of TME-related genes, immune scores/stromal scores/ESTIMATE scores and abundance of 24 types of immune cells stratified by the TMERS in TCGA-BLCA cohort. Yellow indicates upregulated and blue indicates downregulated. The relationship between TMERS and each clinicopathological characteristics, which was divided into the indicated group, was measured with the χ2 test. The asterisks represented the statistical p value (*P < 0.05; **P < 0.01; ***P < 0.001). b Immune cells interaction within TME. Cell cluster-A, blue; Cell cluster-B, yellow; Cell cluster-C, red; Cell cluster-D, brown. The size of each cell represents the impact on OS of each immune cell type, calculated used the formula log10 (Log-rank test p value). The lines connecting immune cells were representative of cellular interactions. The thickness of the line indicates the degree of correlation estimated by Spearman correlation analysis. The red indicated positive correlation, while blue indicated negative correlation. c Correlation matrix of TMERS, TME-related genes, immune score/stromal score/ESTIMATE score and the abundance of 24 types of immune infiltration cell. The blue indicated positive correlation, while yellow indicated negative correlation. Shading colour and asterisks represents the value of corresponding correlation coefficients (*P < 0.05; **P < 0.01; ***P < 0.001). d Violin plots visualizing the correlation between TMERS and 24 types of immune infiltration cells. Within each group, the scattered dots represent TMERS in each patient
TMERS is highly negative correlated with TMB
As BLCA is a disease that features highly somatic alterations, we next determined the distributions of somatic mutations and whether a correlation existed between the TMERS and mutation load. The total mutation load and mutation distribution derived from the TCGA-BLCA cohort were obtained by analysing the mutect2 mutation annotation files (Supplementary Fig. 13). Then, we found that TMB was associated with good OS and patients with lower TMERS displayed higher TMB level (Log-rank test, p = 0.00031, Fig. 4a; Fisher’s exact test, p value = 0.005335, Fig. 4b). Moreover, a significant negative correlation was found between the TMERS and the TMB (spearman correlation analysis, p = 0.022, Fig. 4c; Student t test, p = 0.019, Fig. 4d).The top 30 highly mutated genes were selected to determine the mutation distribution based on the TMERS. We found that mutation, fusion and focal amplification in FGFR3, focal amplification in PPARγ as well as mutation in KDM6A, which is often altered in NMIBC and low malignancy molecular subtypes, such as luminal, GU, TCGA I and II, markedly increased in the low TMERS group and was rarely observed in the high TMERS group (Figs. 1e, 4e left). Moreover, we did see the mutation frequency of TP53 and RB1, which is always mutated and acted as driver in MIBC, was upregulated in high TMERS group compared with low TMERS group (Figs. 1e, 4e right). All of these again verified that the TMERS was positively correlated with tumour malignancy from a different viewpoint. Moreover, our analyses provided a new perspective for the formation of the TME and its relationship with tumour mutation, as well as its role in onco-immunology and immunotherapy. As patients with a high TMB robustly responded to ICIs targeting immunotherapy, we inferred that the TMERS may also be useful for predicting response benefits in MIBC patients treated with ICIs.

TMERS is significantly negative correlated with TMB. a KM survival curves for high and low TMB groups stratified at median cut-off in TCGA-BLCA cohorts (Log-rank test, p = 0.00031). b The proportion of patients with low/high TMB based on TMERS stratification (Fisher’s exact test, p value = 0.005335). c Scatter plots depicting the negative correlation between TMERS and TMB. The spearman correlation between TMERS and TMB was shown (p = 0.022). d Box plot showing the correlation of TMB with TMERS (Student t test, p = 0.015). e The waterfall plot of tumour somatic mutation displayed distribution of top 30 highly variant mutated genes correlated with TMERS. The mutational type include frame shift del, frame shift ins, in frame del, in frame ins, missense mutation, multi hit, nonsense mutation and splice site. Each column represented individual patients. The upper bar plots showed TMERSscore, TMB and OStime, The number on the left indicated the mutation frequency in each gene. The right bar plot showed the proportion of each variant type. The TMERS, TMB, vital status and Lund molecular subtypes were used as patient annotations
The TMERS was a predictive biomarker for immunotherapy
We next aimed to explore whether the TMERS could predict immunotherapeutic benefits in MIBC. The TIDE algorithm was utilized to calculate the immunotherapy response rates in the TCGA-BLCA cohort. Then, we found that high TMERSscore was associated with disease progression when receiving immunotherapy, while low TMERSscore was related with better outcome (Student t test, p < 2.2e–16, Fig. 5d). And the proportion of immunotherapy responders was significantly high among low TMERS patients compared with high TMERS patients (Fisher’s exact test, p value < 0.00001, Fig. 5m right and Supplementary Tables 19).

The TMERS and TMB alone were efficient in prediction the immunotherapy response. a Survival analyses for low (117 cases) and high (117 cases) TMERS groups in IMvigor210 (mUC) cohort using KM curves (Log-rank test, P = 0.0441). b Survival analyses for low (114 cases) and high (120 cases) TMB groups in IMvigor210 (mUC) cohort using KM curves (Log-rank test, P = 0.0016). c Alluvial diagram showing the changes of TMERS, indicated molecular subtypes, vital status, overall response and binary response in IMvigor210 (mUC) cohort. d, e Distribution of TMERSscore (Student t test, p < 2.2e−16, d) and TMB (Student t test, p = 0.11, e) in distinct immunotherapy clinical response groups in TCGA-BLCA cohort. f, g Distribution of TMERSscore (Student t test, p = 0.0485, f) and TMB (Student t test, p = 3.7e−7, g) in distinct immunotherapy clinical response groups in IMvigor210 (mUC) cohort. h–j Differences in TMERSscore between different molecular subtypes. The upper and lower ends of the boxes represented interquartile range of values. The lines in the boxes represented median value. One-way ANOVA test was used to compare the statistical difference between Lund1 (h), Lund2 (i) and TCGA (j) molecular classification system (p < 2.2e–16). k–i The proportion of Lund molecular subtypes in the low or high TMERS and TMB groups in TCGA-BLCA cohort (k) and IMvigor210 (mUC) cohort (l). UroA subtype, blue; UroB subtype, brown; Genomically unstable subtype, red; Infiltrated subtype, green; Basal/SCC-like subtype, yellow. The statistical difference was measured with the Fisher’s exact test. Differences of TMB between Lund molecular subtypes in TCGA-BLCA cohort (p = 0.184495) (k left); Differences of TMERS between Lund molecular subtypes in TCGA-BLCA cohort (p < 0.0001) (k right); Differences of TMB between Lund molecular subtypes in IMvigor210 (mUC) cohort (p = 0.000944) (l left); Differences of TMERS between Lund molecular subtypes in IMvigor210 (mUC) cohort (p < 0.0001) (l right). m, n The proportion of patients with response to immunotherapy in low or high TMERS and TMB groups in TCGA-BLCA cohort (m) and IMvigor210 (mUC) cohort (n). Responder or CR/PR, red; Non-responder or SD/PD, blue. The statistical difference was measured with the Fisher’s exact test. Differences of TMB between overall response in TCGA-BLCA cohort (p = 0.556367) (m left); Differences of TMERS between overall response in TCGA-BLCA cohort (p < 0.00001) (m right); Differences of TMB between overall response in IMvigor210 (mUC) cohort (p < 0.00001) (n left); Differences of TMERS between overall response in IMvigor210 (mUC) cohort (p = 0.011359) (n right)
Then, four independent transcriptomic cohorts with immunotherapy data were retrieved to validate the ability of the TMERS to predict immunotherapeutic benefits. The KM curve revealed that low TMERS patients had significantly longer PFS than high TMERS patients in the IMvigor210 (mUC) cohort (Log-rank test, p = 0.0441, Fig. 5a). We also found a survival benefit of low TMERS in the GSE78220 cohort (Log-rank test, p = 0.21, Supplementary Fig. 14a). Furthermore, we found that all the patients in the low TMERS group were more likely to be immunotherapy responders than those in the high TMERS group in the IMvigor210 (mUC) (Student t test, p = 0.0485, Fig. 5f; Fisher’s exact test, p = 0.011359, Fig. 5n right), as well as GSE78220, GSE35640, GSE117358 cohorts (GSE78220, left, Student t test, p = 0.055, right, Fisher’s exact test, p = 0.1201, Supplementary Fig. 14b; GSE35640, Student t test, p = 0.087, Supplementary Fig. 14c; GSE117358, Student t test, p = 9.3e−7, Supplementary Fig. 14d). Moreover, the ROC curve showed that the TMERS performed well in prediction of the ICIs response in the IMvigor210 (mUC) (Fig. 6k), GSE78220, GSE35640 and GSE117358 cohorts (Supplementary Fig. 14c, e, g). As all the patients in the IMvigor210 (mUC) cohort are suffering from metastatic urothelial carcinoma, we also measure the relationship between TMERS and molecular subtypes, the results were consistence with TCGA-BLCA cohort (Fig. 5c; one-way ANOVA test, p = 2.2e−16, Fig. 5h–j; Fisher’s exact test, p < 0.00001, Fig. 5l right and Supplementary Fig. 15a).

Combination of TMERS and TMB was an efficiency tool for immunotherapy response prediction. a The correlation chord chart showed the mutual correlation between TMERS and several prominent immune-checkpoint-relevant genes (CTLA-4, LAG-3, GAL9, PD-1, PD-1LG2, PD-L1, TIM-3 and TIGIT). b Violin plots indicated the significantly correlation between TMERSscore and immune-checkpoint-relevant genes. c The waterfall plot of tumour somatic mutation established by stratified with TMERS and TMB. Each column represented individual patient. Group1, TMBhigh/TMERSlow, blue; Group2, TMBlow/TMERSlow, red; Group3, TMBlow/TMERShigh, yellow; Group4, TMBhigh/TMERShigh, green. The mutational type include frame shift del, frame shift ins, in frame del, in frame ins, missense mutation, multi hit, nonsense mutation and splice site. The upper bar plots showed TMERSscore, TMB and OStime. The Lund molecular subtypes and vital status were used as patient annotations. d Survival analyses for four groups stratified by combining TMERS with TMB in TCGA-BLCA cohort. Kaplan–Meier curves with Log-rank p value < 0.0001 showed a significant survival difference among different groups. The group1 showed significantly better overall survival than the other groups, especially to group3. e–f The proportion of patients with response to immunotherapy (e) or Lund molecular subtypes (f) in TCGA-BLCA cohort stratified by combining with TMB and TMERS. Responder, red; Non-responder, blue. UroA subtype, blue; UroB subtype, brown; Genomically unstable subtype, red; Infiltrated subtype, green; Basal/SCC-like subtype, yellow. The statistical difference was measured with the Fisher’s exact test. Differences of indicated groups between overall response in TCGA-BLCA cohort (p < 0.00001) (e); Differences of indicated groups between Lund molecular subtypes in TCGA-BLCA cohort (p < 0.00001) (f). g ROC curves measuring the predictive value of the TMERS, TMB, and combination of TMERS and TMB in the TCGA-BLCA cohort. h Survival analyses for four groups stratified by combining TMERS with TMB in IMvigor210 (mUC) cohort. Kaplan–Meier curves with Log-rank p value = 0.0069 showed a significant survival difference among different groups. The group1 showed significantly better overall survival than the other groups, especially to group3. (i-j) The proportion of patients with response to immunotherapy (i) or Lund molecular subtypes (j) in IMvigor210 (mUC) cohort stratified by combining with TMB and TMERS. CR/PR, red; SD/PD, blue. UroA subtype, blue; UroB subtype, brown; Genomically unstable subtype, red; Infiltrated subtype, green; Basal/SCC-like subtype, yellow. The statistical difference was measured with the Fisher’s exact test. Differences of indicated groups between overall response in IMvigor210 (mUC) cohort (p = 0.000015) (i); Differences of indicated groups between Lund molecular subtypes in IMvigor210 (mUC) cohort (p < 0.00001) (j). k ROC curves measuring the predictive value of the TMERS, TMB, and combination of TMERS and TMB in IMvigor210 (mUC) cohort
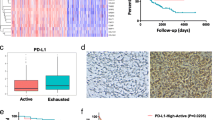
As mentioned above, the expression of immune checkpoints, such as PD-L1 on IHC examination, was used to predict immunotherapeutic benefits in multiple malignancies. Therefore, we selected CD274 (PD-L1), CTLA-4, LAG-3, LGALS9 (GAL9), HAVCR2 (TIM-3), PDCD1 (PD-1), PDCD1LG2 (PD-1LG2) and TIGIT as immune checkpoint-related candidate genes to assess their relationships with our TMERS. The results showed that the TMERSscore was strongly positive associated with the expression of critical immune checkpoints (CTLA-4, LAG-3, PD-1, PD-1LG2, PD-L1, TIM-3 and TIGIT) (Fig. 6a and Supplementary Table 20–22). In addition, we found that the above immune checkpoints were all upregulated in the high TMERS group, indicating that the cause of the poor prognosis of high TMERS patients might be due to the tumour immunosuppressive microenvironment (Fig. 6b and Supplementary Table 20–22).
Considering the potential ability of the TMERS to assess the immunotherapy response and their negative relationship, we investigated whether the combination of the TMERS and TMB, which represent nonsynonymous variants, could be a more powerful predictive biomarker for the ICIs response. We found that patients with high TMB lived significantly longer than patients with low TMB in the IMvigor210 (mUC) cohort, which was consistent with the results in the TCGA-BLCA cohort (Log-rank test, p = 0.0016, Fig. 5b). Furthermore, patients with high TMB were more likely to be ICIs responders in the IMvigor210 (mUC) cohort (Student t test, p = 3.7e–7, Fig. 5 g; Fisher’s exact test, p < 0.00001, Fig. 5n left). However, we could not determine the effect of TMB in the TCGA-BLCA cohort (Student t test, p = 0.11, Fig. 5e; Fisher’s exact test, p = 0.556367, Fig. 5m left). Moreover, IMvigor210 study demonstrated that patients within GU subtype in Lund classification system was more tend to be immunotherapy responders, while the effect of infiltrated subtype was completely opposite. Fortunately, we found that the proportion of GU subtype was significantly higher in low TMERS and high TMB group, while the infiltrated subtype displayed the opposite distribution (Fig. 5c; TCGA-BLCA, TMERS, Fisher’s exact test, p < 0.00001, TMB, Fisher’s exact test, p = 0.184495, Fig. 5k; IMvigor210 (mUC), TMERS, Fisher’s exact test, p < 0.00001, TMB, Fisher’s exact test, p = 0.000944, Fig. 5l left and Supplementary Fig. 15a).
Next, we integrated the TMERS and the TMB to stratify all the samples into high TMB/low TMERS (group 1), low TMB/low TMERS (group 2), low TMB/high TMERS (group 3), and high TMB/high TMERS (group 4) groups. The waterfall plots showed that group1 displayed highest mutation burden and characterized as high mutation in FGFR3 and KDM6A, as well as low mutation in RB1, while group3 demonstrated opposite distribution (Fig. 6c and Supplementary Fig. 15b, c). Moreover, group1 patients have the largest number of GU subtypes and lowest number of infiltrated subtype, and group3 displayed an opposite distribution in both TCGA and IMvigor210 (mUC) cohorts (TCGA-BLCA, IMvigor210 (mUC), Fisher’s exact test, p < 0.00001, Fig. 6f; Fisher’s exact test, p < 0.00001, Fig. 6j and Supplementary Fig. 15b-c). Furthermore, patients in group1 exhibited significantly clinical benefits and a markedly prolonged survival compared patients in group3 in both cohorts (TCGA-BLCA, Log-rank test, p < 0.0001, Fig. 6d; IMvigor210 (mUC), Log-rank test, p = 0.0069, Fig. 6 h). Moreover, the number of immunotherapy responders were highest in group 1 and lowest in group 3 in both cohorts (TCGA-BLCA, Fisher’s exact test, p < 0.00001, Fig. 6e; IMvigor210 (mUC), Fisher’s exact test, p = 0.000015, Fig. 6i). Then, we observed a predictive advantage of TMERS compared with TMB via ROC analysis in the TCGA-BLCA cohort, whereas the opposite results were observed in the IMvigor210 (mUC) cohort (Fig. 6 g, k). Moreover, we found that compared with the TMB or TMERS alone, combining the TMB and TMERS improved the predictive value in the IMvigor210 (mUC) cohort (Fig. 6 k). Although the predictive value was slightly higher in the combination group than that in the TMERS alone group (Fig. 6e), TMB predictive value was not very high in the TCGA-BLCA cohort, again validating that the combination of the TMB and TMERS could be a more effective biomarker for predicting the ICIs immunotherapy response and prognosis.
Discussion
Instead of focusing only on tumour cells, increasing attention has recently been directed towards the TME, which consists of stromal cells, immune cells and the factors that they release around tumour cells. Tumour cells can reconstruct their surroundings to build the TME. Thus, the TME can not only interact with tumour cells to allow them to proliferate and protect them from apoptosis and metastasis but also play a vital role in therapeutic efficacy [39]. TME-mediated drug resistance is not usually permanent unless perpetual crosstalk occurs between the TME and tumour cells. When the components within the TME altered, drug sensitivity change. To kill tumour cells, effector immune cells must be released from the multiple immunosuppressive milieus, which constitute the TME, to counteract their own activation barriers. This effect has been achieved by ICIs, which target key inhibitory factors at the core of these networks.
ICIs exert a beneficial effect on survival in patients with advanced and metastatic MIBC, which was one of the first cancers to be recognized as immunogenic [40], highlighting the potential role for these immunotherapeutic drugs in facilitating potent anti-tumour immune responses. Notably, most patients with MIBC did not respond to ICIs or benefit from them, the mechanisms remain incompletely understood. In recent years, transcriptomic signatures representative of the TME have been identified, and their potential role in MIBC has been evaluated. Researchers have conducted immunogenomic analyses of the tumour immune microenvironment (TIME), its ability to predict levels of tumour inflammation and patient survival, as well as its correlation with the expression of immune checkpoints. Thus, many studies have also shown that infiltration of stromal cells, such as cancer-associated fibroblasts (CAFs), can exhaust immune cells and consequently reverse the survival benefit of immune infiltration through upregulation of immune checkpoints [41]. However, the roles of stromal cells and TME signature-related immune response status in MIBC have not yet been explored.
In the current study, we comprehensively elucidated the TME, including stromal cells status, via ESTIMATE algorithm by using transcriptomic data. Immune scores and stromal scores represent the diversity of immune and stromal cells within the TME in MIBC. The results showed that stromal scores were highly positive correlated with malignancy and negatively correlated with prognosis in MIBC patients, demonstrating the indispensable role of stromal cells within the TME in tumorigenesis from different perspectives. By merging the DEGs obtained from either immune or stromal cells, 788 TME DEGs were identified as candidate genes representing the TME patterns in MIBC. Furthermore, KM survival analyses and a machine-learning method—LASSO Cox regression analysis—were used to screen the optimal selected genes to establish a TMERS, which showed high accuracy in predicting mortality risks and the immunotherapy response in MIBC patients.
We found that patients in the high TMERS group had a markedly shorter OS and DFS than patients in the low TMERS group in the TCGA-BLCA cohort, and these findings were next validated in several independent cohorts. The ROC curves also revealed that our TMERS had a beneficial effect on prognosis prediction. Furthermore, we found that high TMERS was strongly positive correlated with high malignancy molecular subtypes including basal, TCGA III, IV, infiltrated, basal/SCC-like, while low TMERS concentrated on low malignancy and better prognostic molecular subtypes, such as luminal papillary, TCGA I, II, uroA, and GU. Moreover, the TMERSscores were highly associated with all prognostic malignancy clinicopathological characteristics, such as TNM stage and lymph node metastasis. The stratified survival analyses demonstrated that TMERS was an efficient independent prognostic predictor in all stratified clinicopathological features. Additionally, we found that the TMERS remained as an independent prognostic factor when compiling with clinicopathological characteristics in TCGA-BLCA and IMvigor210 (mUC) cohorts. All of these results suggested that the TMERS, which is representative of TME status, played a pro-oncogenic role and was capable of improving the prognostic accuracy of traditional clinical features in MIBC. The constructed nomogram offers a complementary perspective for specifically evaluating individual patients and may be a promising tool for clinical use in the future.
For the GSVA based on the TMERS, we observed that angiogenesis, EMT, inflammatory response, complement and IL2-STAT5 signalling, which are considered immunosuppressive and harmful to patients, were enriched in the high TMERS group. In addition, the DNA repair signalling pathway was enriched in the low TMERS group, indicating beneficial survival and immunotherapy responses. Functional annotation again demonstrated that the TMERS could be representative of TME patterns and was significantly associated with immune infiltration status in MIBC.
According to the consequences of clinical trials with ICIs, in situ immune cell infiltration was identified as critical retrospective information for tumour development and lateral immunotherapy responses [42, 43]. Thus, ssGSEA was performed to depict the TME immune landscape, namely, the infiltration status of 24 immune cell types in MIBC. Amazingly, we found that in patients with high TMERS, almost all immune cells were enriched, except for NK CD56bright cells, which are vigorous innate immune cells that can eradicate tumour cells [44]. Moreover, no relationship was found between the TMERS and the numbers of Th17 and CD8 + T cells, which are adaptive immune cells that are beneficial for patient survival. However, the results showed that effector T cells and cytotoxic cells were upregulated in the high TMERS group, which was unexpected and inconsistent with previous data. However, we cannot easily draw a conclusion as immune cell infiltration is dynamic. In our exploration of the immune landscape, we found that the numbers of robustly immunosuppressive cells, macrophages, pDCs and Tregs were markedly increased in the high TMERS group, far exceeding the numbers of T cells and cytotoxic cells. Macrophages, pDCs and Tregs can cooperate with each other to produce local IL-10 and TGF-β, deprive costimulatory ligands, neutralize the function of APCs, and directly or indirectly inhibit the actions and activation of T cells and NK cells within the TME, leading to immune response suppression and tumorigenesis. Notably, even if tumour-infiltrating lymphocytes (TILs) recognize tumour-specific antigens, the loss of co-stimulation or suppression of various pathways prevents effective TILs from clonal expansion or exerting anti-tumour function [45]. Therefore, we inferred that the activity of T cells, CD8 + T cells and cytotoxic cells, which played a central role in anti-tumour immunity among the TILs, was forcefully suppressed by immunosuppressive cells within the TME. Thus, we observed that the levels of Treg cells were significantly positively correlated with all the other immune cells, supporting our speculation and indicating the existence of a negative regulation process leading to an immunosuppressive milieu within the TME in MIBC. Accordingly, a supportive TME status was found to be crucial for initiating the adaptive immunity cascade to inhibit tumorigenesis. The poor prognosis of patients in the high TMERS group may be a result of this tumour immunosuppressive microenvironment. This assumption could be validated that high TMERS group mainly accumulated in infiltrated and basal/SCC-like molecular subtype, which is characterized by suppressive immune cells infiltration and activation of EMT/TGFβ pathway. Our TMERS provided an individual scoring system to expand the TME status in MIBC patients.
In addition to immune cell infiltration, some inhibitory molecules, which act as rheostats in the immune response, are also involved in priming TIL activation or function and restoring immune surveillance. These inhibitory molecules, namely, immune checkpoints, including CTLA-4 and PD-1/PD-L1, have been well described [46, 47]. The results demonstrated that nearly all immune checkpoints were upregulated in the high TMERS group, which was consistent with the survival analyses, prompting us to determine whether our TMERS could predict the response to ICIs. Encouragingly, the TMERSscore was proven to be predictive of immunotherapeutic benefits in the TCGA-BLCA and IMvigor210 (mUC) cohorts. These findings were further validated in cohorts of human metastatic melanoma patients receiving anti-PD-1 and anti-MAGE-A3 immunotherapy and mice receiving antibodies against CTLA-4 and PD-L1. Although the difference was statistically significance only in the mouse cohort, the TMERSscore was negatively associated with the immunotherapy response in all cohorts. And more immunotherapeutic responders were noticed in low TMERS groups than in the high TMERS groups. All of these results indicated that TMERS is a potent biomarker for predicting the immunotherapy response.
With the development of genome sequencing and bioinformatics, the mutational landscape is beginning to be regarded as a potent biomarker for responses to immunotherapies as initiation of the adaptive tumour immunity cascade relies on recognition of neo-antigens induced by somatic nonsynonymous coding mutations [48]. As the overall neo-antigen load is difficult to measure, the TMB, which can be easily detected and used to assess the neo-antigen load, has been proven to serve as either an indicator of clinical benefits or a prognostic factor with the potential to predict the ICI response [16, 17]. Considering their negative correlation, the predictive value for immunotherapy efficiency of the TMB alone or in combination with the TMERS was evaluated in the TCGA-BLCA and IMvigor210 (mUC) cohorts. The results showed that a high TMB was a favourable prognostic and predictive factor for survival and the ICIs treatment response in IMvigor210 (mUC) cohort. But we have noticed that high TMB could not predict immunotherapy response in TCGA-BLCA cohort. The reason might be relied on the distribution of Lund molecular subtypes. As mentioned above, patients within GU subtype were more likely to be immunotherapy responders, while response rate of patients within infiltrated subtype was lowest. We can see that high TMB group displayed a high proportion of GU subtype, but there is no significantly difference of infiltrated subtype between low and high TMB group in TCGA-BLCA cohort. Moreover, the GU and infiltrated subtypes were both differential distributed in low and high TMB group in IMvigor210 (mUC) cohort. And different distribution of GU and infiltrated subtypes in TMERS was significant in both cohorts. Furthermore, we surprisingly found that the combination of TMERS and TMB could stratify the MIBC patients more clearly and precisely, demonstrating that the prognosis of patients with high TMB/low TMERS was the best, while the prognosis of patients with low TMB/high TMERS was the worst in both cohorts. Moreover, the number of immunotherapy responders was highest in the high TMB/low TMERS group and lowest in the low TMB/high TMERS group. And more GU subtype and less infiltrated subtype located in high TMB/low TMERS group, which is consistent with the results described before. However, the ROC curves revealed that the TMERSscore had a predictive advantage over the TMB in the TCGA-BLCA cohort, while the opposite has been seen in the IMvigor210 (mUC) cohort. According to the predictive efficiency of the combination, we found that the TMERS contributed more in the TCGA-BLCA cohort, while the TMB gave greater contributions in IMvigor210 (mUC) cohorts. This indicated that as the patients varied, the predictive value of the TMERS and TMB was entirely different. All of these findings emphasize the importance of merging the TMERS and TMB to produce a more effective individual biomarker to predict prognosis and ICIs response.
Our study has some limitations as all cohorts utilized were retrospective. Therefore, the efficiency of the TMERS should be further validated in prospective studies. In addition, the platforms were very diverse, and using standard gene expression values through nanostring technologies is appropriate to avoid the consequences of applying uncertain cut-off values. Moreover, we should incorporate more clinical parameters into the TMERS scoring system to improve the prediction accuracy, as not all patients with a high TMB/low TMERS showed a favourable response to immunotherapy.
Conclusion
In conclusion, we comprehensively estimated the TME infiltration patterns associated with the cellular, molecular, and mutation features of MIBC and established a prognostic and predictive TMERS that could predict the response of ICIs when combined with the TMB. These findings have improved our understanding of immunotherapies and may provide useful guidance for their clinical use.
Abbreviations
- aDCs:
-
Activated DCs
- AUC:
-
Area under curve
- BCG:
-
Bacillus Calmette−Guerin
- BLCA:
-
Bladder cancer
- CAF:
-
Cancer associated fibroblasts
- CI:
-
Confidence interval
- DC:
-
Dendritic cells
- DCA:
-
Decision curve analysis
- CTLA-4:
-
Cytotoxic T-lymphocyte-associated protein 4
- DEGs:
-
Differential expression genes
- DFS:
-
Disease free survival
- ECM:
-
Extracellular matrix
- EMT:
-
Epithelial-mesenchymal transition
- ESTIMATE:
-
Estimation of STromal and Immune cells in MAlignant Tumours using Expression data
- FDA:
-
Food and Drug Administration
- FDR:
-
False discovery rate
- GAL9:
-
LGALS9
- GEO:
-
Gene Expression Omnibus
- GSEA:
-
Gene Set Enrichment Analysis
- GSVA:
-
Gene Set Variation Analysis
- GU:
-
Genomically unstable
- HE:
-
Hematoxylin and eosin
- HR:
-
Hazard ratio
- ICIs:
-
Immune-checkpoint inhibitors
- IHC:
-
Immunohistochemistry
- KM:
-
Kaplan–Meier
- LASSO:
-
Least absolute shrinkage and selection operator
- MIBC:
-
Muscle-invasive bladder cancer
- mUC:
-
Metastatic urothelial cancer
- NES:
-
Normalized enrichment score
- NKs:
-
Natural killer cells
- NMIBC:
-
Non-muscle-invasive bladder cancer
- OS:
-
Overall survival
- pDCs:
-
Plasmacytoid DC
- PD-1:
-
Programmed death-1
- PD-L1:
-
Programmed death-ligand-1
- ROC:
-
Receiver operating characteristic curve
- ssGSEA:
-
Single-sample gene set enrichment analysis
- TAM:
-
Tumour associated macrophages
- TCGA:
-
The Cancer Genome Atlas
- Tcm:
-
T central memory
- Tem:
-
T effector memory
- Th1:
-
T helper 1
- Th2:
-
T helper 2
- Th17:
-
T helper 17
- TIDE:
-
Tumour Immune Dysfunction and Exclusion
- TILs:
-
Tumour-infiltrated lymphocytes
- TIM-3:
-
HAVCR2
- TIME:
-
Tumour immune microenvironment
- TMB:
-
Tumour mutation burden
- TME:
-
Tumour microenvironment
- TMERS:
-
TME-related signature
- TNM:
-
Tumour Node Metastasis
- Treg:
-
Regulatory T cells
- Tγδ:
-
T follicular helper
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68(6):394–424. https://doi.org/10.3322/caac.21492
Morales A, Eidinger D, Bruce AW (1976) Intracavitary Bacillus Calmette-Guerin in the treatment of superficial bladder tumors. J Urol 116(2):180–183. https://doi.org/10.1016/s0022-5347(17)58737-6
Veeratterapillay R, Heer R, Johnson MI, Persad R, Bach C (2016) High-risk non-muscle-invasive bladder cancer-therapy options during intravesical BCG shortage. Curr Urol Rep 17(9):68. https://doi.org/10.1007/s11934-016-0625-z
Sanli O, Dobruch J, Knowles MA, Burger M, Alemozaffar M, Nielsen ME, Lotan Y (2017) Bladder cancer. Nat Rev Dis Prim 3:17022. https://doi.org/10.1038/nrdp.2017.22
Aine M, Eriksson P, Liedberg F, Hoglund M, Sjodahl G (2015) On molecular classification of bladder cancer: out of one, many. Eur Urol 68(6):921–923. https://doi.org/10.1016/j.eururo.2015.07.021
Sjodahl G, Lauss M, Lovgren K, Chebil G, Gudjonsson S, Veerla S, Patschan O, Aine M, Ferno M, Ringner M, Mansson W, Liedberg F, Lindgren D, Hoglund M (2012) A molecular taxonomy for urothelial carcinoma. Clin Cancer Res 18(12):3377–3386. https://doi.org/10.1158/1078-0432.ccr-12-0077-t
Damrauer JS, Hoadley KA, Chism DD, Fan C, Tiganelli CJ, Wobker SE, Yeh JJ, Milowsky MI, Iyer G, Parker JS, Kim WY (2014) Intrinsic subtypes of high-grade bladder cancer reflect the hallmarks of breast cancer biology. Proc Natl Acad Sci USA 111(8):3110–3115. https://doi.org/10.1073/pnas.1318376111
Choi W, Porten S, Kim S, Willis D, Plimack ER, Hoffman-Censits J, Roth B, Cheng T, Tran M, Lee IL, Melquist J, Bondaruk J, Majewski T, Zhang S, Pretzsch S, Baggerly K, Siefker-Radtke A, Czerniak B, Dinney CP, McConkey DJ (2014) Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell 25(2):152–165. https://doi.org/10.1016/j.ccr.2014.01.009
Cancer Genome Atlas Research N (2014) Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 507(7492):315–322. https://doi.org/10.1038/nature12965
Ugel S, De Sanctis F, Mandruzzato S, Bronte V (2015) Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest 125(9):3365–3376. https://doi.org/10.1172/jci80006
Shalapour S, Karin M (2015) Immunity, inflammation, and cancer: an eternal fight between good and evil. J Clin Invest 125(9):3347–3355. https://doi.org/10.1172/jci80007
Topalian SL, Drake CG, Pardoll DM (2015) Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 27(4):450–461. https://doi.org/10.1016/j.ccell.2015.03.001
Chen R, Zinzani PL, Lee HJ, Armand P, Johnson NA, Brice P, Radford J, Ribrag V, Molin D, Vassilakopoulos TP, Tomita A, von Tresckow B, Shipp MA, Lin J, Kim E, Nahar A, Balakumaran A, Moskowitz CH (2019) Pembrolizumab in relapsed or refractory Hodgkin lymphoma: 2-year follow-up of KEYNOTE-087. Blood 134(14):1144–1153. https://doi.org/10.1182/blood.2019000324
Robert C, Ribas A, Schachter J, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil CM, Lotem M, Larkin JMG, Lorigan P, Neyns B, Blank CU, Petrella TM, Hamid O, Su SC, Krepler C, Ibrahim N, Long GV (2019) Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): post hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol 20(9):1239–1251. https://doi.org/10.1016/s1470-2045(19)30388-2
Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, Dawson N, O’Donnell PH, Balmanoukian A, Loriot Y, Srinivas S, Retz MM, Grivas P, Joseph RW, Galsky MD, Fleming MT, Petrylak DP, Perez-Gracia JL, Burris HA, Castellano D, Canil C, Bellmunt J, Bajorin D, Nickles D, Bourgon R, Frampton GM, Cui N, Mariathasan S, Abidoye O, Fine GD, Dreicer R (2016) Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 387(10031):1909–1920. https://doi.org/10.1016/s0140-6736(16)00561-4
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, Ibrahim F, Bruggeman C, Gasmi B, Zappasodi R, Maeda Y, Sander C, Garon EB, Merghoub T, Wolchok JD, Schumacher TN, Chan TA (2015) Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348(6230):124–128. https://doi.org/10.1126/science.aaa1348
Gibney GT, Weiner LM, Atkins MB (2016) Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol 17(12):e542–e551. https://doi.org/10.1016/s1470-2045(16)30406-5
Pitt JM, Marabelle A, Eggermont A, Soria JC, Kroemer G, Zitvogel L (2016) Targeting the tumor microenvironment: removing obstruction to anticancer immune responses and immunotherapy. Ann Oncol 27(8):1482–1492. https://doi.org/10.1093/annonc/mdw168
Hanahan D, Coussens LM (2012) Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 21(3):309–322. https://doi.org/10.1016/j.ccr.2012.02.022
Gajewski TF, Schreiber H, Fu YX (2013) Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 14(10):1014–1022. https://doi.org/10.1038/ni.2703
Gautier L, Cope L, Bolstad BM, Irizarry RA (2004) affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20(3):307–315. https://doi.org/10.1093/bioinformatics/btg405
Robertson AG, Kim J, Al-Ahmadie H, Bellmunt J, Guo G, Cherniack AD, Hinoue T, Laird PW, Hoadley KA, Akbani R, Castro MAA, Gibb EA, Kanchi RS, Gordenin DA, Shukla SA, Sanchez-Vega F, Hansel DE, Czerniak BA, Reuter VE, Su X, de Sa Carvalho B, Chagas VS, Mungall KL, Sadeghi S, Pedamallu CS, Lu Y, Klimczak LJ, Zhang J, Choo C, Ojesina AI, Bullman S, Leraas KM, Lichtenberg TM, Wu CJ, Schultz N, Getz G, Meyerson M, Mills GB, McConkey DJ, Network TR, Weinstein JN, Kwiatkowski DJ, Lerner SP (2017) Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell 171(3):540–556. https://doi.org/10.1016/j.cell.2017.09.007
Cao R, Yuan L, Ma B, Wang G, Qiu W, Tian Y (2020) An EMT-related gene signature for the prognosis of human bladder cancer. J Cell Mol Med 24(1):605–617. https://doi.org/10.1111/jcmm.14767
Mayakonda A, Lin DC, Assenov Y, Plass C, Koeffler HP (2018) Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res 28(11):1747–1756. https://doi.org/10.1101/gr.239244.118
Gu Z, Eils R, Schlesner M (2016) Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32(18):2847–2849. https://doi.org/10.1093/bioinformatics/btw313
Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, Schrock A, Campbell B, Shlien A, Chmielecki J, Huang F, He Y, Sun J, Tabori U, Kennedy M, Lieber DS, Roels S, White J, Otto GA, Ross JS, Garraway L, Miller VA, Stephens PJ, Frampton GM (2017) Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 9(1):34. https://doi.org/10.1186/s13073-017-0424-2
Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W, Trevino V, Shen H, Laird PW, Levine DA, Carter SL, Getz G, Stemke-Hale K, Mills GB, Verhaak RG (2013) Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 4:2612. https://doi.org/10.1038/ncomms3612
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43(7):e47. https://doi.org/10.1093/nar/gkv007
Friedman J, Hastie T, Tibshirani R (2010) Regularization Paths for Generalized Linear Models via Coordinate Descent. J Stat Softw 33(1):1–22
Heagerty PJ, Zheng Y (2005) Survival model predictive accuracy and ROC curves. Biometrics 61(1):92–105. https://doi.org/10.1111/j.0006-341x.2005.030814.x
Hanzelmann S, Castelo R, Guinney J (2013) GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14:7. https://doi.org/10.1186/1471-2105-14-7
Iasonos A, Schrag D, Raj GV, Panageas KS (2008) How to build and interpret a nomogram for cancer prognosis. J Clin Oncol 26(8):1364–1370. https://doi.org/10.1200/jco.2007.12.9791
Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A, Bruneval P, Fridman WH, Becker C, Pages F, Speicher MR, Trajanoski Z, Galon J (2013) Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 39(4):782–795. https://doi.org/10.1016/j.immuni.2013.10.003
Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, Li Z, Traugh N, Bu X, Li B, Liu J, Freeman GJ, Brown MA, Wucherpfennig KW, Liu XS (2018) Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med 24(10):1550–1558. https://doi.org/10.1038/s41591-018-0136-1
Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R, Jhunjhunwala S, Banchereau R, Yang Y, Guan Y, Chalouni C, Ziai J, Senbabaoglu Y, Santoro S, Sheinson D, Hung J, Giltnane JM, Pierce AA, Mesh K, Lianoglou S, Riegler J, Carano RAD, Eriksson P, Hoglund M, Somarriba L, Halligan DL, van der Heijden MS, Loriot Y, Rosenberg JE, Fong L, Mellman I, Chen DS, Green M, Derleth C, Fine GD, Hegde PS, Bourgon R, Powles T (2018) TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554(7693):544–548. https://doi.org/10.1038/nature25501
Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, Seja E, Lomeli S, Kong X, Kelley MC, Sosman JA, Johnson DB, Ribas A, Lo RS (2016) Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 165(1):35–44. https://doi.org/10.1016/j.cell.2016.02.065
Ulloa-Montoya F, Louahed J, Dizier B, Gruselle O, Spiessens B, Lehmann FF, Suciu S, Kruit WH, Eggermont AM, Vansteenkiste J, Brichard VG (2013) Predictive gene signature in MAGE-A3 antigen-specific cancer immunotherapy. J Clin Oncol 31(19):2388–2395. https://doi.org/10.1200/jco.2012.44.3762
Zemek RM, De Jong E, Chin WL, Schuster IS, Fear VS, Casey TH, Forbes C, Dart SJ, Leslie C, Zaitouny A, Small M, Boon L, Forrest ARR, Muiri DO, Degli-Esposti MA, Millward MJ, Nowak AK, Lassmann T, Bosco A, Lake RA, Lesterhuis WJ (2019) Sensitization to immune checkpoint blockade through activation of a STAT1/NK axis in the tumor microenvironment. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aav7816
Wu T, Dai Y (2017) Tumor microenvironment and therapeutic response. Cancer Lett 387:61–68. https://doi.org/10.1016/j.canlet.2016.01.043
Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, Bellmunt J, Burris HA, Petrylak DP, Teng SL, Shen X, Boyd Z, Hegde PS, Chen DS, Vogelzang NJ (2014) MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 515(7528):558–562. https://doi.org/10.1038/nature13904
Thommen DS, Schumacher TN (2018) T cell dysfunction in cancer. Cancer Cell 33(4):547–562. https://doi.org/10.1016/j.ccell.2018.03.012
Lee JM, Lee MH, Garon E, Goldman JW, Salehi-Rad R, Baratelli FE, Schaue D, Wang G, Rosen F, Yanagawa J, Walser TC, Lin Y, Park SJ, Adams S, Marincola FM, Tumeh PC, Abtin F, Suh R, Reckamp KL, Lee G, Wallace WD, Lee S, Zeng G, Elashoff DA, Sharma S, Dubinett SM (2017) Phase I trial of intratumoral injection of CCL21 gene-modified dendritic cells in lung cancer elicits tumor-specific immune responses and CD8(+) t-cell infiltration. Clin Cancer Res 23(16):4556–4568. https://doi.org/10.1158/1078-0432.ccr-16-2821
Hegde PS, Karanikas V, Evers S (2016) The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition. Clin Cancer Res 22(8):1865–1874. https://doi.org/10.1158/1078-0432.ccr-15-1507
Wagner JA, Rosario M, Romee R, Berrien-Elliott MM, Schneider SE, Leong JW, Sullivan RP, Jewell BA, Becker-Hapak M, Schappe T, Abdel-Latif S, Ireland AR, Jaishankar D, King JA, Vij R, Clement D, Goodridge J, Malmberg KJ, Wong HC, Fehniger TA (2017) CD56bright NK cells exhibit potent antitumor responses following IL-15 priming. J Clin Invest 127(11):4042–4058. https://doi.org/10.1172/jci90387
Li Z, Chen G, Cai Z, Dong X, Qiu L, Xu H, Zeng Y, Liu X, Liu J (2019) Genomic and transcriptional Profiling of tumor infiltrated CD8(+) T cells revealed functional heterogeneity of antitumor immunity in hepatocellular carcinoma. Oncoimmunology 8(2):e1538436. https://doi.org/10.1080/2162402x.2018.1538436
Dempke WCM, Fenchel K, Uciechowski P, Dale SP (2017) Second- and third-generation drugs for immuno-oncology treatment-The more the better? Eur J Cancer 74:55–72. https://doi.org/10.1016/j.ejca.2017.01.001
Le Mercier I, Lines JL, Noelle RJ (2015) Beyond CTLA-4 and PD-1, the generation Z of negative checkpoint regulators. Front Immunol 6:418. https://doi.org/10.3389/fimmu.2015.00418
Fancello L, Gandini S, Pelicci PG, Mazzarella L (2019) Tumor mutational burden quantification from targeted gene panels: major advancements and challenges. J Immunother Cancer 7(1):183. https://doi.org/10.1186/s40425-019-0647-4
Acknowledgements
The authors are grateful for the invaluable support and useful discussions with other members of the Department of Urology. We thank the TCGA and GEO databases as well as IMvigor210 package for the availability of the data.
Funding
This work was supported by grants from National Natural Science Foundation of China (Grant No. 81802550), China Postdoctoral Science Foundation (Grant No. 2019M660041) and Beijing Postdoctoral Research Foundation (Grant No. ZZ2019‐04).
Author information
Authors and Affiliations
Contributions
All authors read and approved the final manuscript. RC, LY made substantial contributions to conception and design of the research. RC, LY and BM integrated and analysed the data. RC, LY and YT wrote the paper. RC, LY, BM, GW and TY edited the manuscript and provided critical comments. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Informed consent
All the analysed data used in our research were collected from a public database, such as TCGA and GEO as well as IMvigor210 package and so on; therefore, informed consent was not required for this analysis.
Ethical approval
Since this was a retrospective study and all the data were collected from a public database TCGA and GEO or IMvigor210 package and so on, therefore, ethical approval was not required.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Cao, R., Yuan, L., Ma, B. et al. Tumour microenvironment (TME) characterization identified prognosis and immunotherapy response in muscle-invasive bladder cancer (MIBC). Cancer Immunol Immunother 70, 1–18 (2021). https://doi.org/10.1007/s00262-020-02649-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-020-02649-x